

Abstract
Nogo-A is one of the most potent oligodendrocyte-derived inhibitors for axonal regrowth in the injured adult CNS. However, the physiological function of Nogo-A in development and in healthy oligodendrocytes is still unknown. In the present study, we investigated the role of Nogo-A for myelin formation in the developing optic nerve. By quantitative real-time PCR, we found that the expression of Nogo-A increased faster in differentiating oligodendrocytes than that of the major myelin proteins MBP (myelin basic protein), PLP (proteolipid protein)/DM20, and CNP (2′,3′-cyclic nucleotide 3′-phosphodiesterase). The analysis of optic nerves and cerebella of mice deficient for Nogo-A (Nogo-A−/−) revealed a marked delay of oligodendrocyte differentiation, myelin sheath formation, and axonal caliber growth within the first postnatal month. The combined deletion of Nogo-A and MAG caused a more severe transient hypomyelination. In contrast to MAG−/− mice, Nogo-A−/− mutants did not present abnormalities in the structure of myelin sheaths and Ranvier nodes. The common binding protein for Nogo-A and MAG, NgR1, was exclusively upregulated in MAG−/− animals, whereas the level of Lingo-1, a coreceptor, remained unchanged. Together, our results demonstrate that Nogo-A and MAG are differently involved in oligodendrocyte maturation in vivo, and suggest that Nogo-A may influence also remyelination in pathological conditions such as multiple sclerosis.
Introduction
In the CNS, the myelinating cells are the oligodendrocytes that derive from oligodendrocyte precursor cells (OPCs) generated in the ventricular zone (Kessaris et al., 2006). As oligodendrocytes differentiate and produce myelin sheets, they start expressing high levels of myelin basic protein (MBP) and proteolipid protein (PLP) constituting most of the myelin protein content. The central role of those proteins in myelination is illustrated by defect in structure stability of the myelin sheath in MBP or PLP knock-out (KO) mice (Popko et al., 1987; Griffiths et al., 1998). In contrast, little is known about the functional role of other myelin proteins in oligodendrocyte differentiation.
Nogo-A is a high molecular weight transmembrane protein that was initially identified as a potent myelin-associated inhibitor for axonal growth expressed mostly by oligodendrocytes (Caroni and Schwab, 1988; Chen et al., 2000). In the injured spinal cord, treatments with neutralizing antibodies against Nogo-A or Nogo-A gene deletion stimulate long-range axonal growth and improve locomotor functions (Schwab, 2004; Dimou et al., 2006). Nogo-A binds to a multisubunit receptor complex including the Nogo66 receptor NgR1 (Fournier et al., 2001), the adaptor molecule Lingo-1 (Mi et al., 2004), and the effector components p75/Troy (Shao et al., 2005). The stimulation of this complex at the surface of neurons leads to the intracellular activation of the small GTPase RhoA that mediates actin depolymerization and thereby the collapse or retraction of neurites (Yamashita et al., 2005). The same molecular cascade can be elicited by MAG and oligodendrocyte myelin glycoprotein (OMgp), which are structurally different from Nogo-A but bind also to the NgR–p75 receptor complex (Wang et al., 2002a).
Molecules related to Nogo-A signaling, such as MAG and Lingo-1, have been shown to be involved in oligodendrocyte development. Lingo-1 was recently shown to be a negative regulator of oligodendrocyte differentiation in vitro and in vivo (Mi et al., 2005). MAG, a cell surface Ig superfamily protein, is present at the periaxonal membrane of oligodendrocytes where it mediates axon–oligodendrocyte adhesion (Owens et al., 1990; Owens and Bunge, 1991). Surprisingly, constitutive MAG−/− mice showed only a slight delay in myelination and subtle abnormalities in the myelin structure (Li et al., 1994; Montag et al., 1994). This could be attributable to compensating mechanisms involving, for example, Nogo-A, which, like MAG, can bind the NgR complex. In the first 2 postnatal weeks, Nogo-A protein and mRNA levels were detected in many neurons including retinal ganglion cells, in addition to oligodendrocytes (Huber et al., 2002; Wang et al., 2002b). This period corresponds to oligodendrocyte differentiation and to the beginning of myelination. It is thus possible that the developmentally regulated expression of Nogo-A participates to axoglial contact and/or oligodendrocyte maturation.
In this study, we investigated the differentiation of oligodendrocytes and myelination in knock-out animals deficient for Nogo-A or MAG or in double knock-outs (DOKOs). The ablation of Nogo-A caused a severe but transient impairment of oligodendrocyte differentiation and production of myelin. In contrast to MAG, however, the lack of Nogo-A did not alter the structure of myelin or the node of Ranvier.
Materials and Methods
Animals: knock-out animal generation.
Animal procedures were performed in agreement with the guidelines of the Veterinary Office of the Canton of Zurich. Nogo-A knock-out mice were generated by homologous recombination of exons 2 and 3 in Nogo-A gene as described previously (Simonen et al., 2003). The MAG-null mice were obtained by homologous recombination of mag gene from exons 5–8 including the region encoding the signal sequence (Montag et al., 1994). For single and double knock-out mice, the strain background reached >99% C57BL/6. Double knock-out mutants were obtained by crossing Nogo-A−/− and MAG−/− mice. The transgenic strain was routinely genotyped by PCRs using the following primer sequences for Nogo-A−/− phenotype: 5′-AGT GAG TACCCAGCT GCA C-3′, 5′-CCT ACC CGG TAG AAT ATC GAT AAG C-3′, 5′-TGC TTT GAA TTA TTC CAAGTA GTC C-3′; and for MAG−/− phenotype: 5′-TTG GCG GCG AAT GGG CTG AC-3′, 5′-GCA GGG AAT GGA GAC ACA CG-3′, 5′-CAC CCT GCC GCT GTT TTG GAT AAT-3′.
Quantitative real-time PCR.
At different ages ranging from postnatal day 0 (P0) to adulthood, cerebella, optic nerves, or retinas were rapidly dissected in RNAlater solution (Ambion) after cervical dislocation. The tissues were placed in Eppendorf tubes, flash frozen in liquid nitrogen, and stored at −80°C until RNA extraction. The RNeasy RNA isolation kit (QIAGEN) was used for retinal and cerebellar samples, whereas optic nerve RNA was prepared with the RNeasy Micro kit (QIAGEN). Residual genomic DNA was digested by DNase treatment. For reverse transcription, the same amounts of total RNA were transformed by oligo-dT and M-MLV reverse transcriptase (Promega). For all tissues, cDNAs corresponding to 5 ng of total RNA were amplified with specific primers designed to span intronic sequences or cover exon–intron boundaries. Primers specific for CNP (forward, 5′-AGGAGAAGCTTGAGCTGGTC; reverse, 3′-CGATCTCTTCACCACCTCCT), GAPDH (forward, 5′-CAGCAATGCATCCTGCACC; reverse, 3′-TGGACTGTGGTCATGAGCCC), LINGO1 (forward, 5′-AAGTGGCCAGTTCATCAGGT; reverse, 3′-TGTAGCAGAGCCTGACAGCA), MAG (forward, 5′-GATGATATTCCTCGCCACC; reverse, 3′-ACTGACCTCCACTTCCGTT), MBP (forward, 5′-CACACACGAGAACTACCCA; reverse, 3′-GGTGTTCGAGGTGTCACAA), NgR1/RTN4R (forward, 5′-CTCGACCCCGAAGATGAAG; reverse, 3′-TGTAGCACACACAAGCACCAG), NogoA/RTN4 (forward, 5′-CAGTGGATGAGACCCTTTTTG; reverse, 3′-GCTGCTCCTTCAAATCCATAA), PLP/DM20 (forward, 5′-TCAGTCTATTGCCTTCCCTAGC; reverse, 3′-AGCATTCCATGGGAGAACAC). Gene expression was analyzed by real-time reverse transcription (RT)-PCR with a polymerase ready mix (LightCycler 480; SYBR Green I Master; Roche) and a thermocycler (LightCycler; Roche Diagnostics). The analysis of the melting curve of each amplified PCR product and the visualization of the PCR amplicons on 1.5% agarose gels allowed to control the specificity of the amplification. For relative quantification of gene expression, mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the comparative threshold cycle (ΔΔCT) method. For calibration, a control sample was used to calculate the relative values, as indicated in the figure legend. Each reaction was done in triplicate.
Immunohistochemistry.
At different ages (P0 to adult), mice were killed by injecting an overdose of anesthetic intraperitoneally. After intracardiac perfusion with PBS (0.1 m) and 4% paraformaldehyde (PFA), the eyes and the brain were rapidly dissected and postfixed in 4% PFA for 48 h at 4°C. The tissues were cryoprotected in 30% sucrose overnight, and then frozen in OCT (optimal cutting temperature) compound (Tissue-Tek; Sakura Finetek) with a liquid nitrogen-cooled bath of 2-methylbutane. Longitudinal brain sections (20 μm), optic nerves sections (10 μm), and retinal sections (12 μm) were cut with a cryostat and collected on Superfrost Plus slides (Menzel-Glaser). For immunohistochemistry procedure, tissue slices were blocked with 5% BSA, 3% normal serum, 0.3% Triton X-100 in PBS for 1 h at room temperature to avoid unspecific crossreactivity. Then, primary antibodies were applied in 5% BSA, 3% normal serum, 0.3% Triton X-100 in PBS overnight at 4°C. After PBS washes, sections were incubated with the appropriate secondary antibody for 1 h at room temperature and mounted with MOWIOL anti-fading medium [10% Mowiol 4–88 (w/v) (Calbiochem) in 100 mm Tris, pH 8.5, 25% glycerol (w/v), and 0.1% DABCO (1,4-diazabicyclo[2.2.2]octane)]. Primary antibodies were as follows: rabbit anti-Nogo-A [Laura; Rb173A (Oertle et al., 2003)] serum (1:200), rabbit anti-MBP (1:1000; Millipore Bioscience Research Reagents; AB980), mouse anti-green fluorescent protein (GFP) (clone 3E6; Invitrogen), rabbit anti L-MAG (1:500; gift from Dr. N. Schaeren-Wiemers, University Hospital Basel, Pharmacenter, Basel, Switzerland), rabbit anti-olig2 (1:4000–1:10,000; provided by Dr. C. Stiles, Harvard University, Boston, MA), mouse anti-adenomatous polyposis coli (APC) (1:1000; clone CC-1; Calbiochem), guinea pig anti-NG2 (1:500; given by Dr. W. Stallcup, Burnham Institute for Medical Research, Cancer Research Center, La Jolla, CA). Immunofluorescent labelings were analyzed with Zeiss Axioskop 2 Plus microscope (Carl Zeiss) and images were taken with a CCD video camera.
Paranodin/sodium channel labeling.
Alternatively, for paranodin/Caspr and sodium channel double stainings in the optic nerve, fresh frozen cryosections were cut immediately after animal decapitation and placed at −80°C. Slides were thawed and dried at room temperature, fixed in absolute methanol for 20 min at −20°C, and washed three times with PBS. As described previously, the preincubation was done for 30 min to 1 h at room temperature. Mouse anti-pan sodium channel (1:50–1:100; clone K58/35; Sigma-Aldrich) and rabbit anti-paranodin (1:500; gift from Dr. J.-A. Girault, Université Pierre et Marie Curie, Paris, France) were applied on slices and left overnight at 4°C. For sodium channel (NaCh) staining, a horse anti-mouse biotin-coupled antibody (1:100; Vector) was added for 2 h at room temperature and a third incubation was performed with streptavidin linked to Alexa 488 (1:400; Invitrogen). A goat anti-rabbit antibody (1:1000; Invitrogen) was used to detect paranodin. Image stacks were acquired by confocal microscopy in the chiasmal region (caudal) of the optic nerve. Two to four picture stacks were sampled in a slice volume of 119.05 × 119.05 × 5 μm per animal and for the different groups. Calculations of paranodin and NaCh clusters were made on merged pictures in NIH software.
Western blot.
Mice were killed by cervical dislocation and retinas, cerebellum, or optic nerve were quickly dissected out and flash frozen in liquid nitrogen. Tissues were stored at −80°C until extraction in lysis buffer (10 mm Tris-HCl, 1% Nonidet 40, 150 mm NaCl, 0.1% SDS, 1% deoxycholate, pH 7.4) containing protease inhibitors (Complete Mini; Roche Diagnostics). The samples were homogenized on ice for 30 min. After centrifugation for 15 min at 10,000 × g and 4°C, the supernatant was retrieved in new Eppendorf tubes and kept at −80°C or −20°C. Proteins (50–100 μg) were separated by electrophoresis on a 4–12% polyacrylamide gel and transferred to polyvinylidene difluoride Immobilon P membranes. Blots were preincubated in a blocking solution of 5% BSA in 0.2% TBST (0.1 m Tris base, 0.2% Tween 20, pH 7.4) for 1 h at room temperature, incubated with primary antibodies overnight at 4°C and after washing, with a horseradish peroxidase-conjugated anti-rabbit antibody (1:10,000–1:25,000; Pierce Biotechnology). Primary antibodies were rabbit anti-MBP (1:250–1:500; Millipore Bioscience Research Reagents), rabbit anti-NgR1 (1:250–1:500; R&D Systems), rabbit anti-Lingo-1 (1:500; Abcam), and anti-GAPDH (1:10,000; Abcam). Protein bands were detected by adding SuperSignal West Pico Chemiluminescent Substrate (Pierce) by exposing the blot in a Stella detector (Raytest). Densitometry analysis was performed with NIH software and by normalizing the band intensities to GAPDH values.
Electron microscopy.
Mice were perfused intracardially at P15 and at P28 with a solution containing 2% of PFA and 2% of glutaraldehyde diluted in Sorensen buffer (0.067 m). Immediately after perfusion, optic nerves were dissected and postfixed for 3 d in the same fixative mixture at 4°C. The midoptic nerves were cut in two pieces with scissors and then incubated for 3 h in 2% osmium tetroxide (PB; 0.1 m) at room temperature under gentle rotary agitation. The tissues were dehydrated in graded baths of ethanol, immersed in propylene oxide, and embedded in epoxy resin. Polymerization was made at 60°C for 5 d. Semithin cross-sections of 0.7 μm in thickness were cut for light microscopy inspection, whereas ultrathin sections of 50 nm were cut for electron microscopy analysis. Pictures were taken randomly in central and peripheral optic nerves at 4400 or 8800×.
Results
Nogo-A is developmentally regulated in neurons and oligodendrocytes
We monitored Nogo-A mRNA expression changes in glial and neuronal cells by quantitative RT-PCR (qRT-PCR) in the developing mouse optic nerve and in the retina, respectively (Fig. 1). The visual system was ideal to discriminate neuronal from glial Nogo-A because the optic nerve is deprived of neuronal cell bodies and the retina does not contain oligodendrocytes in rodents. Nogo-A mRNA expression increased 5- to 10-fold after P3 in the optic nerve, preceding that of the main structural myelin proteins MBP, PLP, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) by 2–3 d (Fig. 1A). The maximal mRNA expression for all these oligodendrocyte proteins was reached at P15. Only Nogo-A mRNA was maintained at a high level in the adult, whereas MBP, PLP/DM20, CNP, and MAG dropped by ∼60, ∼72, ∼84, and ∼85%, respectively. The elevation of MBP mRNA by RT-PCR was correlated with the detection of MBP protein by immunohistochemistry, in the rostral optic nerve (data not shown). MBP immunostaining then spreads in a rostral-to-caudal direction as previously reported (Foran and Peterson, 1992). Interestingly, the increase of Nogo-A mRNA in the optic nerve (>400%) between birth and adulthood was paralleled by a ∼50% decrease in the retina (Fig. 1B). By immunohistochemistry at P8, Nogo-A was detected in many presumptive oligodendrocytes of P8 optic nerves (Fig. 1C,E,F,H, arrowheads), whereas only a few of those cells coexpressed the myelin proteins L-MAG (Fig. 1D,E, stars) or CNP (Fig. 1G,H, stars). The detection of Nogo-A protein by immunohistochemistry corroborates the qRT-PCR results suggesting that Nogo-A expression precedes other oligodendrocytic markers during development. Moreover, Nogo-A was located in the retinal ganglion cell layer and in optic axons at P1, whereas in the adult the Nogo-A signal was restricted to the innermost layer of the retina and became undetectable in the oligodendrocyte-free part of the optic nerve (Fig. 1I). Control stainings on Nogo-A−/− sections or by omitting the incubation with the primary antibody on wild-type (WT) slices failed to provide a signal (data not shown). Nevertheless, as oligodendrocyte differentiation was proceeding at P10, Nogo-A coexisted in unmyelinated axons and in the processes of differentiating, PLP-GFP-positive oligodendrocytes in the optic nerve (Fig. 1J), suggesting that the neuron-to-glia switch of Nogo-A expression occurs progressively. As expected, MAG was only observed in oligodendrocyte cell bodies and processes (Fig. 1K). Together, these results show that glial Nogo-A upregulation occurs rapidly and early at the time of oligodendrocyte differentiation, whereas in neurons Nogo-A expression is progressively downregulated.

Nogo-A is inversely regulated in oligodendrocytes and retinal neurons during development. A, The mRNA expression of myelin-associated proteins was followed by qRT-PCR in the optic nerve from birth until adulthood (Ad). The elevation of Nogo-A mRNA preceded that of other markers for oligodendrocytes by a few days. B, The transcripts of Nogo-A dramatically increased after P3 in the oligodendrocytes of the optic nerve but underwent a parallel downregulation in the ganglion cell layer of the oligodendrocyte-free retina. Error bars indicate SEM. C–F, Single-scan confocal pictures of P8 optic nerve sections showed that Nogo-A was present in typical rows of interfascicular oligodendrocytes. Only a few of these immature Nogo-A-positive cells also showed a staining for L-MAG (D) or for CNP (G) as shown by superimposing the two labelings (E, H, stars). Most of the Nogo-A expressing cells were not yet expressing L-MAG or CNP staining (arrowheads). I, By immunohistochemistry, the signal of Nogo-A was very intense in the retinal ganglion cell layer and in the optic nerve (ON) axons at P1. In the adult retina, the weak Nogo-A labeling was restricted to the innermost layer of the retina. J, Double immunostainings for Nogo-A (red) and PLP-GFP (green) in P15 optic nerve are shown by single-scan confocal microscopy. The superimposition of the two stainings shows the presence of Nogo-A in axon fascicles (small arrows) and the coexpression of Nogo-A and PLP in differentiated oligodendrocytes (yellow, large arrowhead) and their processes. K, Unlike Nogo-A, L-MAG (red) and PLP-GFP (green) are exclusively found in oligodendrocytes (yellow) in the optic nerve. ON, Optic nerve. Scale bars: C–H, 100 μm; I, 200 μm; J, K, 50 μm.
Nogo-A and/or MAG suppression does not prevent oligodendrocyte precursor cell migration in the optic nerve
Because Nogo-A was early detected in the retinal ganglion cell axons and oligodendrocytes, we speculated that Nogo-A may influence the migration of OPCs. In the CNS, the optic nerve is a place of choice to observe OPC invasion occurring from the optic chiasm to the optic nerve head (Small et al., 1987; Colello et al., 1995; Ueda et al., 1999; Sugimoto et al., 2001). OPCs were visualized by using an antibody directed against Olig2, a bHLH (basic helix-loop-helix) transcription factor specifically expressed in OPC nuclei (Lu et al., 2000; Zhou et al., 2000). On longitudinal sections, a small number of Olig2-positive [Olig2-(+)] cells emerged from the optic chiasm at embryonic day 17.5 (E17.5) and then invaded the optic nerve in a chiasma-to-retina gradient (Fig. 2A). As expected, OPCs did not penetrate into the retina and stopped migrating in the optic nerve head by P4. Until P6, Olig2 was found in glial precursors immunostained for NG2 (data not shown). It persisted in differentiated oligodendrocytes that were recognized by PLP-GFP staining at P10 or APC antigen at P15 (data not shown). At the different ages, the density of olig2-labeled cells per square millimeter of optic nerve was evaluated in WT, Nogo-A−/−, MAG−/−, and DOKO animals (Fig. 2B). In WT, the density of Olig2-(+) cells increased fourfold between P0 and P6 and peaked at P10. Then, the density of Olig2-(+) cells steadily declined at P15 and P28. In single knock-out mice, increase and decline of Olig2-(+) cells clearly followed that of WT. In contrast, the density of Olig2-(+) cells in DOKO plateaued after P6 but reached WT levels at P15 and P28 (Figs. 1A, 2B, insets). Quantitatively, the reduction of labeled cells in DOKO represented ∼26% at P8 and ∼29% at P10 of the WT group values.

Effects of Nogo-A and MAG deletion on oligodendrocyte precursor cell number and migration in the optic nerve. A, Optic nerve longitudinal sections (10 μm thick) were labeled with an antibody recognizing Olig2 in oligodendrocyte precursor cells and in mature oligodendrocytes between E17.5 and P28. Olig2-stained OPC cells migrated from the optic chiasm (OC) at E17.5 and colonized the optic nerve until the optic nerve head (ONH) at later stages. Inset, Comparative examination of Olig2 staining in the rostral region of the WT and DOKO optic nerve showed a lower density of cells in DOKO than WT at P8 but not at P15. B, Quantitative analysis of the density of Olig2-labeled cells in the optic nerve of WT, Nogo-A−/−, MAG−/−, and DOKO mice between P0 and P28. The average density (mean ± SEM) of cells was normalized to the surface of the optic nerve. For each time point, two to seven animals were examined. Scale bars: A, 400 μm; inset, 100 μm.
Deletion of Nogo-A or MAG induces a delay in oligodendrocyte differentiation in the CNS
To observe oligodendrocyte differentiation, we followed the expression of the major oligodendrocyte and myelin markers MBP, PLP, and CNP in optic nerves and cerebella by immunohistochemistry, qRT-PCR, and Western blot analysis (Fig. 3). In the optic nerve of Nogo-A−/− or MAG−/− P8 mouse pups, MBP immunostaining was severely decreased compared with WT animals and an even more dramatic reduction was found in the DOKO group, in which MBP staining was restricted to the most rostral part of the optic nerve (Fig. 3A). Consistent with this, Nogo-A−/− and MAG−/− cerebella presented less MBP-positive axons in the granule cell layer and reduced staining of the white matter at P15 compared with WT mice. Again, an even more striking reduction was observed in the DOKO pups that showed only a few MBP-positive fibers in the granule cell layer (GCL) (Fig. 3B). Surprisingly, the qRT-PCR analysis revealed that the Nogo-A−/− mice had the most marked reductions of MBP, PLP/DM20, and CNP mRNA levels, whereas MAG−/− animals presented virtually no difference relative to the WT group in the cerebellum and in the optic nerve at P10. In the DOKO animals, myelin protein mRNA levels were similar to those of MAG−/− mice in the optic nerve, but equal to Nogo-A−/− mice in the cerebellum (Fig. 3C,D). Western blot analysis showed a strong reduction of the four MBP isoforms in the cerebella of Nogo-A−/−, MAG−/−, and DOKO mice at P15 (Fig. 3E). These results suggest that the absence of Nogo-A compromised myelin protein synthesis, whereas absence of MAG may primarily affect deposition and myelin formation or stability. At P35, the amount of MBP reached comparable levels in all four groups, suggesting that oligodendrocyte differentiation is delayed rather than prevented by the absence of Nogo-A or MAG.

Oligodendrocyte differentiation and myelin protein synthesis are impaired in Nogo-A- and MAG-deficient mice. A, B, Oligodendrocyte differentiation was visualized by MBP immunostaining in longitudinal sections of optic nerves at P8 (A) and of cerebella at P15 (B). The intensity of MBP was substantially weaker in mutant optic nerves and cerebella than in WT tissues. MBP staining was the lowest for the DOKO group in which the signal was restricted to the optic nerve head (arrowhead) and was particularly low in the granule cell layer of the cerebellum (GCL) and in the white matter. C, D, The relative mRNA levels of MBP, PLP, and CNP were analyzed at P10 in the optic nerves and in the cerebella by qRT-PCR. At least three animals per group were analyzed in triplicate. For each myelin protein, the mRNA level was normalized using GAPDH as internal standard and represented in percentage of WT values. The most noticeable reductions of myelin protein mRNAs appeared in Nogo-A−/− tissues. Error bars indicate SEM. E, MBP protein isoforms were strongly reduced in all three KO cerebella at P15 as seen by Western blots (50 μg proteins/lane). At P35, no difference was noted. Scale bars: A, B, 200 μm.
Developing optic nerves of Nogo-A−/−, MAG−/−, and DOKO mice are hypomyelinated
We analyzed myelin formation in optic nerves by electron microscopy at P15 and P28 (Fig. 4). At P15, 71 ± 4% of WT axons were myelinated in contrast to 28 ± 2% in Nogo-A−/−, 33 ± 1% in MAG−/−, and 21 ± 2% in DOKO mice (Fig. 4A–D,I) (mean ± SEM; N = 3; *p < 0.05, ***p < 0.001, ANOVA). At P28, the density of myelinated axons increased in all groups (Fig. 4E–H) reaching 71 ± 1% in WT, 63 ± 4% in Nogo-A−/−, 63 ± 2% in MAG−/−, and 63 ± 5% in DOKO animals (Fig. 4I) (mean ± SEM; N = 3–4; p > 0.05, ANOVA). The large but transitory deficit of myelinated axons revealed here by the EM analysis is consistent with the oligodendrocyte differentiation defects described above.

Nogo-A and MAG influence myelination and growth of optic axons. A–H, At P15 and P28, the myelination of axons was examined by electron microscopy on midway along KO optic nerves. A–D, At P15, less myelinated axons than in WT nerves were detected in Nogo-A−/− and MAG−/− optic nerves and much less in DOKO. E–H, The density of myelinated axons increased in all groups at P28. I, Quantitation of the percentage of myelinated axons at P15 and P28 (***p < 0.001, *p < 0.05, ANOVA; N = 3–4 animals/group). At P15, the number of measured axons per group was 1178 axons in WT, 1873 axons in Nogo-A−/−, 1737 axons in MAG−/−, and 1543 axons in DOKO; at P28, 1549 axons in WT, 1869 axons in Nogo-A−/−, 1806 axons in MAG−/−, and 1570 axons in DOKO were evaluated. J, In the same samples, the spectrum of axon calibers was determined. At P15, the axon diameter profiles were shifted to smaller axon sizes in the KO nerves compared with WT mice. K, Axon calibers were close to the WT values at P28. Error bars indicate SEM. Scale bars: A–H, 3 μm.
Myelination is known to control the axonal diameter growth in normal animals (Colello et al., 1994; Cellerino et al., 1997). To determine whether the axon was also affected in our hypomyelinated mice, the axonal diameter distribution was assessed for each group at P15 and P28. At P15, the axonal caliber profiles of the three KO groups were markedly shifted to smaller dimensions (Fig. 4J). Whereas the mean diameter of WT axons was ∼0.80 μm, the mean axon caliber was ∼0.61 μm in Nogo-A−/− and DOKO, and ∼0.67 μm in MAG−/− mice, with sizes up to 2.8 μm. All KO groups had an axon caliber significantly different from WT mice at P15 (*p < 0.05, ANOVA). At P28, the axon size distribution was not statistically different between KO and WT mice (p > 0.05, ANOVA): the mean axon diameter was ∼0.7 μm in Nogo-A−/−, ∼0.75 μm in DOKO, and ∼0.8 μm in MAG−/− and WT optic nerves (Fig. 4K). Interestingly, the deletion of Nogo-A caused a stronger reduction of the axonal growth at P15 than the deletion of MAG.
Mature optic nerves of Nogo-A−/−/MAG−/− double KO mice show myelin abnormalities
The number of myelinated axons in KO mice was not distinguishable from WT animals at P28, but this does not mean that the myelin structure is normally formed. We thus analyzed the myelin abnormalities in the KO and WT optic nerves at P28 (Fig. 5). DOKO optic nerves showed different kinds of myelin abnormalities such as myelin outfolds (Fig. 5B, arrows), multiple myelin rings (Fig. 5B, arrowhead), uncompacted myelin sheaths, and lack or excess of cytoplasm in the periaxonal space. The quantitation of those abnormalities revealed that DOKO had significantly more aberrances than WT optic nerves (Fig. 5D) (*p < 0.05, ANOVA). Interestingly, the myelin structure of Nogo-A−/− axons appeared to be not different from WT axons, whereas MAG−/− axons exhibited a similar number of abnormalities as DOKO axons (Fig. 5D). This suggests that myelin abnormalities encountered in DOKO are mostly a consequence of the absence of MAG.

The myelin structure presents abnormalities in MAG−/− and DOKO but not in Nogo-A−/− optic nerves at P28. Electron micrographs representing myelinated axons in WT and DOKO optic nerves at P28. A–C, Compared with WT axons (A), many DOKO axons (B, C) displayed abnormalities such as myelin sheath outfolds (B, arrows), multiple myelin sheaths (B, arrowhead), and myelin compaction defects around the axons (C, arrow). D, The quantification of abnormally myelinated axons consisted of counting blindly the aberrant myelin outfolds, multiple myelin ensheathment, the uncompacted myelin sheath, and lack or excess of cytoplasm in the periaxonal space. For that, 692 axons in WT, 532 axons in Nogo-A−/−, 407 axons in MAG−/−, and 379 axons in DOKO were randomly analyzed in three to four different optic nerves. The percentage of abnormalities was significantly increased in DOKO optic nerves (37.9 ± 5.6%) compared with WT (17.5 ± 3.7%) (*p < 0.05, ANOVA). However, Nogo-A−/− (12.1 ± 5.6%) and WT, or MAG−/− (32.2 ± 6.1%) and DOKO were not statistically different. This suggests that Nogo-A deletion does not affect the myelin structure and that the elevated abnormalities found in DOKO optic nerves are mostly attributable to the absence of MAG. Error bars indicate SEM. Scale bars: A, B, 2 μm; C, 0.6 μm.
Formation of nodes of Ranvier
Ranvier nodes are crucial for the function of myelinated axons. They are characterized by a very high density of voltage-gated sodium channels, surrounded on each side by a band containing paranodin/Caspr in the paranodal segments (Menegoz et al., 1997; Dupree et al., 1999). MAG−/− mice are known to have an abnormal distribution of paranodin and sodium channels at the nodes of Ranvier (Marcus et al., 2002). When we stained P15 WT optic nerve sections for paranodin and sodium channels, the typical narrow bands of sodium channels were flanked on each side by paranodin-positive clusters (Fig. 6A). The paranodin/sodium channel triplets had a normal appearance and density in Nogo-A−/− optic nerves (Fig. 6B). In contrast, MAG−/− and DOKO mice showed many heminodes and single paranodin or sodium channel clusters (Fig. 6C,D). The counting of nodes, heminodes, and single paranodes revealed a similar number of nodal patterns in WT and Nogo-A−/− nerves and confirmed that paranodin and sodium channel localization is disrupted in absence of MAG (Fig. 6E) (**p < 0.01, ANOVA). The DOKO nerves were intermediate between WT/Nogo-A−/− and MAG−/− nerves. These results suggest that, in contrast to MAG, Nogo-A does not play a central function in the establishment of the nodes of Ranvier.

Distribution of paranodin and sodium channels at the nodes of Ranvier in Nogo-A−/−, MAG−/−, and DOKO optic nerves. A–D, Optic nerve sections were double-stained for paranodin/Caspr (red) and sodium channels (NaCh; green) at P15. By confocal microscopy, image stacks were captured and merged. A, The WT optic nerve shows NaCh clusters typically flanked by two paranodin/Caspr-labeled paranodes. B, In Nogo-A−/− nerves, the pattern of NaCh/paranodin organization was indistinguishable to that of the WT. C, D, In contrast, MAG−/− and, to a lesser extent, DOKO mice often presented separate paranodin and NaCh clusters. E, The distribution of paranodin-positive elements in nodes, heminodes, and single paranodes was calculated. In MAG−/− optic nerves, only ∼11% of the paranodes were fully assembled (**p < 0.01, ANOVA) compared with ∼42% in WT, ∼47% in Nogo-A−/−, and ∼25% in DOKO (*p < 0.05, ANOVA) optic nerves. Paranodin/Caspr clusters were more frequently found as single paranodes in MAG−/− (∼65%) and DOKO (∼52%) optic nerves in comparison with WT (∼38%) and Nogo-A−/− (∼32%) animals. Error bars indicate SEM. Scale bar: 50 mm; 16.7 μm (insets).
Developmental time course and KO-induced changes of Nogo-A/MAG receptor components
Recent studies demonstrated that Lingo-1, a membrane protein that can associate with the Nogo-A and MAG binding protein NgR, in neurons (Lee et al., 2007) or glial cells (Mi et al., 2005) can exert an inhibitory effect on oligodendrocyte maturation. The relative importance of neuronal versus glial Lingo-1 in this process is unclear. We first looked at the expression of Lingo-1 by qRT-PCR in the optic nerve, in the retina, and in the cerebellum during oligodendrocyte development. Lingo-1 was dramatically downregulated in the WT optic nerve after P6, a time coinciding with the beginning of oligodendrocyte differentiation (Fig. 7A). The level of Lingo-1 mRNA in the retina was lower than in the optic nerve and it decreased only after P15. In the cerebellum, Lingo-1 expression increased until P10, started decreasing at P15, and was low in the adult (Fig. 7B). Thus, the downregulation of Lingo-1 in glia coincided in time with the onset of oligodendrocyte maturation (optic nerve) more than in neurons (retina).

Developmental time course of Lingo-1 in WT and KO mice. A, The time course of Lingo-1 mRNA was assessed in oligodendrocyte-free retinal samples and in neuron cell body-free, oligodendrocyte-containing optic nerves by qRT-PCR. The amount of Lingo-1 transcripts dropped after P6 in the optic nerve and after P15 in the retina. B, In the cerebellum, Lingo-1 mRNA peaked at P10 and then decreased to low adult levels. In A, 100%, retinal level at P0; in B, P10 value. C, Lingo-1 mRNA levels did not differ between KO and WT cerebella at P10. All qRT-PCR measurements were normalized using GAPDH as internal reference. Error bars indicate SEM. D, By Western blot analysis, the levels of Lingo-1 protein (100 μg/lane) were undistinguishable between the groups. The experiments were repeated three times with three animals per group and experiment.
We then wanted to determine whether the elimination of Nogo-A and/or MAG resulted in a compensatory change of Lingo-1. The levels of Lingo-1 mRNA by qRT-PCR and Lingo-1 protein by Western blots did not show differences between Nogo-A−/−, MAG−/−, or DOKO and WT cerebellar samples (Fig. 7C,D). Defects in oligodendrocyte maturation in Nogo-A and MAG-deficient animals is therefore unlikely attributable to Lingo-1.
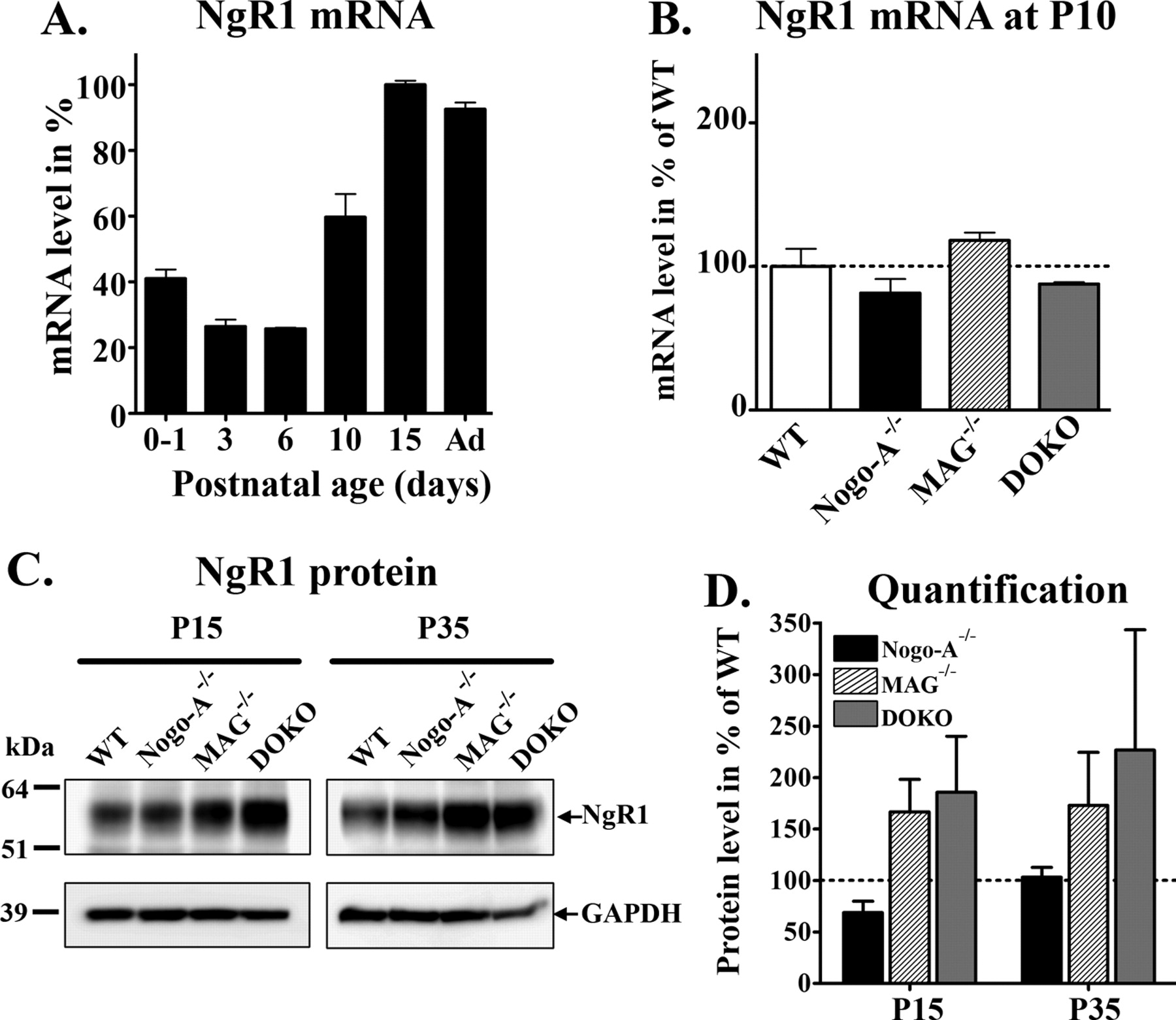
NgR1 is a receptor component associated with Lingo-1 to which MAG and the Nogo66 sequence can bind. In WT cerebella, the level of NgR1 mRNA increased from P6, peaked at P15, and remained high in adults (Fig. 8A). At P10, NgR1 mRNA levels were not different between mutant and WT animals (Fig. 8B). At P15 and P35, however, Western blot analysis showed a robust upregulation of the NgR1 protein level in MAG−/− and DOKO cerebella, whereas in Nogo-A−/− mice, NgR1 appeared unchanged compared with WT animals (Fig. 8C). Densitometric quantification revealed that NgR1 bands in MAG−/− and DOKO samples were ∼1.7–2 times as strong as in WT extracts (Fig. 8D). The specific increase of NgR1 protein in MAG−/− and DOKO tissues suggests that the regulation of NgR1 expression in neurons is MAG-dependent and Nogo-A-independent.

NgR1 is upregulated in absence of MAG but not of Nogo-A. A, Development of NgR1 mRNA levels in the cerebellum of WT mice. B, Comparisons of NgR1 mRNA levels at P10 among WT, Nogo-A−/−, MAG−/−, and DOKO. C, Western blots showing upregulation of NgR1 protein in MAG−/− and DOKO samples of cerebella at P15 and P35 (100 μg protein/lane). No change was seen in Nogo-A−/− animals. D, The intensity of NgR1 bands was quantified by densitometry and normalized to GAPDH protein. The relative amount of NgR1 in the mutants was represented in percentage of WT values (dotted line). Each bar is the average (±SEM) of three repeated experiments realized with three different animals/group.
Nogo-A as well as MAG can bind to NgR1 (Fournier et al., 2001; Liu et al., 2002). To check whether the absence of Nogo-A and MAG induces a compensatory upregulation of the other molecule, we looked at MAG and Nogo-A mRNA levels in Nogo-A−/−, MAG−/−, and DOKO mice. In Nogo-A−/− mice, MAG expression was decreased by ∼23% compared with the WT level (**p < 0.01, ANOVA). This statistically significant reduction of MAG is very likely a consequence of the delay of oligodendrocyte differentiation described above. In addition, the separation of MAG transcripts by agarose gel electrophoresis revealed that the predominant splice version of MAG switched from L-MAG in P6–P15 optic nerve to S-MAG in adult optic nerves; this switch occurred in a similar manner for WT and Nogo-A−/− mice (data not presented). In contrast to the reduction of MAG in Nogo-A−/− animals, in MAG−/− mice the expression of Nogo-A was identical with that of the WT control. These measurements show that MAG or Nogo-A deletion does not induce a compensating upregulation of the other ligand for NgR1/Lingo-1 and further points to separate functions played by Nogo-A and MAG on oligodendrocyte development.
Discussion
Our study shows that Nogo-A is involved in oligodendrocyte development in vivo. Nogo-A was expressed in the optic nerve before the major myelin proteins MBP, PLP, and CNP. MAG−/−, Nogo-A−/−, and DOKO all showed a delay of oligodendrocyte differentiation and severe hypomyelination at P15. The hypomyelination in the optic nerve recovered by P28. Nevertheless, MAG−/− and DOKO, but not Nogo-A−/− animals, displayed abnormalities of the myelin structure and the nodes of Ranvier. The Nogo receptor subunit NgR1 was upregulated in MAG−/− and DOKO but not in Nogo-A−/− tissues.
Nogo-A expression is involved in oligodendrocyte differentiation
The expression of Nogo-A in the optic nerve increased before that of constitutive myelin proteins such as PLP and MBP and may therefore be an important regulatory factor for oligodendrocyte differentiation and myelination. In contrast to MAG, Nogo-A is expressed not only by oligodendrocytes but also by retinal ganglion cell axons, before OPCs colonize the optic nerve. Thus, axonal and/or glial Nogo-A could influence oligodendrocyte maturation via axon-to-oligodendrocyte or oligodendrocyte-to-oligodendrocyte interactions. The latter mechanism is supported by a recent study showing that neutralizing antibodies against Nogo-A blocked the differentiation of oligodendrocytes cultured in the absence of neurons (Zhao et al., 2007). Whether Nogo-A participates to the spatial distribution and architecture of oligodendrocytes in white matter through glia-to-glia interactions remains to be investigated. The migration of OPCs was unchanged in MAG−/− or Nogo-A−/− optic nerves, although the migration of neuronal precursors in the cortex was previously shown to be affected when Nogo-A/B/C proteins were ablated (Mingorance-Le Meur et al., 2007).
Myelin formation is differently impaired by Nogo-A and MAG deletion
The myelin formation was markedly delayed in both Nogo-A−/− and MAG−/− mice, and even more in DOKO mice at P15. Axon diameters were concomitantly reduced, probably as a result of the myelination defect (Colello et al., 1994). Interestingly, we observed obvious structural differences between MAG−/− and Nogo-A−/− mice, especially with regard to defects of Ranvier node formation that occurred in the MAG−/− but not in the Nogo-A−/− mice. The combined deletion of Nogo-A and MAG in DOKO did not enhance the abnormalities of myelin structure or Ranvier nodes compared with MAG−/− mice, and MAG or Nogo-A deletion did not induce the mutual upregulation of the other protein. All these results suggest that these two myelin proteins fulfill distinct functions in the myelin formation. Nogo-A seems to be mostly involved in oligodendrocyte differentiation, whereas MAG appears to be implicated in the myelin structure formation and in the axoglial contact.
The hypomyelination observed at P15 in KO tissues almost completely disappeared at P28. Previous studies have shown the powerful ability of the CNS to compensate for the deletion of important structural myelin proteins or when OPCs are partially ablated during the first postnatal week: in absence of the principal myelin protein PLP, oligodendrocytes still assemble compact myelin sheaths in the CNS (Klugmann et al., 1997). If oligodendrocytes are eliminated within the first postnatal week of life, newly generated oligodendrocytes can fully compensate for the myelination deficit by day 45 (Collin et al., 2004). Given this remarkable compensatory capacity of the CNS for myelination defects, it is not surprising to find a normal proportion of myelinated axons in our Nogo-A−/−, MAG−/−, and DOKO optic nerves at 1 month. It will be interesting to study which molecular pathways are involved in this compensatory effects in the Nogo-A−/− and MAG−/− animals.
MAG and Nogo-A activate distinct molecular pathways
The differences in the myelin defects in MAG−/− and Nogo-A−/− animals point to distinct underlying molecular mechanisms. For example, the mRNA levels for PLP, MBP, and CNP were markedly reduced in the Nogo-A−/− cerebella at P10 but not in the corresponding MAG−/− tissues. Surprisingly, at the same age, the level of myelin protein transcripts was similar in DOKO as in MAG−/− mice. This suggests that MAG deletion may activate compensatory mechanisms that bring myelin protein transcripts to normal levels despite the absence of Nogo-A. Such a compensatory change specific for MAG removal was in fact observed by the upregulation of NgR1 in MAG−/− and DOKO but not in Nogo-A−/−. This is particularly astonishing because NgR1 is a common binding partner for MAG and Nogo-A. It was previously reported that MAG ablation caused N-CAM overexpression, which could partially compensate for the missing adhesive functions of MAG (Montag et al., 1994). A possibility is that NgR1 is not increased in Nogo-A−/− mice because of the Nogo-B upregulation (Simonen et al., 2003). Nogo-B also contains the NgR1 binding region Nogo66 (Fournier et al., 2001; Simonen et al., 2003). This would indicate, however, that the myelin phenotype encountered in young Nogo-A−/− mice could be caused by an NgR1-independent signaling pathway. Beside the Nogo66 fragment that stimulates NgR1, a unique domain in the middle portion of Nogo-A has been shown to strongly inhibit cell spreading and neurite growth by interacting with a yet-uncharacterized receptor (Oertle et al., 2003). The changes of NgR1 in the MAG−/− mice suggest that NgR1 may be necessary for MAG function in myelination. An intracellular signal triggered by MAG is the increased phosphorylation of the tyrosine kinase Fyn, which can then drive MBP expression (Umemori et al., 1999). However, MAG can interact with various molecules on the axonal membrane such as NgR1 or NgR2, gangliosides, and OMgp (Chen et al., 2006; Laurén et al., 2007). Downstream intracellular mediators could involve the small GTPases of the Rho family such as RhoA, Cdc42, and Rac (Thurnherr et al., 2006; Zhao et al., 2007). Interestingly, the NgR1 interacting membrane protein Lingo-1 has been shown to inhibit oligodendrocyte differentiation (Mi et al., 2005). How and whether the NgR1/Lingo-1 complex serves as a key receptor for the Nogo-A and MAG-mediated effects on oligodendrocytes as observed in the present study remains to be analyzed in detail.
Clinical relevance of Nogo-A in demyelinating diseases
Our findings showing that Nogo-A plays an important role for oligodendrocyte differentiation is particularly relevant in the context of myelin repair [e.g., in experimental autoimmune encephalomyelitis (EAE), in multiple sclerosis, or after CNS trauma]. The absence of Nogo-A, or antibodies directed against Nogo-A were shown to block or dampen the appearance of the EAE symptoms, possibly by decreasing local inflammatory processes (Karnezis et al., 2004). Our results showing the positive effects of Nogo-A for oligodendrocyte differentiation should be taken into account when designing treatment schedules; Nogo-A neutralization may have a beneficial effect early during the inflammatory process or for regenerating axons after CNS trauma, but it could be negative for the process of myelin repair at a later stage.
Footnotes
-
This work was supported by Swiss National Science Foundation Grant 31-63633.00. We are grateful to Coni Imsand and Oli Weinmann for technical assistance. We thank the colleagues and laboratories who provided antibodies and tissues.
- Correspondence should be addressed to Dr. Vincent Pernet, Brain Research Institute, University of Zurich/Swiss Federal Institute of Technology, Winterthurerstrasse 190, Room 55J34a, CH-8057 Zurich, Switzerland. pernet{at}hifo.uzh.ch





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}