

Abstract
Interferon-α (IFNα) is a pleomorphic cytokine produced by nucleated cells in response to viral infection. In patients, treatment with IFNα has side effects including cognitive impairment resembling subcortical dementia, which is a hallmark of human immunodeficiency virus (HIV)-associated dementia (HAD). IFNα is increased in the CSF of HAD patients compared with HIV patients without dementia. In this study, blocking IFNα in a HIV encephalitis (HIVE) mouse model with intraperitoneal injections of IFNα neutralizing antibodies (NAbs) significantly improved cognitive function compared with untreated or control antibody-treated HIVE mice during water radial arm maze behavioral testing. Treatment with IFNα NAbs significantly decreased microgliosis and prevented loss of dendritic arborization in the brains of HIVE mice. Furthermore, treatment of primary neuron cultures with IFNα resulted in dose-dependent loss of dendritic arborization that was blocked with IFNα NAb treatment and partially blocked with NMDA antagonists [AP5 and MK801 (dizocilpine maleate)] indicating glutamate signaling is involved in IFNα-mediated neuronal damage. These results show that IFNα has a major role in the pathogenesis of HIVE in mice and is likely important in the development neurocognitive dysfunction in humans with HIV. Blocking IFNα could be important in improving cognitive and pathological developments in HAD patients and may be clinically important in other neuroinflammatory diseases as well.
Introduction
Over 33 million people worldwide are infected with the human immunodeficiency virus (HIV) (World Health Organization, 2007). Approximately 10% of them will develop HIV-associated dementia (HAD), a devastating neurological disease, and as many as 30–40% will develop mild cognitive motor dysfunction (MCMD) (McArthur, 2004; Valcour et al., 2004b; Letendre et al., 2007; Robertson et al., 2007; Wong et al., 2007). Because HIV does not infect neurons, the pathogenesis of HAD is thought to be centered on the production of putative neurotoxins that damage neurons (Fischer-Smith and Rappaport, 2005). These toxins include HIV proteins, cytokines, chemokines, and metabolites. Identifying the major contributors to the development of HAD is critical for the development of future treatments for HAD because highly active antiretroviral therapy is only partially effective.
Interferon-α (IFNα) is a potent antiviral cytokine produced in response to viral infection and inflammation by a wide variety of nucleated cells in the CNS, or that can enter the CNS, including astrocytes, microglia, neurons, macrophages, and T-lymphocytes (Dafny, 1998; Samuel, 2001; Sas et al., 2007). Because of its antiviral and immunoregulatory properties, IFNα has been used as part of treatment regimens for chronic viral infections such as hepatitis C and herpes (Pavol et al., 1995; Valentine et al., 1998). IFNα also has a neuroregulatory role in the CNS. Too much IFNα in the CNS can cause reversible cognitive dysfunction that dissipates when IFNα therapy is stopped (Dafny, 1998; Valentine et al., 1998). In three separate studies, IFNα was significantly increased in the CNS of HIV patients with dementia compared with those without dementia (Rho et al., 1995; Krivine et al., 1999; Perrella et al., 2001). These studies also suggested that IFNα in the CNS was correlated with the severity of dementia (Rho et al., 1995), viral load in the CNS (Krivine et al., 1999), and increased atrophy in the frontal cortex of HAD patients (Perrella et al., 2001). Gene array analysis on postmortem tissues showed that IFNα was significantly upregulated in the frontal cortex of HAD patients (Masliah et al., 2004).
To gain a better understanding of the role of IFNα in the pathogenesis of HAD, we used a SCID mouse model of HIV encephalitis (HIVE) that recapitulates many of the behavioral and pathological features seen in humans with HIVE (Tyor et al., 1993; Persidsky et al., 1996; Avgeropoulos et al., 1998; Cook-Easterwood et al., 2007; Sas et al., 2007). HIVE mice have consistently exhibited cognitive deficits in both the Morris water maze (a reference memory task) (Avgeropoulos et al., 1998; Griffin et al., 2004; Cook-Easterwood et al., 2007) and shown cognitive problems in the water radial arm maze (WRAM) (a working memory and reference memory task) (Sas et al., 2007). HIVE mouse pathology includes HIV-positive human mononuclear phagocytes, multinucleated giant cells, increased mouse mononuclear phagocytes, increased astrogliosis, loss of dendritic arborization, and increased expression of proinflammatory cytokines [interleukin-1 (IL-1), IL-6, and tumor necrosis factor-α (TNFα)] (Tyor et al., 1993; Persidsky et al., 1996, 1997; Avgeropoulos et al., 1998; Cook-Easterwood et al., 2007). A previous study from this laboratory showed that IFNα levels in the brains of HIVE mice were strongly correlated with cognitive deficits (Sas et al., 2007). In the present study, we tested the hypothesis that blocking IFNα in the HIVE mouse model with an IFNα neutralizing antibody (NAb) would ameliorate cognitive and pathological effects.
Materials and Methods
Human monocyte cultures and HIV infection.
Human monocytes were cultured as previously described (Sas et al., 2007). Primary human macrophages were obtained from Dr. H. Gendelman (University of Nebraska Medical Center, Omaha, NE). Cells were cultivated in DMEM (Invitrogen) with 10% human serum, glutamine supplement (Invitrogen), penicillin–streptomycin (Sigma-Aldrich), and M-CSF (monocyte–colony stimulating factor) (Sigma-Aldrich) at 37°C with 5% CO2 in Teflon-coated culture flasks (Corning Life Sciences). After 7 d, macrophages were divided into two cultures, resuspended in 9 ml of media consisting of DMEM with 10% human serum, glutamine supplement, and penicillin–streptomycin. One culture was infected with 1 ml of HIV-1ADA (0.1 multiplicity of infection; obtained from Dr. H. Gendelman), an R5 strain of HIV that uses the chemokine receptor CCR5 as a coreceptor. The other control culture had 1 ml of DMEM added to the flask. After 1 h of infection, the virus was washed off, and both cultures were resuspended in 25 ml of media. Cells were collected 2 weeks after HIV infection. Approximately 1 × 107 monocytes/culture were resuspended in PBS 2 weeks after viral infection for inoculation into mice. Under xylazine (5 mg/kg) and ketamine (95 mg/kg) anesthesia, HIV-infected or uninfected macrophages (105) were injected intracranially into the right frontal lobe of 5-week-old SCID mice.
Mice.
B6.CB17-Prkdcscid/SzJ male mice (4 weeks of age; The Jackson Laboratory) were single housed in microisolator cages within isolator cubicles (biosafety level 3 equivalent) and were given at least 1 week to acclimate to this environment before experimentation. All animal protocols have been approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina.
Antibody treatment.
Antibody treatment injections immediately followed intracranial cell inoculation. Control and HIVE mice were placed in three groups: control treatment (n = 10/group), control antibody treated (n = 4/group), and IFNα NAb treated (n = 10/group), for a total of six groups of mice. Control and HIVE mice received intraperitoneal injections of 300 μl of saline (control treatment), 100 μg/kg isotype-matched control rabbit polyclonal antibody (control antibody; Vector Laboratories), or 100 μg/kg rabbit polyclonal anti-mouse IFNα NAbs (PBL). All mice received their treatments once every 72 h.
The WRAM.
Behavioral testing began 6 d after cell inoculation. The WRAM is a spatial working and reference memory task. WRAM behavioral testing was performed as previously described (Sas et al., 2007). In brief, the maze was an eight-arm maze, with hidden platforms submerged below the surface of the water in four of the arms. Each subject kept the same platform locations throughout testing. The test room contained spatial cues that remained constant throughout testing.
A subject was released in the start arm and had 2 min to locate a platform. If the allotted time expired, the mouse was guided to the nearest available platform. Once the platform was found, the mouse remained on it for 15 s, and then was returned to its heated cage for 30 s until its next trial. During this intertrial interval, the just-located platform was removed. The mouse was then placed back in the start arm and allowed to locate one of the remaining platforms. A daily session repeated this process until all four platforms were found. Thus, for each animal, a daily test consisted of four trials, with the number of arms containing a platform reduced by one with each additional trial. Behavioral testing took place each day between 9:00 A.M. and 12:00 P.M. Each mouse was tested for 12 d. Day 1 was an acquisition day and did not count toward scoring of the animal's performance in the task.
There were the following three error types: (1) reference memory (RM), a first-time entry in an arm that never has a platform; (2) repeat reference memory error (RRM), repeated entries into arms that have never had a platform; (3) working memory correct error (WMC), entries into arms that previously had a platform that the mouse found, but the platform has been removed and the mouse continues to enter that arm.
Tissue sectioning and immunocytochemistry.
After behavioral testing was completed, the mice were killed (18 d after intracranial inoculation of the cells), and their brains were removed for pathological analysis. Tissue sectioning and immunocytochemistry were performed as described previously (Sas et al., 2007). Briefly, mouse brains were snap frozen in tissue-freezing medium, and 5 μm coronal sections were taken starting at the frontal lobe through the temporal-parietal lobe. Immunoperoxidase staining included human macrophages (1:50 EBM11; Dako), HIV (1:20 p24; Dako), astrocytes (1:750 glial fibrillary acidic protein; Millipore Bioscience Research Reagents), microglia (1:20 F4/80; Caltag), IFNα NAbs, polyclonal antibodies (1:100; Vector Laboratories), and neuronal dendrites [1:200 microtubule-associated protein-2 (MAP2); Millipore Bioscience Research Reagents], providing 10 slides for each antibody per brain. Slides were then reviewed using light microscopy (Olympus microscope), and pathology was quantitated using densitometry analysis for microgliosis, astrogliosis, and dendritic arborization as previously described (Sas et al., 2007).
RNA extraction and cDNA synthesis.
Intervening sections not used for pathological analysis were collected and RNA was extracted according to the TriReagent (Ambion) protocol. Approximately 1 μg of RNA was used for cDNA synthesis. First-strand cDNA synthesis was performed using the high-capacity cDNA Archive kit (Applied Biosystems). RNA was diluted 1:10 in DEPC-treated water to obtain the appropriate concentration of RNA for the reaction. RNA was added to the provided master mix and run in a thermal cycler (Eppendorf) at 25°C for 10 min immediately followed by 37°C for 50 min. The cDNA was stored at −20°C until use.
Real-time PCR.
Levels of HIV, mouse IFNα, and mouse ISG15 were analyzed using real-time PCR with ABI 7000 Prism (Applied Biosystems) according to the suggested protocol of ABI. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a housekeeping gene, was used as the endogenous control for all experiments. HIV and GAPDH primers were designed using Assays-by-Design (Applied Biosystems). Mouse IFNα4 and mouse IFNα inducible gene 15 were designed using Assays-on-Demand (Applied Biosystems). cDNA samples were added to TaqMan Assay mix (Applied Biosystems) and the primer/probe mix. All unknown samples were compared with their respective GAPDH levels, and this ratio was then normalized against a highly infected sample to determine a RQ (relative quantification) value.
Rat dissociated neuron cultures.
Twenty-day-gestation Sprague Dawley rat pups (Charles River Laboratory) were killed. Brains were removed and placed into 10 ml of dissecting media (50 ml glucose/sucrose, 28 ml of HEPES, 50 ml of saline solution, filtered pH 7.3–7.4). Meninges and blood vessels were removed from brain, and then frontal cortex was separated and minced in a new 35 mm dish. Two milliliters of 0.125% trypsin solution in DMEM was placed in the dish and was incubated at 37°C 5% CO2 for 15 min. The tissue was transferred into a tube containing 20 ml of plating media (77 ml of MEM, 10 ml of FBS, 10 ml of heat-inactivated horse serum, 1 ml of penicillin–streptomycin, 1 ml of l-glutamine, 1 ml of B-27 Neurobasal supplement; filtered) and triturated through a small-bore pipette until mostly dissociated. This cell solution was filtered through a gauze pad into a fresh tube. Cells were counted, diluted, plated in a poly-l-lysine-coated dish at 8 × 106/dish, and placed in an incubator at 37°C and 5% CO2. After 24 h, 1 ml of media was exchanged [97 ml of neural basal media, 1 ml of l-glutamine, and 2 ml of B-27 supplement (Invitrogen)]. Two days after plating, 10 μl of 10 μm cytosine-β-d-arabinofuranoside (ARC) (Fluka) was added to the cultures. Four days after initial plating, ARC was replaced with fresh media. Media were exchanged every 7 d.
Rat IFNα administration to rat neuronal cultures.
Purified rat IFNα (PBL) was diluted in 0.09% saline to 100 and 25 IU/μl, and was administered directly to the cultures between days 14 and 16. A time course analysis of IFNα effects on neurons was used to determine the length of time the cultures were to be exposed to IFNα. Control cultures received saline treatment.
Immunohistochemistry of neuron cultures.
Immunohistochemistry after control or IFNα exposure was performed using 16- to 18-d-old cultures. After PBS wash, the cells were fixed with ice-cold methanol (−20°C) for 5 min, followed by two PBS washes. Dishes were incubated at room temperature for 1 h in 2% serum blocking buffer (1% goat serum, 1% horse serum in PBS). Two PBS washes followed. MAP2 primary antibody (1:200; Millipore Bioscience Research Reagents) was added for 1 h. Dishes were washed two times in PBS, and then cells were incubated for 30 min in the dark with secondary antibody at 1:100 (FITC-labeled goat anti-rabbit; Vector Laboratories). Dishes were washed one time with PBS and one time with distilled water, and then coverslipped with Vectashield fluorescence protection (Vector Laboratories). Slides were viewed under an Olympus BH-2 microscope with fluorescence.
Sholl analysis.
To analyze the length of the neuronal dendrites and the number of dendritic branches, a Sholl analysis was used (Sholl, 1953; Silva-Gómez et al., 2003). Dendrites stained with MAP2 were examined for their length and the number of branches from them by placing a measured concentric circle (10 μm intervals) overlay on the neurons at 20×. The center of the overlay was placed on the soma of the neuron. The length of a main dendrite extending from the soma was measured by the number of rings the dendrite crossed. Neuronal dendrite branching was measured by manually counting the number of dendrite offshoots produced from a main dendritic growth.
Statistics.
For WRAM, the 11 testing days were blocked into the following two phases: block 1, consisting of testing days 2–7; and block 2, consisting of days 8–12. For WMC and RRM measures, the data for each block were analyzed using a one between (HIV status), two within (days and trials) repeated-measures ANOVA. Trial 1 was not included in the WMC analyses because it is not possible to make a WMC error on the first trial. For RM, each phase was analyzed using a one between (HIV status), one within (days) repeated-measures ANOVA. Analysis of trials was done with a post hoc Fisher's PLSD test. Significance was set at p < 0.05 for all tests.
Analysis of PCR results, densitometry readings, ELISA data, and Sholl data was completed using a two-way ANOVA. Analysis between groups or treatments was done with a post hoc Fisher's PLSD test. Significance was set at p < 0.05 for all analyses.
Results
HIVE mice treated with IFNα neutralizing antibody display improved behavioral performance
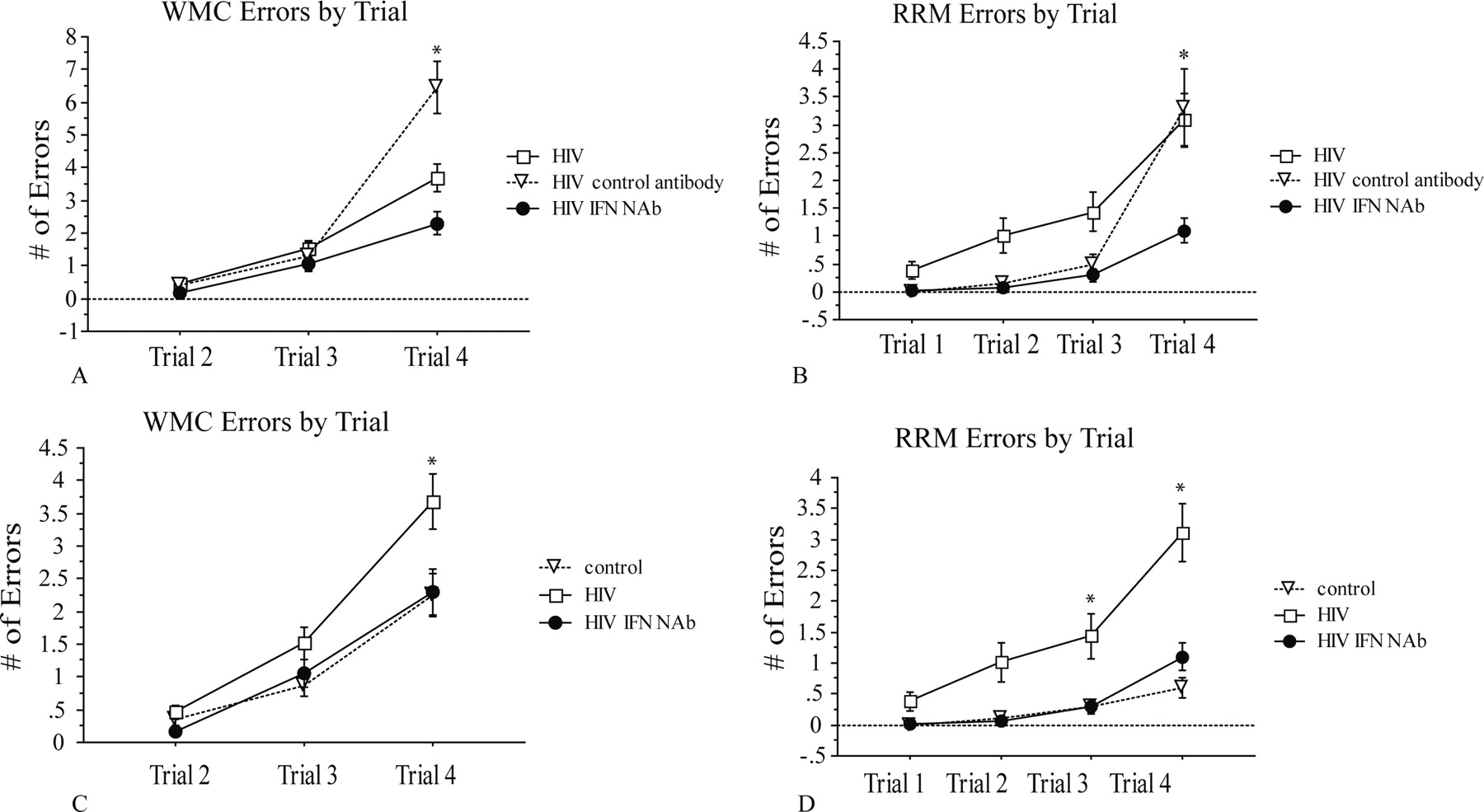
Mice inoculated intracranially with control or HIV-infected human macrophages were separated into three treatment groups. HIVE and control mice received one of three treatment regimens every 3 d starting immediately after intracranial inoculation with human macrophages: intraperitoneal injections of saline, intraperitoneal injections of isotype-matched control antibodies, or intraperitoneal injections of mouse IFNα NAbs. During treatment, mice were tested in a WRAM, which is designed to test spatial and working memory. The WRAM is designed so that the task increases in complexity within a day of testing. Completion of trial 1 requires the least amount of memory recall, and trial 4 requires the most amount of memory recall. During block 2 (days 8–12) of testing, all HIVE mice performed equally well in the early trials (trials 1 and 2) when memory load was low; however, as memory load increased, HIVE mice that received IFNα NAbs were more successful in completing the task compared with other HIVE groups, and performed similar to the control groups (Fig. 1). When comparing only the performance of the HIV-infected mice, HIVE mice with IFNα NAbs made significantly fewer working memory correct errors (WMC) on trial 4 and significantly fewer repeat reference memory errors (RRM) on trials 3 and 4 than HIVE mice that received control antibody or saline injections (Fig. 1A,B). When comparing the performance of the HIVE control-treated and HIVE IFNα NAb mice to uninfected controls, HIVE mice with IFNα NAbs performed comparable with control mice in both WMC and RRM errors throughout all trials, even when memory load was highest (Fig. 1C,D). Confirming that the control antibodies and vehicle had no effect on cognition, HIVE mice treated with isotype-matched control antibodies performed similarly to HIVE mice with saline injections (Fig. 1A,B), and control mice treated with isotype-matched control antibodies performed similarly to control mice injected with saline for WMC and RRM errors (data not shown) (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material).

WMC and RRM errors by trial for HIVE and control saline-treated mice. HIVE mice that received IFNα NAbs made less WMC (p = 0.0017) and RRM (p = 0.013) errors as memory load increased during trials than HIVE mice that received no treatment or control antibodies (A, B). HIVE mice with IFNα NAbs were able to maintain performance similar to control mice without treatment as memory load increased in WMC and RRM errors (C, D). HIVE mice without treatment made significantly more WMC (p = 0.016) and RRM (p = 0.0002) errors as the memory load increased compared with control and HIVE IFNα NAb mice (C, D). Error bars indicate SEM. *indicates statistical significance described in each instance above, except for trial 3 in D where p < 0.01.
IFNα NAb treatment improves pathological markers in HIVE mice
Immunohistochemical staining was used to determine whether intraperitoneally injected antibodies were present in the brains of the antibody-treated mice. Control antibodies and IFNα NAbs were both detectable in the brain tissue of mice inoculated with either control or HIV-infected macrophages. Consistent with results from our previous study (Sas et al., 2008), the antibody staining was primarily localized around the endothelium and in the brain parenchyma (data not shown).
Consistent with previous studies (Cook-Easterwood et al., 2007; Sas et al., 2007), densitometry analysis of the brains showed that HIVE mice had increased astrogliosis, microgliosis, and significant loss of dendritic arborization surrounding HIV-infected cells compared with mice inoculated intracranially with control human macrophages. HIVE mice that received IFNα NAbs had significantly less microgliosis compared with HIVE mice that received other control treatments (Fig. 2A). However, there was no statistical difference in astrogliosis in the brains of HIVE mice injected with IFNα NAbs compared with HIVE mice that received no treatment or control antibody treatment (Fig. 2B). MAP2 staining to visualize dendritic arborization revealed that untreated HIVE mice showed a significant loss of dendritic arborization in the frontal cortex compared with mice inoculated intracranially with control macrophages (Fig. 2C). HIVE mice injected with IFNα NAbs presented with an intermediate loss of dendritic arborization indicating that blocking IFNα prevented some loss of dendritic arborization.

Densitometry analysis of microgliosis, astrogliosis, and dendritic arborization in all mice. Densitometry analysis of microgliosis in all control and HIV mouse groups (A). Mice treated with IFNα NAbs had significantly less microgliosis compared with other HIVE mouse groups (*p < 0.01). Densitometry analysis of astrogliosis in all control and HIV mouse groups (B). There was no statistical difference in astrogliosis between HIV groups (*p < 0.05 compared to control). Densitometry analysis of dendritic arborization (MAP2 staining) in all control and HIV mouse groups (C). HIVE mice treated with IFNα NAbs did not have a significant loss of dendritic arborization compared with control mice. *p < 0.05 compared to control). Ir represents isotype control Ab. Error bars indicate SEM.
IFNα NAbs inhibit IFNα signaling in the brain
To assess the nature of the IFNα NAbs to block IFNα-mediated actions, real-time reverse transcription (RT)-PCR was used to measure mRNA for IFNα (IFNα4) and IFNα stimulated gene 15 (ISG15). Mouse IFNα4 was detectable in mice inoculated intracranially with control human macrophages and was significantly elevated in HIVE mice (Fig. 3A). Within the HIVE mouse treatment groups, IFNα4 mRNA levels were not significantly different (Fig. 3A). ISG15 mRNA was also elevated in HIVE mice compared with control mice (Fig. 3B) (for all controls, see supplemental Fig. 2, available at www.jneurosci.org. as supplemental material). However, mice that received IFNα NAbs had significantly less ISG15 compared with HIVE mice that received saline or control antibody injections (Fig. 3B).

Real-time RT-PCR analysis of IFNα4 and ISG15 mRNA in control, HIVE control, HIVE control Ab, and HIVE IFNα NAb mice (A, B). All three treatments of HIV-infected mice expressed higher levels of IFNα4 compared with control mice, but there was no significant difference between the three groups (*p < 0.05 for all three compared to control). HIV-infected mice treated with IFNα NAbs produced significantly less ISG15 compared with other HIV-infected mice (*p < 0.03 in both HIV and HIV Ir Ab compared to control) (B). Error bars indicate SEM. Ir represents isotype control Ab.
In agreement with the PCR data, immunohistochemical analysis of protein kinase R (PKR), an IFNα-activated protein, was decreased in brain tissue of HIVE mice treated with IFNα NAbs compared with other HIVE mouse groups (Fig. 4A–D). Quantitation of PKR by densitometry analysis showed that HIVE mice treated with IFNα NAbs had significantly less PKR staining compared with other HIVE mice (Fig. 4E). Together with the results from the RT-PCR experiments, these results show that IFNα is produced in response to the HIV infection in all mice groups, but that intraperitoneally injected IFNα NAbs can inhibit downstream IFNα-mediated signaling events in brain.

Immunohistochemistry staining for PKR in mouse brain tissue. PKR staining in control mouse (A), PKR staining in HIV mouse injected with saline (B), PKR staining in HIV mouse injected with control Ab (C), and PKR staining in HIV mouse injected with IFNα NAbs (D) are shown. Densitometry analysis of PKR staining in untreated control mice and all groups of HIVE mice (E). HIVE mice treated with IFNα NAbs had significantly less PKR present in brain tissue compared with other HIVE mice (*p < 0.0001). Error bars indicate SEM. Ir represents isotype control Ab.
Viral load not affected by IFNα neutralizing antibody treatments
Although IFNα NAbs improved the cognitive performance and decreased pathological manifestations in HIVE mice, inhibition of the antiviral actions of IFNα could lead to increased HIV in these mice. However, analysis of HIV by RT-PCR showed no statistical difference in the viral load of HIVE mice treated with IFNα NAbs compared with saline-treated or control antibody-treated HIVE mice (Fig. 5A). As part of the pathology analysis, the number of human macrophages and the number of p24-positive macrophages were counted in the brains of the HIV-infected mice. The percentage of p24-positive macrophages counted in the brains of HIVE mice was similar between the three treatment groups, supporting the HIV PCR data (Fig. 5B). One reason that the level of HIV was not significantly affected by IFNα NAbs may be attributable to increases in other interferons. ELISA analysis of IFNβ in the brain tissue of HIVE and control mice showed significantly elevated IFNβ in HIVE mice (mean, 43.75 ± 13.1 pg/mg) compared with control mice (mean, 10 ± 6.5 pg/mg; p = 0.03).

Real-time RT-PCR analysis of HIV Gag transcripts in the brains of HIV-infected mice (n = 4 for each) (A). Percentage of p24-positive human macrophages counted in the brains of HIV-infected mice (B). There was no difference in Gag or HIV p24-positive macrophages in the brains of the three HIVE mouse groups. Error bars indicate SEM. Ir represents isotype control Ab.
IFNα administration has dose-dependent effects on neuron dendritic morphology
To begin to determine the mechanism(s) of IFNα toxicity on neurons, cortical neuron cultures were exposed to increasing doses of exogenous IFNα for 48 h. Sholl analysis of IFNα-treated cultures showed a loss of dendritic arborization that was dependent on the concentration of IFNα used (Fig. 6A–C). Significant effects of IFNα on dendritic length and dendritic branching were observed at concentrations >100 IU of IFNα. At very high levels of IFNα (>500 IU), many of the neurons examined had little to no dendritic projections. The effects of 300 IU of IFNα on dendritic length and branching were mostly reversed by cotreatment with IFNα NAbs (Fig. 6D,E). To determine whether neurotoxicity mediated by NMDA glutamate receptors was involved in the IFNα-mediated loss of dendritic arborization, cultures were treated with 300 IU of IFNα and an NMDA antagonist [dizocilpine maleate (MK801) or AP5] (Fig. 6D,E). Both NMDA antagonists protected against IFNα-mediated loss of dendritic length (Fig. 6D). However, these agents did not significantly prevent the loss of dendritic branching (Fig. 6E). Analysis of cell viability by trypan blue staining showed that IFNα produced a dose-dependent increase in cell death (data not shown).

Immunofluorescence stain of dendritic arbors (MAP2) during a dose–response of changes in dendritic arborization to increasing doses IFNα (A). Increasing the dose of IFNα in the neuron culture decreased dendritic arborization of neurons. Dendrite length decreases as the amount of IFNα increases in the neuron cultures (n = 100 neurons/dose) (B). A total of 200 IU of IFNα is enough to significantly decrease the length of dendrites (*p < 0.0003). Dendritic branches decrease as the amount of IFNα increases in the neuron cultures (n = 100 neurons/dose) (C). A total of 200 IU of IFNα is enough to decrease the branching of dendrites (*p < 0.0004). Dendritic length and branching measurements of neuron cultures treated with 300 IU of IFNα and a blocking treatment of IFNα NAbs or an NMDA antagonist (MK801 or AP5) (D, E). NMDA antagonists protected against loss of dendritic length (*p < 0.0001) (D); however, they were not able to prevent loss of dendritic branching in the neuron cultures (E). Error bars indicate SEM.
Discussion
The major finding of this study is that systemic treatment of HIVE mice with IFNα NAbs prevents the cognitive deficits and some pathological markers of HIVE. Importantly, HIV viral load was not significantly affected in HIVE mice treated with IFNα NAbs. Although a complete understanding of IFNα actions is not known, in vitro studies of neurons showed that IFNα decreases dendritic arborization via a process that involves NMDA receptors. Given the importance of these receptors in processes of synaptic plasticity, learning and memory, it can be suggested that the IFNα-mediated loss of dendritic arborization results in cognitive impairment observed in the behavioral memory tasks.
In frontal cortex of HAD patients, IFNα is elevated as measured by increased expression of the type I IFN genes, including IFNα and IFNα-inducible genes (Masliah et al., 2004). IFNα has consistently been significantly higher in the CSF of HIV-infected patients with dementia compared with HIV patients without dementia or HIV-negative individuals (Rho et al., 1995; Krivine et al., 1999; Perrella et al., 2001). The amount of IFNα in the CSF of HAD patients was indicative of the severity of dementia (Rho et al., 1995) and was directly correlated with the severity of cortical atrophy measured by magnetic resonance imaging (Krivine et al., 1999). In addition, the amount of IFNα in the CSF of HAD patients correlated with viral load in the CSF (Perrella et al., 2001). Patients with high levels of IFNα in the CNS have cognitive deficits (Pavol et al., 1995; Valentine et al., 1998; Hoffman et al., 2003), similar to patients with HAD. Together with the results of the present study, these data strongly suggest that IFNα is critically involved in the pathogenesis of HAD in humans (Rho et al., 1995; Krivine et al., 1999; Perrella et al., 2001; González-Scarano and Martín-García, 2005).
HIVE mice that received NAbs exhibited a superior ability to remember numerous items of information than other HIVE mice and were able to perform the cognitive task similar to control mice. Spatial working memory maze tasks, such as the WRAM used in the current study, depend on the hippocampus and frontal cortex (Jarrard, 1993; Hyde et al., 1998; Bimonte-Nelson et al., 2003). Blocking IFNα in the CNS of the HIVE mice improved memory so that mice receiving this treatment did not differ from controls, suggesting IFNα NAb treatment reversed the HIV-induced memory impairment. These effects were seen on two different measures of working memory. IFNα-induced cognitive dysfunction has been well documented in rodents and humans (Dunn and Crnic, 1993; Valentine et al., 1998; Campbell et al., 1999; Makino et al., 2000; Capuron and Miller, 2004). In patients, high doses of IFNα cause loss of memory, and even frank, subcortical dementia (Dafny, 1998; Valentine et al., 1998; Capuron and Miller, 2004), very similar to the type of dementia seen in HAD patients. IFNα has profound effects on the hippocampus, basal ganglia, and frontal cortex, regions of the brain important in memory function and adversely effected in HIVE mice and HAD patients (Dafny, 1998; Valentine et al., 1998; Campbell et al., 1999; McArthur, 2004; Dafny and Yang, 2005; Sas et al., 2007). These data further support the evidence that IFNα is an important cause of cognitive dysfunction in neuroinflammatory conditions.
Pathologically, HIVE mice treated with NAbs exhibited less loss of dendritic arborization in the frontal cortex, compared with other HIVE mouse groups. Consistently, dendritic arborization analysis of the HIVE mouse model has shown dendritic simplification surrounding HIV-infected cells (Cook et al., 2005; Sas et al., 2007), and that dendritic simplification can be detected far from the HIV-infected cell injection site in the hippocampus (Poluektova et al., 2005). Furthermore, loss of dendritic arborization is associated with cognitive deficits in behavioral testing (Cook-Easterwood et al., 2007). Previously, we have seen that IFNα levels in the brains of HIVE mice directly correlate with the number of errors made during WRAM testing (Sas et al., 2007). The behavioral deficits seen in the HIVE mouse model are likely linked with increased IFNα and dendritic loss. Blocking IFNα with NAbs improved cognitive function and prevented loss of dendritic arborization. IFNα toxicity affecting dendritic arborization was further supported by the in vitro data showing that IFNα has a dose-dependent effect on dendritic simplification. We have further demonstrated that this IFNα toxicity is at least partially mediated by NMDA-related effects. Determining the exact molecular mechanism(s) by which IFNα exerts its neurotoxic effects is an important future goal of our research.
IFNα NAb treatment decreased the amount of microgliosis compared with HIVE mice that received control antibodies or saline. IFNα activates microglia in the brain (Paul et al., 2007). IFNα-activated microglia, in turn, both produce and stimulate IL-1, IL-6, TNFα, and reactive oxygen species (hydrogen peroxide, superoxide, nitrogen oxide) by mononuclear phagocytes, microglia, and astrocytes (Paul et al., 2007). Therefore, ultimately IFNα stimulates the production of a wide variety of putative neurotoxins that may be involved in HIVE (Rosenberg and Fauci, 1990; Tyor et al., 1992; Hansson et al., 2004; Fischer-Smith and Rappaport, 2005; Scumpia et al., 2005; Dantzer et al., 2008). Interestingly, it has been shown in sooty mangabeys infected with simian immunodeficiency virus (SIV) that those without IFNα production lack a generalized inflammatory immune response including reduced production of IL-6 and TNFα. This results in a nonprogressive SIV infection in these animals, emphasizing the importance of IFNα in disease progression (Mandl et al., 2008). Similarly, IFNα NAb-treated mice had significantly less activated microglia compared with untreated HIVE mice and thus are likely to have less of the aforementioned inflammatory cytokines, chemokines, metabolites, and reactive oxygen species that can damage neurons. IFNα NAb treatment likely prevents loss of cognitive function by reducing the loss of dendritic arborization caused by the direct effects of IFNα on neurons, and also reducing the synthesis of inflammatory neuromodulators in HIVE mice, therefore preventing potential indirect effects of IFNα caused by inflammatory cytokines and other possible neurotoxic substances.
The pathogenesis of HIV-associated cognitive disorders has centered around the idea that neurotoxins, produced by mononuclear phagocytes and astrocytes, are responsible for neuronal dysfunction and eventually neuronal death (Lipton and Gendelman, 1995; McArthur, 2004; Fischer-Smith and Rappaport, 2005). A plethora of putative neurotoxins have been proposed for this role (Tyor et al., 1992; McArthur, 2004; Fischer-Smith and Rappaport, 2005), although to date, no definitive identification of neurotoxins that are dominant in this process has been made. We believe the data in this study combined with substantial corroborative data in the literature reviewed above points to IFNα as a prominent neurotoxin involved in the pathogenesis of HIV-associated cognitive disorders. The neurotoxic actions of IFNα along with HIV proteins gp120 and Tat may be particularly important early in the development of cognitive deficits, such as in MCMD, by causing glutamate excitotoxicity and dendritic shortening when neuronal death is not a major feature and when cognitive dysfunction is still reversible (Kaiser et al., 1990; McArthur, 2004; Valcour et al., 2004a; Aksenova et al., 2006). As stated, IFNα signaling also can lead to increased production of other inflammatory cytokines IL-1, IL-6, and TNFα, along with the generation of free radicals (Hansson et al., 2004; Dantzer et al., 2008). Perhaps later, during HAD and its advanced stages, neuronal death is caused by the combination of significant neuronal toxicity by IFNα and one or more of these other putative neurotoxins including TNFα, metabolic free radicals, and HIV proteins gp120 and tat described in the literature (Price et al., 1988; Rosenberg and Fauci, 1990; McArthur, 2004; Fischer-Smith and Rappaport, 2005).
This study demonstrates that systemically administered IFNα NAb treatment is effective and safe. Based on these results, clinical trials involving blocking IFNα directly or its downstream effects may be indicated in patients with relatively early forms of HIV-associated cognitive dysfunction. Furthermore, because IFNα is produced in significant amounts in other neuroinflammatory conditions such as CNS lupus, various infectious and noninfectious encephalidities, and multiple sclerosis (Rho et al., 1995; Dafny, 1998; Paul et al., 2007; Mathian and Koutouzov, 2008), it is conceivable that blocking IFNα action may prevent or reduce cognitive dysfunction that is often seen in these disorders.
Footnotes
-
This work was supported by National Institutes of Health Grants F31 NS054592-01A2, C06 RR015455, and AA009986, by Veterans Affairs Merit Award 0007, and by grants from the Medical University of South Carolina Institute of Neuroscience and the Medical University of South Carolina Research Committee.
-
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. William R. Tyor, Neurology Service, Atlanta Veterans Affairs Medical Center, 1670 Clairmont Road, Decatur, GA 30033. william.tyor{at}va.gov





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}