

Abstract
The Biogenesis of Lysosome-Related Organelles Complex 1 (BLOC-1) is a protein complex containing the schizophrenia susceptibility factor dysbindin, which is encoded by the gene DTNBP1. However, mechanisms engaged by dysbindin defining schizophrenia susceptibility pathways have not been quantitatively elucidated. Here, we discovered prevalent and novel cellular roles of the BLOC-1 complex in neuronal cells by performing large-scale Stable Isotopic Labeling of Cells in Culture (SILAC) quantitative proteomics combined with genetic analyses in dysbindin-null mice (Mus musculus) and the genome of schizophrenia patients. We identified 24 proteins that associate with the BLOC-1 complex, many of which were altered in content/distribution in cells or tissues deficient in BLOC-1. New findings include BLOC-1 interactions with the COG complex, a Golgi apparatus tether, and antioxidant enzymes peroxiredoxins 1–2. Importantly, loci encoding eight of the 24 proteins are affected by genomic copy number variation in schizophrenia patients. Thus, our quantitative proteomic studies expand the functional repertoire of the BLOC-1 complex and provide insight into putative molecular pathways of schizophrenia susceptibility.
Introduction
Schizophrenia is a psychotic disorder where genetic factors account for 80% of disease susceptibility (Tandon et al., 2008). The identity of these genetic factors has been under scrutiny leading to the discovery of DTNBP1, the gene encoding dysbindin (Ross et al., 2006; Allen et al., 2008; Sun et al., 2008; Talbot et al., 2009; Ghiani and Dell'Angelica, 2011; Mullin et al., 2011). Dysbindin expression is reduced in hippocampi and cortical areas of schizophrenia patients highlighting the relevance of DTNBP1 and dysbindin in molecular pathways leading to schizophrenia (Talbot et al., 2004, 2011; Tang et al., 2009a; Mullin et al., 2011). Dysbindin is a subunit of the endosome-localized complex Biogenesis of Lysosome-Related Organelles Complex 1 (BLOC-1), an octamer composed of dysbindin, pallidin, muted, snapin, cappuccino, and BLOS1–3 subunits (Li et al., 2004; Di Pietro and Dell'Angelica, 2005; Wei, 2006; Raposo and Marks, 2007; Dell'Angelica, 2009; Lee et al., 2012). Buds and vesicles containing BLOC-1 and AP-3 participate in a cellular route that culminates with the delivery of cargo between early endosomes and late endosomal/lysosomal compartments (Dell'Angelica et al., 1998; Li et al., 2004; Di Pietro and Dell'Angelica, 2005; Borner et al., 2006; Di Pietro et al., 2006; Wei, 2006; Raposo and Marks, 2007; Setty et al., 2007; Dell'Angelica, 2009; Salazar et al., 2009). In neurons, BLOC-1 and AP-3 also define a route that delivers membrane proteins from cell body endosomes to the synapse (Larimore et al., 2011). This route would explain, in part, why null alleles in BLOC-1 alter the composition of synaptic vesicles and the surface expression of neurotransmitter receptors (Talbot et al., 2006; Iizuka et al., 2007; Ji et al., 2009; Newell-Litwa et al., 2009; Tang et al., 2009b; Marley and von Zastrow, 2010; Newell-Litwa et al., 2010), which in turn trigger neurobehavioral phenotypes resembling those found in schizophrenia patients (Hattori et al., 2008; Bhardwaj et al., 2009; Cox et al., 2009; Dickman and Davis, 2009; Talbot, 2009; Cheli et al., 2010; Papaleo et al., 2010). This evidence points toward fundamental vesicle transport processes controlled by dysbindin-BLOC-1 in neurons delineating a schizophrenia susceptibility pathway. However, the expanse of mechanisms controlled by dysbindin and the BLOC-1 complex in neurons remains largely unexplored. Here we approach this question, revealing novel neuronal pathways that intersect with the BLOC-1 complex and their relationship to schizophrenia.
We quantitatively identified dysbindin-BLOC-1 complex interactors and pathways using mass spectrometry and stringent biochemical and genetic criteria. We identified the subunits of the BLOC-1 and AP-3 complexes as well as the Golgi tether, the COG complex (Smith and Lupashin, 2008), and the antioxidant enzymes peroxiredoxins 1–2 (Bell and Hardingham, 2011; Finkel, 2011). Importantly, genes encoding one-third of the proteins identified as dysbindin-BLOC-1 interactors are included in genomic copy number variation (CNV) regions identified in schizophrenia individuals (International Schizophrenia Consortium, 2008; Karayiorgou et al., 2010). Our proteomic analysis expanded upon the functional repertoire of the BLOC-1 complex. Our findings suggest that endosome trafficking controlled by the BLOC-1 complex and associated factors constitute a cell-autonomous molecular pathway of schizophrenia susceptibility.
Materials and Methods
Antibodies and cell culture.
Antibodies used in this study are listed in Table 1. SH-SY5Y and HEK293 (ATCC) cells were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS) and 100 μg/ml penicillin and streptomycin (Hyclone) at 37°C in 10% CO2. SH-SY5Y and HEK cell lines were transfected with 3x-FLAG Dysbindin (catalog #EX-Mm12550-M12) or 3x-FLAG Muted (catalog #EX-T4795-M14) constructs obtained from Genecopoeia. Both constructs were in a pReceiver vector backbone and sequences were independently confirmed. These stably transfected cell lines were maintained in DMEM media supplemented with 10% FBS, 100 μg/ml penicillin and streptomycin, and 0.2 μg/μl neomycin (catalog #SV30068, Hyclone) at 37°C in 10% CO2. For shRNA-mediated pallidin knockdowns, shRNA in a pLKO.1 vector for lentiviral infection was obtained from Open Biosystems (Clone ID: TRCN0000122781, Item #RHS3979-98828366). Control shRNA in pLKO.1 was obtained from Addgene (vector 1864). SH-SY5Y cells were treated with lentiviral particles for 7 d to obtain efficient knockdown. After day 3 of infection, cells were maintained DMEM media supplemented with 10% FBS and puromycin (2 μg/ml; Invitrogen). In some cases, 12 h before lysis, cells were treated with a 3 mm sodium butyrate solution.
Antibodies used in this study
For SILAC labeling, cells were grown in DMEM media with either “light” unlabeled arginine and lysine amino acids (R0K0) or “heavy” 13C- and 15N-labeled arginine and 13C- and 15N-labeled lysine amino acids (R10K8) supplemented with 10% FBS and 100 μg/ml penicillin and streptomycin, and in some cases 0.2 μg/μl neomycin. Cells were grown for a minimum of six passages ensuring maximum incorporation of the amino acids in the cellular proteins. All reagents for SILAC labeling were obtained from Dundee Cell Products. We confirmed the degree of incorporation of labeled amino acids in the total cellular pool as 97.5%.
Immunoprecipitation and immunoaffinity chromatography.
To assess low-affinity interactions of BLOC-1 subunits, we performed cross-linking in intact cells with dithiobis(succinimidylpropionate) (DSP) as previously described (Craige et al., 2008; Salazar et al., 2009; Zlatic et al., 2010). Briefly, untransfected HEK or SH-SY5Y cells or SH-SY5Y cells stably transfected either with FLAG-dysbindin or FLAG-muted were placed on ice, rinsed twice with PBS, and incubated either with 10 mm DSP (Pierce), or as a vehicle control DMSO, diluted in PBS for 2 h on ice. Tris, pH 7.4, was added to the cells for 15 min to quench the DSP reaction. The cells were then rinsed twice with PBS and lysed in buffer A (150 mm NaCl, 10 mm HEPES, 1 mm EGTA, and 0.1 mm MgCl2, pH 7.4) with 0.5% Triton X-100 and Complete anti-protease (catalog #11245200, Roche), followed by incubation for 30 min on ice. Cells were scraped from the dish, and cell homogenates were centrifuged at 16,100 × g for 10 min. The clarified supernatant was recovered, and at least 500 μg of protein extract was applied to 30 μl Dynal magnetic beads (catalog #110.31, Invitrogen) coated with antibody, and incubated for 2 h at 4°C. In some cases, immunoprecipitations were done in the presence of the antigenic 3x-FLAG peptide (340 μm; F4799, Sigma) as a control. The beads were then washed 4–6 times with buffer A with 0.1% Triton X-100. Proteins were eluted from the beads either with sample buffer or by 2 h incubation with either buffer A alone as a control or 340 μm 3x-FLAG antigenic peptide on ice. Samples were resolved by SDS-PAGE and contents analyzed by immunoblot or silver stain. In the case of the large-scale proteomic analysis, proteins eluted from the beads were combined and concentrated by TCA precipitation. Samples were analyzed for SILAC protein identification by Dundee Cell Products, MS Bioworks, and the Emory Center for Neurodegenerative Diseases' Proteomics Facility.
SILAC labeled samples were separated on a 4–12% Bis-Tris Novex mini-gel (Invitrogen) using the MOPS buffer system. The gel was stained with coomassie and the lane was excised into 20 equal segments using a grid. Gel pieces were processed using a robot (ProGest, DigiLab) with the following protocol. First, slices were washed with 25 mm ammonium bicarbonate followed by acetonitrile; then they were reduced with 10 mm dithiothreitol at 60°C followed by alkylation with 50 mm iodoacetamide at room temperature (RT). Samples were digested with trypsin (Promega) at 37°C for 4 h and quenched with formic acid, and the supernatant was analyzed directly without further processing. Each gel digest was analyzed by nano liquid chromatography with tandem mass spectrometry (LC/MS/MS) with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher LTQ Orbitrap Velos. Peptides were loaded on a trapping column and eluted over a 75 μm analytical column at 350nl/min; both columns were packed with Jupiter Proteo resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with MS performed in the Orbitrap at 60,000 FWHM resolution and MS/MS performed in the LTQ. The 15 most abundant ions were selected for MS/MS. Data were processed through the MaxQuant software v1.0.13.13 (www.maxquant.org), which served data recalibration of MS, filtering of database search results at the 1% protein and peptide false discovery rate, and calculation of SILAC heavy:light ratios. Data were searched using a local copy of Mascot.
Mice. Mocha (STOCK gr +/+ Ap3d1mh/J, here referred to as Ap3d1mh/mh) and its control grizzled (STOCK gr +/+ Ap3d1+/J, here referred to as Ap3d+/+) and pallid (B6.Cg-Pldnpa/J, here referred to as Pldnpa/pa) breeding mouse pairs were obtained from The Jackson Laboratory. Muted mice and their controls (B6C3 Aw-J/A-Mutedmu/J, Mutedmu/mu, and CHMU+/mu; Zhang et al., 2002) were obtained from Dr. Richard Swank (Roswell Park Cancer Institute, Buffalo, NY) and bred in-house. Sandy (Dtnp1dntp1/dtnp1) mice in the C57/B6 background were obtained in breeding pairs from Dr. Konrad Talbot (University of Pennsylvania, Philadelphia, PA) as previously described (Cox et al., 2009). All mice were bred in-house following Institutional Animal Care and Use Committee-approved protocols and used at 6 weeks of age. We used both male and female mice indistinctively.
Preparation of tissue and cell lysates.
Hippocampal tissue was dissected from either wild-type C57B6 mice or from dysbindin deficient Sandy mice and placed in cold PBS. We added 500 μl of lysis buffer [Buffer A, 0.5% Triton X-100 and Complete antiprotease (catalog #11245200, Roche)] to each tissue followed by homogenization by sonication on ice. The sonicated tissue was allowed to rest on ice for 30 min followed by a high-speed spin at 16,100 × g for 10 min at 4°C. The supernatant was then recovered, followed by protein measurements and then aliquoted for biochemical analysis.
Control or pallidin knockdown SH-SY5Y cells were rinsed twice with PBS and lysed in buffer A with 0.5% Triton X-100 supplemented with Complete antiprotease, followed by incubation for 30 min on ice. Cells were scraped from the dish, and cell homogenates were centrifuged at 16,100 × g for 10 min. The clarified supernatant was recovered and measured for total protein content. Samples were then analyzed by immunoblot.
Cell lysates were resolved by sucrose sedimentation in 5–20% sucrose gradients as previously described (Salazar et al., 2009).
Dichlorofluorescin diacetate labeling and flow cytometry analysis.
Control or pallidin knockdown SH-SY5Y cells were grown in selection media (DMEM + 10% FBS + puromycin) for 7 d. Cells were then lifted by trypsin, pelleted, and resuspended in PBS and the hydrogen peroxide-sensitive probe dichlorofluorescin diacetate, 4 μl/ml (D-399, Invitrogen). As a negative control, cells were suspended only in PBS (nonstained control). The cells were incubated at 37°C for 30 min, then pelleted at 800 × g for 5 min and resuspended either in PBS only or PBS supplemented with 2 μm hydrogen peroxide as a positive control (added after resuspending in PBS). The samples were protected from light and analyzed by the LSR II FACScan analyzer (Becton Dickinson) using the 488 nm excitation laser. Data were analyzed using the FlowJo software, version 8.2.2 (Tree Star).
Immunofluorescence labeling for confocal microscopy.
Brain-slice sections were prepared from mice between 6 and 8 weeks of age. Animals were anesthetized with Nembutal, then transcardially perfused with Ringer's solution followed by perfusion of fixative (4% paraformaldehyde with 0.1% gluteraldehyde in PBS). Perfused brains were postfixed with 4% paraformaldehyde, which was replaced with PBS within 12–18 h. Brain tissue was cut into 60-μm-thick sections using a vibrating microtome, and brain-slice sections were stored in antifreeze (0.1 m sodium phosphate monobasic, 0.1 m sodium phosphate dibasic heptahydrate, 30% ethylene glycol, 30% glycerol) at −20°C until immunohistochemical preparation.
Brain sections containing the hippocampus formation were incubated for 20 min at room temperature in 1% sodium borohydride. Sections were rinsed with PBS, then preincubated for 60 min at room temperature in a blocking solution (5% normal horse serum (NHS) and 1% BSA and 0.3% Triton X-100). Sections were incubated overnight at 4°C in primary antibody solutions (1% NHS and 1% BSA with the antibodies described in Table 1). The following day, sections were incubated for 60 min at room temperature in a secondary antibody solution (1% NHS and 1% BSA with 1:500 dilutions of Alexa-conjugated secondary antibodies: anti-mouse 488 or 568, anti-rabbit 488 or 568; Invitrogen). Sections were rinsed and then incubated for 30 min at RT in cupric sulfate (3.854 w/v ammonium acetate, 1.596 w/v cupric sulfate in distilled water, pH 5). Sections were rinsed and mounted on glass coverslips with Vectashield mounting media (Vector Laboratories) for confocal microscopy analysis (Newell-Litwa et al., 2010). Confocal microscopy was performed with an Axiovert 100 M microscope (Carl Zeiss) coupled to an Argon and HeNe1 lasers. Images were acquired using Plan Apochromat 10x/0.5 dry, 20x/0.5 dry, and 40x/1.3 and 63x/1.4 DiC oil objectives. Emission filters used for fluorescence imaging were BP 505–530 and LP 560. Images were acquired with ZEN and LSM 510 software (Carl Zeiss). Fluorescence intensities were determined by MetaMorph software (Newell-Litwa et al., 2010). At least four independent stainings from two animals were performed.
Genetics.
We searched for an overlap between the list of schizophrenia candidate genes identified in our study and all the CNVs reported by the International Schizophrenia Consortium (2008; 3391 patients with schizophrenia and 3181 ancestrally matched controls) to determine whether any one of our candidate genes was included. The complete list of all QC-passing CNVs (BED file format, hg17) can be accessed at http://pngu.mgh.harvard.edu/isc/cnv.html. We used UCSC Genome Browser's “Batch Coordinate Conversion (liftOver)” utility to convert genome coordinate annotations from hg17 to hg19. This tool is publicly accessible at http://genome.ucsc.edu/index.html.
Statistical analysis.
Experimental conditions were compared with the nonparametric Wilcoxon–Mann–Whitney Rank Sum Test or one-way ANOVA, Dunnett's Multiple Comparison using Synergy KaleidaGraph v4.03 (Synergy Software) or StatPlus Mac Built 5.6.0pre/Universal (AnalystSoft). Data are presented as dot or boxplots. The latter display the four quartiles of the data, with the “box” comprising the two middle quartiles, separated by the median.
Results
Identification of novel BLOC-1 interacting protein complexes using quantitative SILAC proteomics
We used a highly sensitive quantitative proteomic approach to identify preponderant BLOC-1 interacting proteins to shed light on the broader functions of this complex. We took advantage of a well established in vivo cellular cross-linking protocol based on the cell permeant homobifunctional crosslinking agent DSP. DSP has an 11Å spacer arm and a disulphide bond allowing a reversal of chemical bridges between proteins by reducing agents (Lomant and Fairbanks, 1976; Zlatic et al., 2010). Upon DSP crosslinking, BLOC-1 proteins are presumably associated with other cellular proteins and therefore, predictably, we see a shift in the molecular sedimentation on a sucrose gradient of BLOC-1 component proteins, pallidin and dysbindin, compared with the BLOC-1 proteins that are not crosslinked (Fig. 1A). We selectively enriched for BLOC-1 interacting proteins once molecular associations were chemically stabilized using immunomagnetic purification of protein complexes. We selected human neuroblastoma cell lines (SH-SY5Y) stably expressing either 3x-FLAG-tagged Dysbindin or 3x-FLAG-tagged Muted as a source of BLOC-1 complexes and interactors. We confirmed that tagged BLOC-1 subunits incorporated into the BLOC-1 complex in neuronal and non-neuronal cell lines (see Fig. 4A,B, and unpublished data). Immunoprecipitation using the FLAG antibody from cross-linked FLAG-dysbindin-expressing cells isolated dysbindin as well as other bands detected by silver stain (Fig. 1B, lane 4 asterisk marks FLAG-dysbindin). However, many of the bands precipitated were common with bands in control lanes (Fig. 1B, compare lane 4 with lanes 2, 3, 5). Therefore, to further purify and specifically enrich for polypeptides that would represent BLOC-1 interacting proteins with minimum background, we used the antigenic FLAG-peptide to selectively elute FLAG-dysbindin and its cross-linked interacting proteins from the immunomagnetic beads (Fig. 1B, lane 7). We termed this method “immunoaffinity chromatography.” The immunoblot in Figure 1B, probed with the FLAG antibody, and the silver stain confirmed that FLAG-dysbindin and associated proteins were efficiently and specifically eluted using the antigenic FLAG peptide since only low levels of dysbindin and dysbindin-interacting polypeptides were detected in the magnetic beads after elution (Fig. 1B, compare lanes 7, 8). We further refined the selectivity of the FLAG immunoaffinity chromatography by coupling it to a two-tier protein identification strategy (Fig. 2A). First, protein identification was based on Stable Isotopic Labeling of Cells in Culture (SILAC)-mass spectrometry, followed by a curation of proteins against a database of polypeptides that nonselectively bind to FLAG antibody-decorated magnetic beads from untransfected, DSP cross-linked cell extracts (Ong et al., 2002; Mann, 2006; Trinkle-Mulcahy et al., 2008).

BLOC-1 immunoaffinity chromatography. A, SHSY5Y cells were treated with vehicle (DMSO) or 1 mm DSP. Clarified detergent soluble extracts from DSP treated and untreated cells were sedimented in 5–20% sucrose gradients, and fractions were resolved by SDS-PAGE gels and analyzed by silver stain or probed with antibodies against the BLOC-1 subunits dysbindin and pallidin. B, Lane 1 contains the total crosslinked cytoplasmic lysates from 3x-FLAG Dysbindin-SH-SY5Y cells. As controls, the lysate was incubated with immunomagnetic beads in the absence of antibody (lane 2), decorated with an irrelevant SV2 antibody (lane 3), or decorated with the FLAG antibody either in the absence (lane 4) or presence of the antigenic FLAG peptide (lane 5). Beads as those in lane 4 instead of being eluted with SDS-PAGE sample buffer were eluted either buffer alone (lane 6) or in the presence of an excess FLAG peptide [lane 7; immunoaffinity chromatography (Imm. Aff. Chr.)] where the bands signify putative BLOC-1 interacting proteins. After the peptide elution depicted in lane 7 beads were further eluted with SDS-PAGE sample buffer to determine the protein profile of the leftover material (lane 8). Immunoblot below was probed with the FLAG antibody. Asterisk (*) marks the dysbindin polypeptide.

SILAC experimental design and preparative immunoaffinity chromatography. A, Schematic for the SILAC experimental design: SH-SY5Y neuroblastoma cells stably transfected with either 3xFLAG-Dysbindin or 3xFLAG-Muted were isotopically labeled with either light (R0K0) or heavy (R10K8) media followed by in vivo chemical crosslinking using DSP. Cytoplasmic lysates were then immunoprecipitated either using FLAG antibody beads (R10K8 lysates) or FLAG antibody beads plus FLAG peptide (R0K0 lysates; out competition control). Both sets of beads were then incubated and associated proteins eluted with the antigenic FLAG peptide (immunoaffinity chromatography). The samples were then combined at a 1:1 ratio and analyzed by nano LC/MS/MS. Peptides that were labeled with the R10K8 amino acids and enriched twofold or more were considered as preponderant BLOC-1 interactors. As an additional control, the entire experiment was repeated with HEK293 cells and untransfected SH-SY5Y cells labeled with R0K0 media to identify cellular proteins that nonspecifically bound to FLAG antibody beads. The Venn diagram represents an overall profile of the number of peptides identified in the experiment. One hundred five R10K8-labeled peptides were enriched (B, lanes 1, 2) and 43 were identified to specifically interact with BLOC-1, of which 24 were enriched twofold or more. B, Preparative immunoaffinity chromatography from 3xFLAG-Dysbindin and 3xFLAG-Muted SH-SY5Y cell lines. The silver stain shows the biochemical profile of a fraction of the sample from both cell types that was analyzed by nano LC/MS/MS following SILAC labeling and immunoaffinity chromatography.
Cells expressing 3x-FLAG-tagged BLOC-1 subunits were stably labeled either with “light media” for controls (R0K0) or “heavy media” (R10K8, Fig. 2A) to generate “light” and “heavy” cell extracts. We incubated light cell extracts with FLAG-decorated magnetic beads in the presence of an excess FLAG peptide. This approach completely prevented the binding of FLAG-dysbindin and associated polypeptides to beads making it a robust control to identify nonspecific proteins bound to beads independent of the FLAG antibody (Fig. 1B, lanes 2, 3, 5). In contrast, heavy cell extracts were incubated only with FLAG-antibody decorated magnetic beads. We scaled-up the FLAG immunoaffinity chromatography for preparative purification from either FLAG-muted or FLAG-dysbindin-expressing cell extracts (Fig. 2B). Light- and heavy-labeled eluted polypeptides were mixed 1:1 for MS/MS analysis. We identified a total of 105 eluted polypeptides associated with FLAG dysbindin (Fig. 2A). Subtraction of these 105 polypeptides with a nonspecific polypeptide library generated from FLAG immunoaffinity chromatography eluates derived from untransfected cells reduced them to 43 specific polypeptides (Fig. 2A). We focused on 24 of these 43 proteins because their heavy/light labeling exceeded a ratio of 2 (Fig. 2A, 3A,B, Table 2). The twofold enrichment is a stringent cutoff criterion for SILAC experiments to reliably detect differences in the protein content between two samples (Ong et al., 2002; Ong and Mann, 2006; Trinkle-Mulcahy et al., 2008). The proteins were independently and reproducibly identified in multiple non-SILAC MS/MS experiments using cell lines transfected with two different subunits of the BLOC-1 complex (FLAG-muted and FLAG-dysbindin, Table 2). Some of the proteins identified were known BLOC-1 binding partners validating the experimental approach. For example, we identified most subunits of the AP-3 adaptor protein complex and all eight subunits of the BLOC-1 complex (Di Pietro et al., 2006; Salazar et al., 2009). Importantly, we identified several novel putative BLOC-1 interacting proteins (listed in Table 2 and depicted in Fig. 3). Those include two clathrin heavy-chain isoforms, members of two tethering complexes, the COG and the exocyst complex; redox enzymes (peroxiredoxins I and II), membrane proteins such as the SNARE Snap29 and the potassium channel KCNQ5, as well as proteins involved in axonal guidance and growth CRMP4 and αN-catenin (Fig. 3C, Table 2; Schmidt and Strittmatter, 2007; Suzuki and Takeichi, 2008).

SILAC identification of preponderant putative BLOC-1 interactors. A, B depict 43 specific polypeptides specified in Table 2 that associate with FLAG-dysbindin after subtraction with the nonspecific polypeptide library obtained from cross-linked nontransfected SH-SY5Y and HEK293 cells. Gray lines depict the twofold enrichment cutoff. Red symbols denote proteins identified from SH-SY5Y cells expressing FLAG-dysbindin, and blue symbols denote FLAG-muted. Note that dysbindin and the AP-3 subunit AP-3 β3A are similarly enriched and represented by a similar number of peptides. In B, protein number corresponds to the entry number in Table 2. C shows that 24 proteins exclusively and reproducibly identified as preponderant BLOC-1 interactors assemble in four protein complexes. Other than subunits of the adaptor protein AP-3, members of the BLOC-1 complex, and clathrin 17 (CHC17), other proteins listed here are novel BLOC-1 interactors. The table depicts the maximal fold enrichment for any polypeptide in three independent SILAC experiments and the sum of polypeptides correspond to all polypeptides identified in three SILAC and one non-SILAC MS/MS experiment.
Summary of the BLOC-1 interactors identified by SILAC proteomics and MS/MS protein identification
Biochemical and phenotypic confirmation of the BLOC-1 interacting proteins
We used biochemical and genetic approaches to further authenticate putative BLOC-1 interactors isolated by FLAG-dysbindin immunoaffinity chromatography, namely, coimmunoprecipitation and analysis of changes in the content or distribution of the candidate BLOC-1 interactors in the dentate gyrus of BLOC-1-null mouse brains (Figs. 4, 5). This combined strategy has previously been used to characterize the interaction between the adaptor protein complex AP-3 and BLOC-1 (Fig. 4; Di Pietro et al., 2006; Salazar et al., 2009; Newell-Litwa et al., 2010). Various subunits of the AP-3 complex coimmunoprecipitated with FLAG-dysbindin either from SH-SY5Y or HEK293 cell extracts (Fig. 4A,B). Importantly, this biochemical interaction manifested itself as a decrease in the AP-3 immunoreactivity in the dentate gyrus of BLOC-1 deficient mice sandy (Dtnbp1sdy/sdy, Fig. 4F, F′) by quantitative confocal microscopy, a finding similar to our previous report in Mutedmu/mu-null dentates (Newell-Litwa et al., 2010). The robustness of this BLOC-1-null phenotype is highlighted by the reciprocal decrease in pallidin immunoreactivity in the dentate gyrus of AP-3 deficient mice (mocha, Ap3d1mh/mh; Fig. 4H, H′).

BLOC-1 interacts biochemically and genetically with the AP-3 complex. A, B, Crosslinked lysates from HEK and SH-SY5Y cells stably transfected with FLAG-dysbindin were immunoprecipitated with the FLAG antibody (lanes 2, 2′). Lanes 1, 1′ contain the total cellular lysate input and as a negative control the immunoprecipitation was done in the presence of the antigenic peptide (out competition control; lanes 3, 3′). Samples were analyzed by immunoblotting with BLOC-1 subunit antibodies and AP-3 subunit antibodies. C–J′, Dentate gyri from wild-type (C, E, G, I, K), AP-3-null (Ap3d1mh/mh, D, H), and BLOC-1-null mouse brains (Pldnpa/pa, M; Mutedmu/mu, J; and Dtnbp1sdy/sdy, L) were stained with antibodies against AP-3 δ and VAMP2 as a control. C′–M′ depict AP-3 δ staining; note the marked alterations in the AP-3 δ staining in the AP-3-null Ap3d1mh/mh brains.
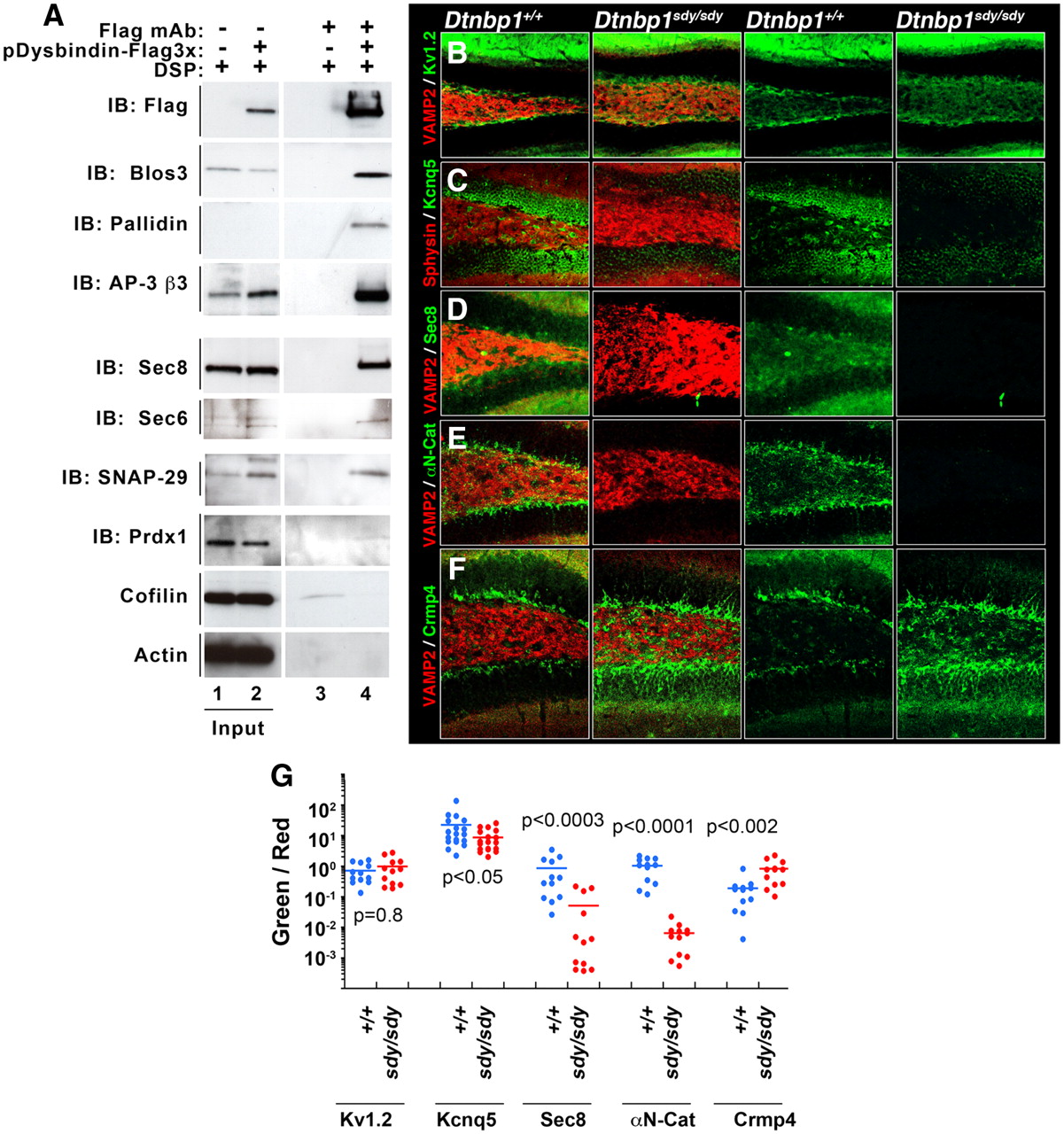
Biochemical and genetic confirmation of preponderant BLOC-1 interactors. A, Immunoaffinity chromatography experiments, using the FLAG antibody and eluting with FLAG peptide, were performed using lysates prepared from either 3xFLAG-Dysbindin SH-SY5Y cells or from untransfected SH-SY5Y cells. Each of the putative BLOC-1 interactors was specifically found only in the experimental lane 4 and not in the control lane 3. Controls were performed with actin and cofilin. B–F, Dentate gyri of wild-type and BLOC-1 deficient mice (Dtnbp1sdy/sdy) were analyzed for alteration in content or distribution of the newly identified BLOC-1 interactors. Immunoreactivity of αN-Catenin, the exocyst subunit Sec8, and the potassium channel KCNQ5 was decreased in BLOC-1 deficient Dtnbp1sdy/sdy mice. In contrast, Crmp4 immunoreactivity is increased in the dentate of Dtnbp1sdy/sdy mice. VAMP2, Synaptophysin (Sphysin) and the potassium channel Kv1.2 antibodies (B) were used as positive controls. G, The total fluorescent pixels for each of the antibodies were quantified by MetaMorph analysis and expressed as a ratio to their corresponding control antibodies, i.e., KCNQ5/Sphysin (C), Sec8/VAMP2 (D), αN-Catenin/VAMP2 (E), and Crmp4/VAMP2 (F). Dot plot depicts the fluorescent ratios in wild-type (Dtnbp1+/+) and BLOC-1 deficient mice (Dtnbp1sdy/sdy).
To test the relevance of novel putative BLOC-1 interactors, we used, as a confidence criterion, either coimmunoprecipitation with tagged dysbindin and/or alteration in antigen distribution/content in the dentate gyrus of dysbindin-null mouse brains (Dtnbp1sdy/sdy). FLAG-dysbindin coprecipitated BLOC-1 (pallidin and Blos3) and AP-3 (AP-3 β3) subunits (Figs. 4A,B, 5A). Importantly, putative novel interactors such as the exocyst subunits (sec6 and sec8), snap29, and peroxiredoxin I also coprecipitated selectively with FLAG-dysbindin. Importantly, abundant proteins such as cofilin and actin were not found in FLAG-dysbindin precipitates (Fig. 5A, compare lanes 3, 4). Other proteins identified by SILAC such as αN-catenin, CRMP4, and KCNQ5 could not be detected reliably in FLAG-dysbindin or FLAG-muted immunoprecipitations. Therefore, we resorted to distribution/content modifications of these antigens in the dentate gyrus of Dtnbp1sdy/sdy mouse brains using quantitative confocal microscopy. The distribution/content of KCNQ5, Sec8, CRMP4, and αN-catenin was specifically altered in the dentate gyrus of BLOC-1 deficient Dtnbp1sdy/sdy mice. While the immunoreactivity of KCNQ5 and αN-catenin was reduced in Dtnbp1sdy/sdy dentate gyri, CRMP4 immunoreactivity increased (Fig. 5B–F). All these phenotypes were selective as evidenced by the colabeling with VAMP2 or synaptophysin (Sphysin) antibodies, which detect two synaptic vesicle protein not affected by BLOC-1 deficiencies (Newell-Litwa et al., 2009). As an additional control for KCNQ5, we analyzed the distribution/content of another unrelated potassium channel, Kv1.2. Kv1.2 was not affected in Dtnbp1sdy/sdy brains (Fig. 5B,G). Overall, our results revealed that our unbiased and quantitative mass-spectrometry identification of BLOC-1 interactors generated a specific database of proteins whose expression can phenotypically distinguish wild-type from Dtnbp1sdy/sdy dentate gyri. Moreover, these results point to two previously unsuspected associations of the BLOC-1 complex: first, with the COG Golgi tethering complex, and second, with peroxiredoxins, enzymes involved in redox metabolism.
Functional interaction between the BLOC-1 and the COG complex Golgi tether
We focused on the COG complex, a hetero-octameric protein complex that acts as a tether for vesicles fusing with the cis-Golgi that regulates the steady-state content of Golgi proteins (Shestakova et al., 2006; Ungar et al., 2006; Smith and Lupashin, 2008). The COG complex is organized into two distinct lobes: lobe A (Cog1–4) and lobe B (Cog5–8; Ungar et al., 2002; Lees et al., 2010). Our proteomic analysis directly identified 2 subunits from lobe B that coisolate with the FLAG dysbindin: Cog 6 and Cog 8 (Fig. 3, Table 2). However, upon further analysis we discovered that FLAG dysbindin coprecipitates subunits of both lobe A (Cog 3 and 4) and lobe B (Cog 5, 6, 7 and 8; Fig. 6A). Using a Cog5 antibody, we reciprocally coimmunoprecipitated FLAG-dysbindin from transfected cell lines (Fig. 6B) as well as the endogenous BLOC-1 subunit pallidin from untransfected cell lines (Fig. 6C). Notably, the chemical crosslinker DSP was not essential to reveal the interaction (Fig. 6B, lane 6; 6D, lane 7) indicating that the COG-BLOC-1 interaction does not require the stringency of our precipitation assay and is maintained even in the absence of chemical stabilization. Importantly, and consistent with previously published data (Ungar et al., 2002), the COG complex isolated with Cog5 antibodies did not coprecipitate the exocyst complex as revealed by the absence of Sec8 in protein complexes precipitated with Cog5 antibodies (Fig. 6C; IB: Sec8). The BLOC-1-COG interaction is conserved regardless of the cell type used (neuronal cell type in Fig. 6B,C, and epithelial cell line in Fig. 6D) suggesting that a ubiquitous pathway between BLOC-1 and the COG tether may exist in all cells.

BLOC-1 complexes associate with the Golgi tether, the COG complex. A, BLOC-1 interactions with COG complex subunits Cog 2–8 were independently confirmed by immunoaffinity chromatography using lysates from untransfected SH-SY5Y cells and 3xFLAG-Dysbindin SH-SY5Y cells. Noninteracting protein controls actin or cofilin can be found in Figure 4A. B, Cog5 antibody reciprocally immunoprecipitates FLAG-dysbindin from 3xFLAG-Dysbindin SH-SY5Y cells or C, the endogenous BLOC-1 subunit pallidin from SH-SY5Y cells (lane 7). The exocyst subunit Sec8 was not coimmunoprecipitated with the Cog5 antibody indicating the specificity of the Cog5-BLOC-1 interaction. D, The BLOC-1-COG interaction exists in a non-neuronal HEK293 cell line both by immunoprecipitation (lanes 7, 8) and by immunoaffinity chromatography (lane 12). Transferrin receptor (TrfR) was used as a control. E, SH-SY5Y cells where the BLOC-1 complex expression is downregulated by treatment with shRNA against pallidin also display a concomitant decrease in the expression of the COG subunits as well as a downstream COG-sensitive Golgi membrane protein CASP. Actin and clathrin heavy chain are used as controls. F, G, COG7 as well as CASP content in hippocampal extracts wild-type (Dtnbp1+/+) and BLOC-1 deficient mice (Dtnbp1sdy/sdy). Dots depict independent determinations performed in six animals.
Loss-of-function of individual subunits of the BLOC-1 complex leads to downregulation of the other subunits of the BLOC-1 octamer, suggesting that a loss of protein expression may be useful as an assay for structural or functional protein associations beyond those of the BLOC-1 complex proper (Zhang et al., 2002; Li et al., 2003; Starcevic and Dell'Angelica, 2004). Thus, we explored whether the downregulation of the BLOC-1 subunit pallidin (Fig. 6E) could alter the content of other BLOC-1 subunits and possibly components of the COG complex. SH-SY5Y cells treated with shRNA directed against pallidin downregulated the BLOC-1 subunits muted and dysbindin, as is the case with murine null alleles of these BLOC-1 subunits (Fig. 6E; Zhang et al., 2002; Li et al., 2003; Starcevic and Dell'Angelica, 2004). Importantly, the content of the COG subunit Cog7 was also partially decreased in pallidin shRNA-treated cells. This effect of BLOC-1 loss-of-function upon Cog7 content was recapitulated in hippocampi from BLOC-1 deficient Dtnbp1sdy/sdy mice (Fig. 6F,G).
To further estimate the downstream effect of BLOC-1 loss of function upon COG function, we took advantage of the previously characterized “COG-sensitive” proteins. There are seven COG mutation-sensitive Golgi resident integral membrane proteins (GEARs) whose levels are downregulated in the absence of the COG complex (Oka et al., 2004). Among those, we used CASP, an alternative splicing variant of the CUX1 gene, as a reporter for COG function (Gillingham et al., 2002; Oka et al., 2004). We hypothesized that downregulation of COG complex subunits by BLOC-1 loss-of-function could, in turn, result in CASP downregulation. Indeed, CASP levels in pallidin shRNA-treated cells were reduced to 77% of shRNA control-treated cells (Fig. 6E; 77.2 ± 5.5%, n = 4, p = 0.021; Wilcoxon–Mann–Whitney Rank Sum Test). Similarly, the content of CASP was reduced in the hippocampal formation of BLOC-1 deficient Dtnbp1sdy/sdy mice (Fig. 6F,G). This evidence supports a biochemical and functional interaction between the BLOC-1 and the COG complex and suggests a novel pathway linking the endosome-localized BLOC-1 protein complex to the Golgi apparatus.
BLOC-1 deficiency affects steady-state peroxide levels
Our FLAG dysbindin proteomic analysis identified an additional novel association with peroxiredoxins I and II. These enzymes are involved in metabolism of peroxide and therefore the prediction is that BLOC-1 could modulate the redox state in cells (Bell and Hardingham, 2011). We tested the peroxiredoxin and BLOC-1 complex interaction analyzing the content/distribution of these enzymes in the dentate gyrus of BLOC-1 deficient Dtnbp1sdy/sdy mice as well as neuroblastoma cells rendered BLOC-1 deficient by shRNA downregulation of another BLOC-1 subunit pallidin. Similar to other proteins identified by FLAG-dysbindin immunoaffinity chromatography, BLOC-1 deficiency either in the dentate gyrus of BLOC-1 deficient sandy Dtnbp1sdy/sdy mice or in pallidin shRNA-treated neuroblastoma cells demonstrated a significant decrease in peroxiredoxins immunoreactivity (Fig. 7A–F). The Dtnbp1sdy/sdy dentate gyrus and pallidin shRNA-induced peroxiredoxin phenotypes provide strong evidence of a functional consequence of the association between these redox enzymes and the BLOC-1 complex. Since peroxiredoxins scavenge hydrogen peroxide, we predicted that downregulated peroxiredoxin I and II levels observed in BLOC-1 deficiencies would induce a steady-state increase in cellular hydrogen peroxide levels. To test this hypothesis, we used a flow cytometry assay, using neuroblastoma cells treated with control or pallidin targeting shRNA where cellular peroxide levels were measured in vivo using the hydrogen peroxide-specific fluoroprobe 2′,7′-dichlorofluorescein (DCF; Myhre et al., 2003; Cossarizza et al., 2009). Fluorescence intensity in control shRNA-treated neuroblastoma cells was increased 1.6-fold by the addition of a physiologically relevant concentration of 2 μm H2O2 (Fig. 7G,I). Notably, shRNA downregulation of one of the BLOC-1 subunit, pallidin, was sufficient to increase DCF fluorescence intensity robustly. The magnitude of this increase in BLOC-1 downregulated cells was comparable to the increase seen with exogenously added H2O2 to control cells (Fig. 7H,I). Thus, these results identify a novel function of the BLOC-1 complex regulating enzymes involved in redox metabolism.

BLOC-1 deficiences modulate peroxiredoxin levels and cell redox state. A–E, Dentate gyri of wild-type (Dtnbp1+/+) and BLOC-1 deficient (Dtnbp1Sdy/Sdy) mice were analyzed for alteration in content or distribution of redox enzymes peroxiredoxin 1 and peroxiredoxin 2 (Prdx1, Prdx2). Fluorescence intensity is significantly reduced in the dentate region of BLOC-1 deficient mice. VAMP2 was used as positive control. E, Box plot depicts the fluorescent ratios in wild-type (Dtnbp1+/+) and BLOC-1 deficient (Dtnbp1sdy/sdy) mice. F, Reduction in Prdx1 and Prdx2 levels is recapitulated in SH-SY5Y cells where BLOC-1 is downregulated by shRNA. G, H, Flow cytometry quantification of cellular hydrogen peroxide in control shRNA and pallidin shRNA-treated cells using a fluorescent probe against hydrogen peroxide (DCF). As controls, cells were either not stained with the probe or treated with 2 μm hydrogen peroxide and stained with DCF. I, Ratios of Experimental and Control means of cell population fluorescence profiles depicted in G, H. Each dot represents an independent experiment performed with three independent shRNA knock-downs. One-way ANOVA, Dunnett's multiple comparison.
Discussion
We explored prevalent cellular roles of the BLOC-1 complex in neuronal cells by performing a large-scale SILAC proteomic analysis. We identified 105 proteins and, after a stringent two-tier set of filters, zeroed in on 24 proteins that associate with the BLOC-1 subunit dysbindin—a schizophrenia susceptibility factor. Of these 24 proteins, 11 were novel BLOC-1 binding partners. Prominent among these 24 proteins are all the subunits of the BLOC-1 and AP-3 adaptor complexes. Each of these 24 proteins was independently confirmed to be a BLOC-1 interactor and many were altered in content/distribution in cells or tissues deficient in BLOC-1 complexes. Major new findings from our studies are BLOC-1 interactions with the COG complex Golgi tether, and biochemical and genetic evidence supporting interactions between BLOC-1 and the antioxidant enzymes peroxiredoxins I and II. Thus, our quantitative proteomic analysis expanded upon the functional repertoire of the BLOC-1 complex and provides insight into molecular pathways of schizophrenia susceptibility, which may now include endosome to TGN retrograde traffic (COG) as well as redox metabolism (peroxiredoxins).
A common way to elucidate the function of proteins or their complexes is to identify other proteins associated with it. However, there are pitfalls to the methods used to define interactors, in particular those associated with BLOC-1 (Ghiani and Dell'Angelica, 2011). To address several of the problems associated with studying the BLOC-1 complex, such as the low expression levels of BLOC-1 subunits, we used a multifold approach. We quantitatively estimated peptides specifically enriched by immunoaffinity chromatography of FLAG-tagged BLOC-1 subunits purified from cells labeled with nonradioactive isotope-tagged amino acids (Mann, 2006). This procedure was coupled with in vivo crosslinking with DSP to stabilize BLOC-1 interactions (Zlatic et al., 2010). Since DSP was used at a substoichiometric level, we likely identified major BLOC-1 interacting proteins as attested by the enrichment of BLOC-1 and AP-3 subunits. The substoichiometric use of DSP was intended to stabilize immediate, or first-order, interactors and to minimize formation of extended crosslinked higher order networks. For example, crosslinked complexes revealed that BLOC-1 co-isolated with either the COG complex or exocyst complex proteins and did not result in a large complex containing BLOC-1, COG, and exocyst subunits together (Ungar et al., 2002). An additional layer of stringency in protein identification was the independent verification of protein interactions in BLOC-1 deficiencies, either using neuroblastoma cells rendered deficient by shRNA or hippocampal tissue from Dtnbp1sdy/sdy mice. We reasoned that if proteins interact with the BLOC-1 complex, then they could reveal novel phenotypes in BLOC-1 deficiencies. One of these phenotypes is the co-downregulation of BLOC-1 subunits in cellular lysates when one of the subunits is absent or reduced (Zhang et al., 2002; Li et al., 2003; Starcevic and Dell'Angelica, 2004). Such a phenotype was observed with peroxiredoxins I and II, as well as one of the COG subunits, Cog7. For another group of markers, we observed that their immunoreactivity was modified in the dentate gyrus of Dtnbp1sdy/sdy mice. Such is the case of AP-3 subunits, KCNQ5, peroxiredoxins I and II, αN-catenin, CRMP4, the exocyst subunit Sec8, and Snap29 (our unpublished observations).
Dysbindin interactions and their functions are frequently considered independent of the BLOC-1 complex. However, several lines of evidence summarized by Ghiani and Dell'Angelica emphasize dysbindin to be an integral component of the BLOC-1 complex (Ghiani and Dell'Angelica, 2011; Mullin et al., 2011). Our data are in rapport with this paradigm. The BLOC-1 interactions reported here are reproducibly obtained with two independent BLOC-1 subunits: dysbindin and muted. Furthermore, phenotypes induced by downregulation of a third BLOC-1 subunit, pallidin, are recapitulated in dysbindin-null, Dtnbp1sdy/sdy mice. This is particularly evident in the downregulation of Cog7 both in pallidin and dysbindin-deficient cells or tissues, respectively. What is the nature of the association between the COG and the BLOC-1 complex? One way we addressed this question was to analyze one of the COG-sensitive integral membrane Golgi proteins, collectively called GEAR proteins. GEAR proteins are reduced in COG-deficient cells and therefore provide a phenotypic readout for COG functions (Oka et al., 2004). CASP, the GEAR protein analyzed here, is an integral Golgi membrane protein (Gillingham et al., 2002). Our analysis demonstrated that when BLOC-1 was downregulated, it led to a decreased level of COG complex proteins and in turn led to a reduction of CASP. These data indicate that the BLOC-1-dependent reduction of a COG complex subunit, although moderate, is sufficient to trigger a COG-dependent phenotype. These findings suggest that the BLOC-1 complex participates in an endosome route back to the Golgi complex delivering membrane proteins resident to or transiting through the Golgi complex (Smith et al., 2009). Since the CASP downregulation observed in BLOC-1 deficiency is subtle, as expected from a moderate Cog7 reduction, we speculate that a subset of COG-dependent vesicles derived from endosomes and bound to the Golgi complex may be uniquely susceptible to BLOC-1 deficiency. These vesicles likely would use the Snap29 SNARE, another new associate of the BLOC-1 complex. In agreement with this model, we have recently identified Snap29 as a direct binding partner of Cog6 protein (V. V. Lupashin, unpublished data).
Another interesting family of proteins identified in our SILAC proteomic analysis is peroxiredoxins. Peroxiredoxins I and II are ubiquitously expressed enzymes that remove low level peroxides generated as a result of steady-state cellular metabolism (Bell and Hardingham, 2011). These two enzymes are downregulated in pallidin shRNA-treated cells and their immunoreactivity is decreased in the dentate gyrus of the hippocampal formation from BLOC-1 deficient Dtnbp1sdy/sdy mice. This decrease in the peroxiredoxins, in turn, resulted in a significant increase in the steady levels of hydrogen peroxide in the range of low micromolar level. Apart from being a byproduct of oxidative metabolism, hydrogen peroxide also participates in cell signaling (Finkel, 2011). Consequently, peroxiredoxins play essential roles in mediating signaling cascades targeted by hydrogen peroxide (Neumann et al., 2009; Finkel, 2011). BLOC-1 may regulate the activity or subcellular location of peroxiredoxins, for example, in signal transduction by tyrosine kinase receptors on endosomes. Alternatively, peroxiredoxins may modulate BLOC-1 function by a redox mechanism, such as by regulating the oxidation status of cysteines either in BLOC-1 subunits (all human BLOC-1 isoforms contain cysteine residues), or in membrane proteins in close proximity of BLOC-1. It is of interest that the levels of peroxiredoxin I are reduced in the frontal cortex of schizophrenia patients (Föcking et al., 2011; Martins-de-Souza et al., 2011). Such a reduction is consistent with the low expression of dysbindin and other BLOC-1 subunits in cortical areas of patients with schizophrenia (Talbot et al., 2004, 2011; Tang et al., 2009a; Mullin et al., 2011). This raises the possibility that schizophrenia pathogenesis hypotheses centered on redox alterations and those linked to dysbindin may converge on a common molecular mechanism.
Part of our interest in BLOC-1 and dysbindin biology stems from its correlation with schizophrenia risk (Talbot et al., 2009; Mullin et al., 2011). We hypothesized that if dysbindin is part of a molecular pathway contributing to or affected by schizophrenia, then genes encoding dysbindin interactors should be significantly represented among those genes with structural variants associated with schizophrenia risk, such as SNAP29 (Malhotra et al., 2011). To test this prediction, we analyzed the International Schizophrenia Consortium database of cases carrying rare chromosomal deletions and duplications that increase risk of schizophrenia (International Schizophrenia Consortium, 2008). This database contains 3391 schizophrenia cases and 3181 controls. Our prediction is strongly supported by CNVs encompassing genes encoding dysbindin interactors (Table 3). Eight of the 24 proteins identified as dysbindin-BLOC-1 interactors are represented among genes within CNVs exclusively found in schizophrenia patients. Among those we found COG and AP-3 complex subunits. None of these eight loci are affected in two genome-wide analyses of unaffected individuals totaling 3853 subjects (International Schizophrenia Consortium, 2008; Buizer-Voskamp et al., 2011). Strikingly, the dysbindin interactors Snap29 and the clathrin heavy-chain isoform CLTCL1 (CHC22) were among those proteins whose genes are most frequently affected in schizophrenia individuals (Table 3). These two genes are located within the chromosome 22q11.2 region. Individuals with haploinsufficiency of this region have 22q11.2 deletion syndrome and develop schizophrenia at rate of ∼30%. These deletions account for as many as ∼2% of de novo schizophrenia cases in the general population (Karayiorgou et al., 2010). The 22q11.2 deletion syndrome also encompasses SEPT5, the gene encoding septin 5, a protein that binds septin 8 and AP-3 complexes both found in our dysbindin proteome (Baust et al., 2008; Nakahira et al., 2010). Thus, 22q11.2 deletion syndrome combines up to three haploinsufficiencies that converge on a pathway defined by the schizophrenia susceptibility factor dysbindin. The molecular associations between a clathrin heavy-chain isoform (CHC22), Snap29, and septin 5 suggest that the 22q11.2 deletion syndrome may have a pronounced deficiency of this pathway. These findings support the concept that quantitative proteomes of a schizophrenia susceptibility factor, such as dysbindin, can define putative schizophrenia susceptibility pathways by revealing unsuspected connections between the disease-associated genomic loci. This hypothesis is supported by the dysbindin interactor Snap29 and the clathrin heavy-chain isoform CLTCL1 (CHC22), which are among those proteins whose genes are frequently affected in schizophrenia individuals carrying CNVs associated to disease (Table 3).
Candidate genes included in CNVs from schizophrenia cases and controls
Pathogenic hypotheses for schizophrenia have tended to emphasize individual genes of “interest” rather than cell-autonomous pathways defined by the molecular interactions of a schizophrenia susceptibility factor (Ross et al., 2006; Tandon et al., 2008). One of those putative pathways is an endosomal hub defined by dysbindin and its protein interactors, many of which remained unknown or were not prioritized based on their abundance in dysbindin isolates (Mead et al., 2010). Based on the work presented here, we propose that defective endosome sorting mechanisms controlled by the BLOC-1 complex may contribute to the pathogenesis of schizophrenia and systemic disorders that characterize the 22q11.2 deletion syndrome.
Footnotes
This work was supported by grants from the National Institutes of Health to V.F. (NS42599, GM077569) and V.V.L. (GM083144). A.G. and J.L. were supported by National Institutes of Health Fellowships in Research and Science Teaching Grant K12 GM000680. This work was supported in part by the Neuronal Imaging Core of the Emory Neuroscience National Institute of Neurological Disorders and Stroke Core Facilities Grant P30NS055077, and by the Flow Cytometry Core Facility of the Emory University School of Medicine. We are indebted to the Faundez laboratory members and Dr. Frances Brodsky for their comments.
- Correspondence should be addressed to Victor Faundez, Department of Cell Biology, Emory University School of Medicine, 615 Michael Street, Room 446, Atlanta, GA 30322. vfaunde{at}emory.edu
References
- Allen et al., 2008.↵
- Baust et al., 2008.↵
- Bell and Hardingham, 2011.↵
- Bhardwaj et al., 2009.↵
- Borner et al., 2006.↵
- Buizer-Voskamp et al., 2011.↵
- Cheli et al., 2010.↵
- Cossarizza et al., 2009.↵
- Cox et al., 2009.↵
- Craige et al., 2008.↵
- Dell'Angelica, 2009.↵
- Dell'Angelica et al., 1998.↵
- Dickman and Davis, 2009.↵
- Di Pietro and Dell'Angelica, 2005.↵
- Di Pietro et al., 2006.↵
- Finkel, 2011.↵
- Föcking et al., 2011.↵
- Ghiani and Dell'Angelica, 2011.↵
- Gillingham et al., 2002.↵
- Hattori et al., 2008.↵
- Iizuka et al., 2007.↵
- International Schizophrenia Consortium, 2008.↵
- Ji et al., 2009.↵
- Karayiorgou et al., 2010.↵
- Larimore et al., 2011.↵
- Lee et al., 2011.↵
- Lees et al., 2010.↵
- Li et al., 2003.↵
- Li et al., 2004.↵
- Lomant and Fairbanks, 1976.↵
- Malhotra et al., 2011.↵
- Mann, 2006.↵
- Marley and von Zastrow, 2010.↵
- Martins-de-Souza et al., 2011.↵
- Mead et al., 2010.↵
- Mullin et al., 2011.↵
- Myhre et al., 2003.↵
- Nakahira et al., 2010.↵
- Neumann et al., 2009.↵
- Newell-Litwa et al., 2009.↵
- Newell-Litwa et al., 2010.↵
- Oka et al., 2004.↵
- Ong and Mann, 2006.↵
- Ong et al., 2002.↵
- Papaleo et al., 2012.
- Raposo and Marks, 2007.↵
- Ross et al., 2006.↵
- Salazar et al., 2009.↵
- Schmidt and Strittmatter, 2007.↵
- Setty et al., 2007.↵
- Shestakova et al., 2006.↵
- Smith and Lupashin, 2008.↵
- Smith et al., 2009.↵
- Starcevic and Dell'Angelica, 2004.↵
- Sun et al., 2008.↵
- Suzuki and Takeichi, 2008.↵
- Talbot, 2009.↵
- Talbot et al., 2004.↵
- Talbot et al., 2006.↵
- Talbot et al., 2009.↵
- Talbot et al., 2011.↵
- Tandon et al., 2008.↵
- Tang et al., 2009a.↵
- Tang et al., 2009b.↵
- Trinkle-Mulcahy et al., 2008.↵
- Ungar et al., 2002.↵
- Ungar et al., 2006.↵
- Wei, 2006.↵
- Zhang et al., 2002.↵
- Zlatic et al., 2010.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}