

Abstract
Activity-dependent bulk endocytosis is the dominant synaptic vesicle retrieval mode during high intensity stimulation in central nerve terminals. A key event in this endocytosis mode is the generation of new vesicles from bulk endosomes, which replenish the reserve vesicle pool. We have identified an essential requirement for both adaptor protein complexes 1 and 3 in this process by employing morphological and optical tracking of bulk endosome-derived synaptic vesicles in rat primary neuronal cultures. We show that brefeldin A inhibits synaptic vesicle generation from bulk endosomes and that both brefeldin A knockdown and shRNA knockdown of either adaptor protein 1 or 3 subunits inhibit reserve pool replenishment from bulk endosomes. Conversely, no plasma membrane function was found for adaptor protein 1 or 3 in either bulk endosome formation or clathrin-mediated endocytosis. Simultaneous knockdown of both adaptor proteins 1 and 3 indicated that they generated the same population of synaptic vesicles. Thus, adaptor protein complexes 1 and 3 play an essential dual role in generation of synaptic vesicles during activity-dependent bulk endocytosis.
Introduction
Neuronal activity stimulates the fusion of neurotransmitter-containing synaptic vesicles (SVs) with the nerve terminal plasma membrane. SVs that are available for exocytosis in central nerve terminals are commonly referred to as the recycling pool (Sudhof, 2000), which can be further subdivided into the readily releasable pool (RRP) and the reserve pool. The RRP contains SVs that are immediately available for fusion, whereas the reserve pool only contributes SVs during periods of intense neuronal activity (Sudhof, 2000).
Neuronal activity is also a key determinant in the triggering of specific SV retrieval modes after exocytosis. Clathrin-mediated endocytosis (CME) is the dominant SV retrieval mode during mild stimulation (Granseth et al., 2006; Zhu et al., 2009), whereas during increased activity activity-dependent bulk endocytosis (ADBE) dominates (Clayton et al., 2008). ADBE is a high capacity SV retrieval mode that forms endosomes directly from large invaginations of the nerve terminal plasma membrane (Clayton and Cousin, 2009a). SVs that are generated from bulk endosomes specifically repopulate the reserve pool (Richards et al., 2000; Cheung et al., 2010), suggesting a functional link between reserve pool mobilization and ADBE triggering (Clayton and Cousin, 2009a; Shupliakov, 2009).
The formation of functional SVs from donor membrane requires the efficient sorting of protein cargo, a role performed at the plasma membrane by the adaptor protein (AP) 2 complex during CME (Royle and Lagnado, 2003). The molecules that mediate SV generation from bulk endosomes during ADBE are still unknown. This process is thought to be clathrin dependent (Heerssen et al., 2008; Kasprowicz et al., 2008), suggesting that AP complexes will also be required. Five different AP complexes have been identified (AP-1 to AP-5) that mediate vesicle formation at specific donor membranes (Boehm and Bonifacino, 2001; Robinson, 2004; Hirst et al., 2011). In both non-neuronal and neuroendocrine cells, endosomal vesicle budding is inhibited by brefeldin A (BFA), which inhibits the GTPase ADP-ribosylation factor 1 (ARF1) (Drake et al., 2000). ARF1 is essential for the recruitment of both AP-1 and AP-3 to membrane, implicating both AP-1 and AP-3 in endosomal vesicle generation (Faundez et al., 1998; Pagano et al., 2004; Newell-Litwa et al., 2007).
A potential role for either AP-1 or AP-3 in SV generation from bulk endosomes is supported by previous studies. Both AP complexes are enriched in central nerve terminals (Glyvuk et al., 2010; Newell-Litwa et al., 2010) and are present on SVs (Takamori et al., 2006). Furthermore, SV endocytosis is sensitive to BFA only during intense activity in neuronal culture (Voglmaier et al., 2006; Kim and Ryan, 2009). Finally, mice lacking the σ subunit of AP-1B display SV recycling defects, a large increase in endosome numbers, and a delay in recycling pool replenishment after strong stimulation (Glyvuk et al., 2010).
We directly examined the role of AP-1 and AP-3 in SV generation from bulk endosomes using recently established morphological and optical assays that specifically track SVs derived from this ADBE-dependent compartment. We uncovered an essential requirement for both AP-1 and AP-3, highlighting a molecular locus in a key SV endocytosis mode triggered during high intensity stimulation.
Materials and Methods
FM1-43, FM2-10, penicillin/streptomycin, phosphate-buffered salts, fetal calf serum, Minimal Essential Medium, and Alexa Fluor 568 goat anti-mouse IgG antibody were purchased from Invitrogen. Glutaraldehyde and osmium tetroxide were from Agar Scientific. ProFection mammalian calcium phosphate transfection system was obtained from Promega. Anti-AP-1γ was purchased from BD Biosciences. Anti-AP-3δ antibody was from Developmental Studies Hybridoma Bank. All other reagents were from Sigma. shRNAs targeting AP-1γ or AP-3δ subunits were designed using the pSUPER vector system (pSUPER neo-GFP, OligoEngine) with the following oligonucleotides: Oligo AP-1a, CAAACGCATTGGCTATTTA; Oligo AP-1b, GGAATGCTATTCTGTATGA; Oligo AP-3a, ACAAAGTGTTCCTCAAGTA; Oligo AP-3b, CTATCATCCTGGAGAATCA. The pSUPER vector was engineered to express mCerulean as described previously (Cheung et al., 2010).
Primary cultures, transfections and immunofluorescence studies of cerebellar granule neurons.
Preparation, transfections, and immunofluorescence studies of rat cerebellar granule neurons (prepared from 7-d-old pups of either sex) were carried out according to previously described protocols (Tan et al., 2003; Clayton et al., 2009). Neurons were used between 8 and 10 d in vitro for all experiments. In some experiments, cultures were transfected using a calcium phosphate precipitation protocol between 5 and 7 d in vitro and used after 72 h. AP-1γ or AP-3δ expression was separately monitored by immunofluorescence intensity. For quantification, the fluorescence intensity in transfected neurons was expressed as a percentage of the averaged expression in untransfected neurons in the same field of view. At least three independent experiments were performed, with five fields of view being assessed for each experiment.
Fluorescence imaging of SV turnover
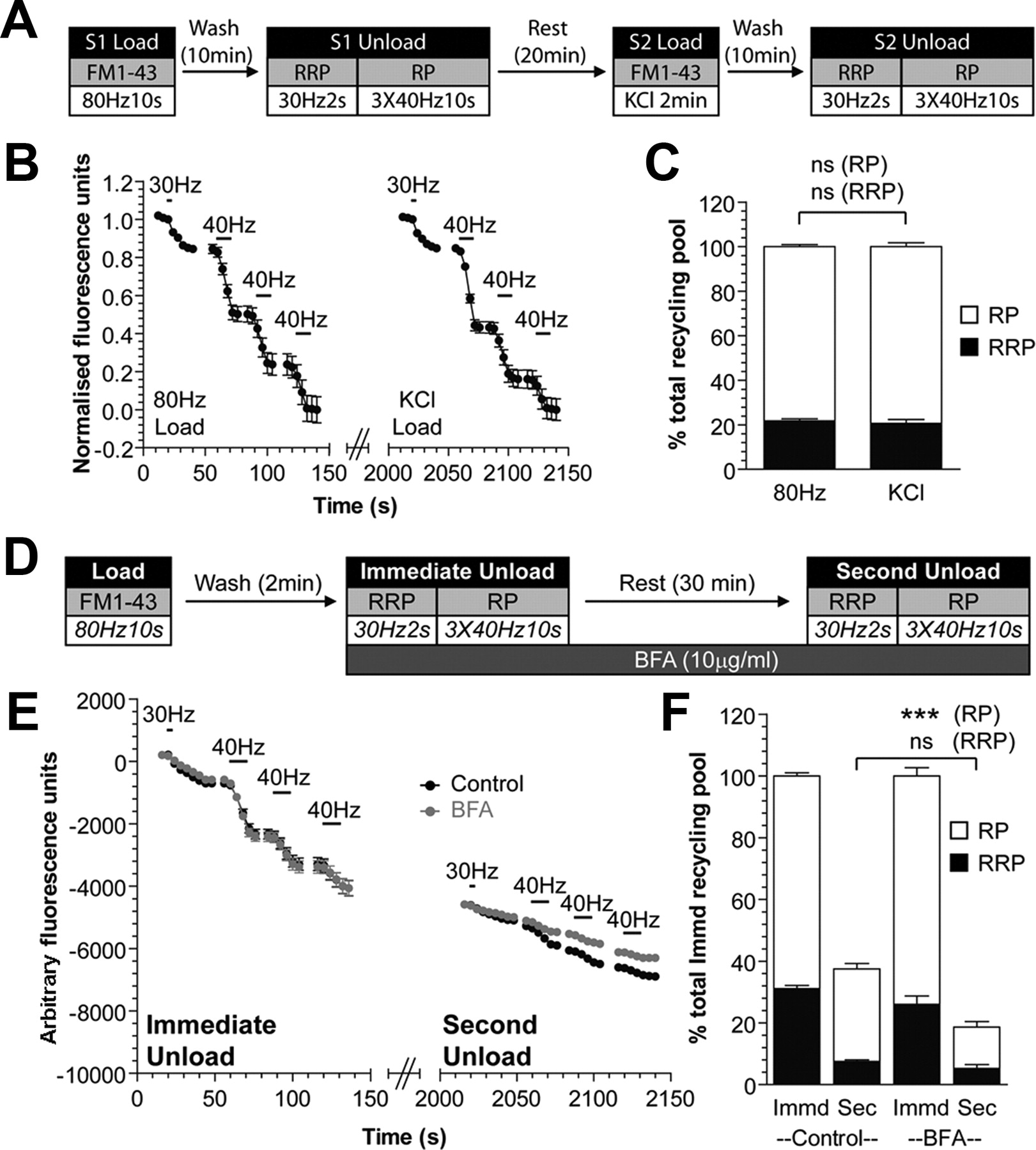
Fluorescence imaging of SV turnover using FM dyes was performed as described previously (Cheung et al., 2010). Briefly, cultures were repolarized for 10 min in incubation medium [in mm: 170 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 TES (N-tris [hydroxyl-methyl]-methyl-2-aminoethane-sulfonicacid), 5 NaHCO3, 5 glucose, 1.2 Na2SO4, 1.2 MgCl2, and 1.3 CaCl2; at pH 7.4]. Cultures were then mounted in an imaging chamber (RC-21BRFS, Warner Instruments). Invaginating membrane was loaded with either FM1-43 (10 μm) or FM2-10 (100 μm) by evoking SV turnover using electrical field stimulation delivered by platinum wires embedded in the imaging chamber (800 action potentials, 100 mA, 1 ms pulse width). After washout of excess FM dye, the dye was unloaded using a series of action potential trains. For FM1-43 experiments, cultures were immediately stimulated (Immediate Unload) with sequential trains of action potentials to first unload the RRP (30 Hz, 2 s) and then the reserve pool (three trains of 40 Hz,10 s). After a 30 min rest period, an identical unloading protocol was repeated (Second Unload). This protocol allows quantification of newly generated SVs that replenish the RRP and reserve pool. The average fluorescence drop for each unloading step was expressed as a percentage of the total SV recycling pool (RRP plus reserve pool) of the Immediate Unload, allowing comparison across multiple experiments. For transfected nerve terminals, this was further normalized to the equivalent drop in untransfected nerve terminals within the same field of view.
S2/S1 experiments were performed using FM2-10 in which cultures underwent two rounds of FM dye loading and unloading, separated by a 20 min rest period. BFA (10 μg/ml) was present before and during the S2 unloading step. The total S2 fluorescence drop (ΔS2) was expressed as a ratio of the total S1 fluorescence drop (ΔS1) for the same nerve terminals. In all cases, at least three independent experiments were performed with 90 nerve terminals from each untransfected culture being assessed. In experiments using cultures transfected with shRNA, 10–30 transfected and untransfected nerve terminals in the same field of view were examined. Only nerve terminals displaying stimulation-dependent dye loading and unloading were selected for analysis. The kinetics of dye unloading were analyzed by normalizing the fluorescence unload between 1 and 0 and calculating the time taken for 50% of the dye to be released (t½).
Labeling of endocytic pathways by horseradish peroxidase.
Cultures were fixed and processed for electron microscopy as described previously (Cheung et al., 2010). Briefly, cultures were placed in incubation medium for 10 min and then stimulated for 2 min with 50 mm KCl in the presence of 10 mg/ml horseradish peroxidase (HRP). After washout of HRP, cultures were immediately stimulated with two consecutive 30 s applications of 50 mm KCl. Cultures were then left to rest for 30 min. Cultures were incubated with BFA (10 μg/ml) during the unload and rest periods. Cultures were fixed in 2% glutaraldehyde in phosphate buffered saline at one of the three fixation time points, either directly after HRP loading, after unloading, or after rest. After washing with 100 mm Tris, pH 7.4, cultures were exposed to 0.1% diaminobenzidine and 0.2% H2O2 in 100 mm Tris until color developed. Cultures were then washed with 100 mm Tris, stained with 1% osmium tetroxide for 30 min, and then dehydrated using ethanol series and polypropylene oxide and embedded using Durcupan. Samples were sectioned, mounted on grids, and viewed using a FEI Tecnai 12 transmission electron microscope. Nerve terminals were included in the analysis providing they contained small SVs, regardless of whether they contained HRP. Intracellular structures that were <100 nm in diameter were arbitrarily designated to be SVs, whereas larger structures were considered endosomes. The average endosome diameter was obtained by taking the average of the longest and shortest diameters of individual endosomes using ImageJ (National Institutes of Health).
Fluorescence imaging of dextran uptake.
Uptake of tetramethylrhodamine-dextran (40 kDa) was monitored as described previously (Clayton et al., 2009). Briefly, transfected cultures were placed in incubation medium for 10 min and then stimulated with a train of 800 action potentials (80 Hz) in the presence of 50 μm tetramethylrhodamine-dextran. Non-internalized dextran was then washed away by perfusion for 2 min. The number of dextran puncta per 100 μm of transfected neurite was determined in each field of view. At least three independent experiments were performed, with at least six fields of view being assessed for each experiment.
Image acquisition, processing, and analysis.
In all experiments, fluorescence signals were visualized using a Zeiss Axio Observer A1 epifluorescence microscope. Transfected neurons expressing mCerulean were identified using 430 nm excitation, whereas FM dye loading and unloading was monitored at 500 nm excitation (in all cases, emission > 535 nm). Immunostaining and dextran uptake was visualized using 550 nm excitation and a bandpass (575–640 nm) emission filter. Experiments using untransfected cultures were performed using a 20× air objective, while experiments with transfected cultures were performed using a 40× oil objective (both from Zeiss). All images were acquired using a Zeiss AxioCam CCD camera controlled by a Zeiss AxioVision Release software. Time-lapse FM images were acquired at 4 s intervals. All immunofluorescence and dextran uptake images were captured as z-stacks and processed as follows. Maximum intensity projections of stacks were generated using an ImageJ Z Project function. Background fluorescence was subtracted using the Rolling Ball Background Subtraction plugin (http://rsbweb.nih.gov/ij/plugins/rolling-ball.html). Total fluorescence intensity of immunostaining was measured from ROIs of identical size over cell bodies. Dextran puncta on transfected neurites were highlighted using the Colocalization Finder plugin (http://rsb.info.nih.gov/ij/plugins/colocalization-finder.html) and counted. The length of transfected neurite in each field of view was measured using the Simple Neurite Tracer plugin (http://pacific.mpi-cbg.de/wiki/index.php/Simple_Neurite_Tracer). All calculations and statistical analyses were performed using Microsoft Excel and GraphPad Prism software.
Results
Brefeldin A arrests SV budding from bulk endosomes
ADBE is the dominant SV retrieval mode during intense neuronal activity in central nerve terminals (Clayton et al., 2008); however, the molecular mechanism responsible for generation of SVs from bulk endosomes is unknown. Recent genetic studies in model systems have suggested a requirement for clathrin in this process (Heerssen et al., 2008; Kasprowicz et al., 2008). To determine whether the endosomal adaptors AP-1 or AP-3 were required, we disrupted their function using the ARF1 inhibitor BFA in primary neuronal cultures. To ensure that we observed SV generation specifically from bulk endosomes, the following experimental protocol was performed. Both CME and ADBE were triggered using a maximal depolarizing stimulus (50 mm KCl) in the presence of the fluid phase marker HRP (Fig. 1A). SVs and bulk endosomes that were generated by CME and ADBE, respectively, are visible as HRP-labeled structures (Fig. 1B). HRP-labeled SVs were then depleted by two sequential stimuli of 50 mm KCl (Fig. 1B,C). Cultures were then left to rest for 30 min, allowing the generation of new HRP-labeled SVs from bulk endosomes (Fig. 1A). This protocol thus allows tracking of HRP-labeled SVs specifically generated from bulk endosomes during a defined time window.

Brefeldin A arrests SV budding from bulk endosomes. A, Cultures were loaded with HRP (10 mg/ml) in the presence of 50 mm KCl for 2 min. HRP was washed away immediately, and neurons were stimulated to release all available SVs using two sequential 30 s KCl (50 mm) stimuli. Cultures were then left to rest for 30 min. Brefeldin A (10 μg/ml) was added to the cultures during the KCl unload and rest period. Cells were fixed either immediately after HRP loading (Load), immediately after KCl unloading (Unload), or after the 30 min rest period (Rest) as indicated by arrows. B, Representative electron micrographs are shown for the treatments described above. Black arrows indicate HRP-labeled SVs, and white arrows indicate HRP-labeled endosomes (scale bar, 500 nm). C, Bar graph displays the mean number of HRP-labeled SVs per nerve terminal for the indicated fixation points. For comparison, the mean number of HRP-labeled endosomes were: Load, 4.0 ± 0.6; Unload-Control, 5.7 ± 0.7; Unload-BFA, 5.6 ± 0.3; Rest-Control, 4.4 ± 0.5; Rest-BFA, 3.7 ± 0.6 (n is number of nerve terminals [N is number of independent experiments]: Load, n = 551 [N = 6]; Unload-Control, n = 227 [N = 4]; Unload-BFA, n = 123 [N = 3]; Rest-Control, n = 232 [N = 4]; Rest-BFA, n = 95 [N = 3]; all ± SEM, ***p < 0.001, **p < 0.01, one-way ANOVA using N). D–F, Frequency distribution of endosome diameter during Load (white bars), Unload (black bars), and Rest (gray bars) periods under control conditions (D), and either Unload (E) or Rest (F) in the presence or absence of BFA (hatched bars) (n is number of HRP endosomes, with total endosome number in parentheses: Load, n = 1980 (3678); Unload-Control, n = 1245 (2320); Unload-BFA, n = 1980 701 (1447); Rest-Control, n = 781 (1336); Rest-BFA, n = 339 (617); all ± SEM, ***p < 0.001, two-way ANOVA).
The immediate unload of HRP-labeled SVs resulted in over a 60% depletion of HRP-labeled SVs in control cultures (Fig. 1C). After the 30 min rest period a large (>3.5-fold) increase in HRP-labeled SVs was then observed (Fig. 1C), indicating that SVs had been generated from bulk endosomes. When HRP-labeled endosomes in control cultures were examined in more detail, there was no change in their diameter during stimulation (Load, 278 ± 5 nm; Unload, 295 ± 8 nm). However, after the 30 min rest period there was a significantly larger population of smaller endosomes, indicating again that SV budding had occurred (Fig. 1D). The mean HRP-endosome diameter was also significantly smaller when compared to HRP-labeled endosomes either before or after the immediate unload stimulus (229 ± 8 nm, p < 0.001, one-way ANOVA).
To determine whether ARF1 recruitment of either AP-1 or AP-3 were required for SV generation specifically from bulk endosomes, cultures were incubated with BFA (10 μg/ml) only after HRP exposure (and endocytosis) was complete (Fig. 1A). Therefore, this protocol ensures that bulk endosome generation is unaffected by the presence of BFA. BFA did not affect the depletion of HRP-labeled SVs (Fig. 1C) or the mean diameter of endosomes during the immediate unload (Control, 295 ± 8 nm; BFA, 290 ± 7 nm; Fig. 1E). In contrast, BFA abolished the generation of new HRP-labeled SVs during the 30 min rest period (Fig. 1C). This inhibition was accompanied by a significant reduction in the population of small HRP-labeled endosomes (mean diameter, 262 ± 11 nm; Fig. 1F). Thus, BFA arrests: (1) generation of HRP-labeled SVs after endocytosis; and (2) a decrease in the diameter of HRP-labeled endosomes. Both lines of evidence indicate a role for the ARF1-dependent recruitment of either AP-1 and/or AP-3 in SV generation from bulk endosomes.
Replenishment of the reserve pool via bulk endosomes is BFA sensitive
Bulk endosome-derived SVs selectively replenish the reserve pool of SVs in nerve terminals (Richards et al., 2000; Cheung et al., 2010). Thus, the replenishment of the reserve pool is an independent indicator of SV production from bulk endosomes. To confirm that BFA also inhibits the replenishment of the reserve SV pool, we performed an experiment in which we loaded cultures with the fluorescent dye FM1-43 using a train of 800 action potentials (80 Hz). This stimulation protocol triggers both CME and ADBE pathways, allowing FM1-43 to label both CME-derived SVs and ADBE-derived bulk endosomes (Clayton et al., 2008) and results in an identical pattern of dye loading to that observed using elevated KCl (Fig. 2A–C). Immediately after the loading of dye, FM1-43-loaded SVs were depleted by sequentially unloading the RRP (60 action potentials at 30 Hz) and then the reserve pool (three 400 action potential trains at 40 Hz) (Cheung et al., 2010). After a rest period of 30 min to allow SV generation from bulk endosomes, cultures were challenged again with identical unloading stimuli (Fig. 2D). Almost all released fluorescence originated from the reserve pool (percentage of second unload: RRP, 20.0 ± 1.5; Reserve, 80.0 ± 4.75; Fig. 2E,F), confirming that the reserve pool is replenished by bulk endosome-derived SVs (Cheung et al., 2010).

Brefeldin A inhibits SV reserve pool replenishment via bulk endosomes. A, Cultures were subjected to an S2/S1 protocol where they were loaded with FM1-43 (10 μm) using a train of 800 action potentials (80 Hz) at S1 and KCl (50 mm for 2 min) at S2. Dye was washed away immediately and unloaded with 30 Hz, 2 s (RRP) and three trains of 40 Hz, 10 s (reserve pool, RP) sequentially in each instance. B, Representative trace of dye unloading during both S1 and S2 unloads is shown. Both Immediate Unload and Second Unload traces are normalized to their respective recycling pool. C, Bar graphs display mean values of the size of the RRP and RP of each unload normalized to the total recycling pool; all n = 3 ± SEM, one-way ANOVA. D, Cultures were loaded with FM1-43 (10 μm) using a train of 800 action potentials (80 Hz). Dye was washed away immediately after stimulation and after 2 min the RRP (30 Hz 2 s) and reserve pool (RP, three trains of 40 Hz, 10 s) were sequentially unloaded (Immediate Unload). The same unloading stimuli were delivered after a 30 min rest period (Second Unload). Brefeldin A (10 μg/ml) was exposed to cultures before the Immediate Unload and was present thereafter. E, Representative traces displaying the unloading of dye during both immediate and second unloads (black, control; gray, BFA). Traces are presented in arbitrary fluorescence units. Bars indicate the period of stimulation. F, Bar graphs display mean values of the size of both the RRP and RP during both unloads, with the second unload normalized to total immediate recycling pool. All experiments, n = 3 ± SEM, ***p < 0.001, one-way ANOVA.
To determine whether ARF1 is required for reserve SV pool replenishment, BFA was applied after washout of FM1-43 (Fig. 2D). BFA had no effect on the fusion of either the RRP or the reserve pool during the immediate unloading challenge (Fig. 2E,F). After the 30 min rest period, however, replenishment of the total recycling pool was significantly reduced (percentage of immediate unload recycling pool: Control, 37 ± 2%; BFA, 19 ± 3%; p < 0.01, Student's t test), suggesting that replenishment of either or both of the RRP and reserve pool was retarded. When this was determined, a specific decrease in the replenishment of the reserve pool was observed in BFA-treated cultures (Fig. 2E,F). Importantly the small replenishment of the RRP was unaltered (Fig. 2E,F). Thus, the inhibition of SV generation from bulk endosomes by BFA translates into a decreased replenishment of the reserve pool by these SVs.
To confirm that the observed effects of BFA were not a direct effect on SV exocytosis, we performed an S2/S1 experiment where the same nerve terminals undergo two identical cycles of dye loading and unloading. To ensure that the assay readout was not confounded by bulk endosome-dependent effects, we used the dye FM2-10, which does not label ADBE (Clayton et al., 2008; Clayton and Cousin, 2008). FM2-10 was loaded at both S1 and S2 with 800 action potentials (80 Hz) and unloaded (ΔS1 and ΔS2) using three trains of 400 action potentials (40 Hz). Preincubation with BFA for 10 min before the second unloading step (ΔS2) had no effect on the extent of FM2-10 unloading (SV exocytosis) when compared to control (ΔS2/ΔS1 Control, 1.05 ± 0.01; BFA, 1.04 ± 0.04; p = 0.82, Student's t test). Thus, BFA does not directly interfere with SV exocytosis, confirming its effect was specific to reserve pool replenishment by bulk endosome-derived SVs.
Bulk endosome budding is impaired by silencing expression of either AP-1 or AP-3
BFA interferes with the ARF1-dependent recruitment of both AP-1 and AP-3 (Drake et al., 2000), implicating either or both AP complexes in the generation of SVs from bulk endosomes. To determine their potential roles, we designed shRNA oligonucleotides specifically targeting the expression of either AP-1γ (oligonucleotides AP-1a and AP-1b) or AP-3δ subunits (oligonucleotides AP-3a and AP-3b). These subunits were targeted since they are essential components of their respective AP complexes (Boehm and Bonifacino, 2001; Robinson, 2004). All oligonucleotides greatly reduced expression of their targeted subunit (Fig. 3). Importantly, no off-target effects on the reciprocal AP was observed (Fig. 3B,D).

shRNAs targeting AP-1γ or AP-3δ subunit specifically decreased their expression. A, C, Cultures were transfected with shRNA vectors that were either empty or expressing oligonucleotides against AP-1γ (AP-1a, AP-1b) or AP-3δ (AP-3a, AP-3b). After 72 h, the expression of AP-1γ or AP-3δ was quantified using immunofluorescence. Representative images are shown for either AP-1γ (A) or AP-3δ (C). Left panels show either AP-1γ (A) or AP-3δ (C) immunostaining (red). Middle panels show transfected neurons (green). Right panels show a merged image with the corresponding bright-field image. White arrows indicate AP-1γ (A) or AP-3δ (C) expression in transfected cells (scale bars, 10 μm). B, D, Bar graphs display mean intensity of either AP-1γ (B) or AP-3δ (D) immunofluorescence in transfected neurons normalized to untransfected neurons in the same field of view (n = 4, 5, 4, 3, and 3 [AP-1], and 4, 3, 3, 5 and 5 [AP-3] independent experiments for empty, AP-1a, AP-1b, AP-3a, and AP-3b vectors; all ± SEM, ***p < 0.001 to empty vector, one-way ANOVA).
The replenishment of the reserve SV pool provides an accurate readout of SV generation from bulk endosomes (Cheung et al., 2010); therefore, we used this assay to determine whether knockdown of either AP-1γ or AP-3δ mimicked the effect of BFA (Fig. 2D–F). Cultures expressing shRNA against either subunit were loaded with FM1-43 using a train of 800 action potentials (80 Hz) followed by an immediate unload, rest period, and second unload in an identical experiment to before (Figs. 4A, 5A). The unloading of both the RRP and reserve pool in the immediate unloading step was unaffected by the silencing of either AP-1γ or AP-3δ, suggesting no direct role for the AP-1 and AP-3 complexes in SV exocytosis (Figs. 4B–E, 5B–E). However, after the 30 min rest period to allow SV generation, the total second unload of dye (reflecting the SV recycling pool) was significantly smaller in neurons where either AP-1γ or AP-3δ were silenced in comparison to empty vector (Figs. 4F, 5F). When contributions from both RRP and reserve pool were determined, a selective inhibition of reserve pool unloading was observed for both AP-1γ and AP-3δ silenced neurons (Fig. 4B–E,G and 5B-E,G). Thus, a reduction in the expression of the essential subunits of either AP-1 complex or AP-3 complex resulted in a specific inhibition of reserve pool replenishment by SVs generated from bulk endosomes.

Reserve pool replenishment is inhibited by silencing AP-1γ expression. Cultures were transfected with shRNA that was either empty or expressing oligonucleotides against AP-1γ (AP-1a, AP-1b). A, After 72 h, cultures were loaded with FM1-43 (10 μm) using an 800 action potential (80 Hz) train. Dye was washed away immediately and after 2 min the RRP (30 Hz 2 s) and reserve pool (RP, 3 trains of 40 Hz 10 s) were sequentially unloaded (Immediate Unload). The same unloading stimuli were delivered after a 30 min rest period (Second Unload). B–D, Representative traces of dye unloading during both the immediate and second unloads for cells transfected (gray) with either empty vector (B), AP-1a (C), or AP-1b (D). Graph also displays unloading from untransfected cells (control, black) from the same field of view. Both Immediate Unload and Second Unload traces are normalized to their respective recycling pool. E–G, Bar graphs display mean values of the size of the RRP and RP of the Immediate Unload (E), the Total Second Unload (F), and the RRP and RP of the Second Unload (G). All values were first normalized to the total immediate recycling pool and then to the equivalent unload of untransfected nerve terminals in the same experiment (Empty, n = 3; AP-1a, n = 3, AP-1b, n = 4 independent experiments; all ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 to empty vector, one-way ANOVA).

Reserve pool replenishment is inhibited by silencing AP-3δ expression. Cultures were transfected with shRNA that was either empty or expressing oligonucleotides against AP-3δ (AP-3a, AP-3b). A, After 72 h, cultures were loaded with FM1-43 (10 μm) using an 800 action potential (80 Hz) train. Dye was washed away immediately and after 2 min the RRP (30 Hz, 2 s) and reserve pool (RP, three trains of 40 Hz, 10 s) were sequentially unloaded (Immediate Unload). The same unloading stimuli were delivered after a 30 min rest period (Second Unload). B–D, Representative traces of dye unloading during both the immediate and second unloads for cells transfected (gray) with either empty vector (B), AP-3a (C), or AP-3b (D). Graph also displays unloading from untransfected cells (Control, black) from the same field of view. Both Immediate Unload and Second Unload traces are normalized to their respective recycling pool. E–G, Bar graphs display mean values of the size of the RRP and RP of the Immediate Unload (E), the Total Second Unload (F), and the RRP and RP of the Second Unload (G). All values were first normalized to the total immediate recycling pool and then to the equivalent unload of untransfected nerve terminals in the same experiment (Empty, n = 3; AP-3a, n = 6, AP-3b, n = 3 independent experiments; all ± SEM, *p < 0.05, **p < 0.01, to empty vector, one-way ANOVA).
To confirm that the observed effects after either AP-1γ or AP-3δ silencing were due to inhibition of SV generation from bulk endosomes, we monitored FM2-10 loading and unloading in shRNA-silenced cultures. This allows the effect of this maneuver to be determined on a population of SVs not generated by ADBE (Clayton et al., 2009). Dye was loaded into cultures using a train of 800 action potentials (80 Hz), and the extent and kinetics of SV exocytosis were monitored using a subsequent unloading stimulus of 800 action potentials (40 Hz, Fig. 6A). The kinetics of dye unloading in all neurons transfected with silencing shRNAs were indistinguishable from those transfected with empty shRNA vectors, confirming no direct effect of AP-1γ and AP-3δ silencing on SV exocytosis (Fig. 6B–E). Furthermore, there was no difference in the extent of dye unloading (Fig. 6F–I), which reflects replenishment of the SV recycling pool by CME-derived SVs (Cheung et al., 2010). Thus, silencing the expression of either AP-1γ or AP-3δ had no effect on the retrieval, recycling, or exocytosis of CME-generated SVs (Fig. 6E,I), confirming their specific role in ADBE.

SV exocytosis and CME are both unaffected by silencing AP-1γ or AP-3δ expression. Cultures were transfected with shRNA that was either empty or expressing oligonucleotides against either AP-1γ (AP-1a, AP-1b) or AP-3δ (AP-3a, AP-3b). A, After 72 h cultures were loaded with FM2-10 (100 μm) using a train of 800 action potentials (80 Hz). Dye was washed away immediately and cells were left to rest for 10 min. Cells were then unloaded with a train of 800 action potentials (40 Hz). B–D, Representative traces of dye unloading in neurons transfected (gray) with either empty (B), AP-1b (C), or AP-3a (D) shRNA after normalization of fluorescence drop between 1 and 0. Unloading from untransfected neurons (control, black) from the same field of view are shown. Curve fit for plateau followed by one-phase decay was performed for each trace and presented by solid lines. E, Bar graph displays mean values of the half-time (T-half) of the fluorescence drop obtained from the fitted curves. Values were normalized to the untransfected nerve terminals in the same experiment (all ± SEM, one-way ANOVA). F–H, Representative traces of dye unloading in neurons transfected (gray) with either Empty (F), AP-1b (G), or AP-3a (H) shRNA. Unloading from untransfected neurons (control, black) from the same field of view are shown. I, Bar graph displays the average total FM2-10 Unload from transfected neurons and normalized untransfected neurons in the same field of view (Empty, n = 5; AP-1a, n = 3; AP-1b, n = 3; AP-3a, n = 4; AP-3b, n = 4 independent experiments; all ± SEM, one-way ANOVA).
The arrest of SV budding and subsequent reserve pool replenishment caused by the knockdown of either AP-1γ or AP-3δ may have resulted from an upstream inhibition in the generation of bulk endosomes. To assess this, we monitored the uptake of high molecular weight fluorescent dextrans (tetramethylrhodamine-dextran, 40 kDa), which are too large to be accumulated by CME (Clayton et al., 2008; Clayton and Cousin, 2009b). Cultures transfected with shRNA against either AP-1γ or AP-3δ were stimulated with a train of 800 action potentials (80 Hz) in the presence of dextran to evoke its internalization by ADBE. In all instances, dextran uptake was comparable to uptake in neurons transfected with empty shRNA vectors (Fig. 7). Thus, the arrest in reserve pool replenishment observed in AP-1γ- or AP-3δ-silenced neurons was due to defective SV generation from bulk endosomes and not the faulty formation of bulk endosomes.

Silencing AP-1γ or AP-3δ expression does not inhibit ADBE. Cultures were transfected with shRNA that was either empty or expressing oligonucleotides against either AP-1γ (AP-1a, AP-1b) or AP-3δ (AP-3a, AP-3b). After 72 h, cultures were stimulated with a train of 800 action potentials (80 Hz) in the presence of 40 kDa tetramethylrhodamine-dextran (50 μm). Dextran was washed away immediately after stimulation. A, Representative images of dextran loading are shown. Left panels show neurons transfected with the shRNA vector (green). Middle panels show dextran uptake (red). Right panels show a merged image of transfected neuron (green) and dextran uptake (red). White arrows indicate dextran puncta on transfected neurites (scale bar, 10 μm). B, Bar graph displaying the mean number of dextran puncta per 100 μm of neurite (Empty, n = 3; AP-1a, n = 4; AP-1b, n = 3; AP-3a, n = 3; AP-3b, n = 3 independent experiments; all ± SEM, one-way ANOVA).
AP-1 and AP-3 generate the same SV population from bulk endosomes
We have shown that the AP-1 and AP-3 complexes are individually required for SV generation from bulk endosomes. There are two possible explanations for this requirement. These protein complexes could either be producing two separate populations of SVs in parallel budding pathways, or be sequentially required in the same budding pathway. To determine this, we simultaneously silenced the expression of AP-1γ and AP-3δ in the same neurons. If AP-1 and AP-3 had essential roles in parallel pathways, this would be visualized as an additive inhibition of reserve pool replenishment, whereas generation of the same population of SVs would produce no additional inhibition.
Cultures were loaded with FM1-43 using 800 action potentials (80 Hz) followed by an immediate unload, rest period, and second unload as before (Fig. 8A). Both RRP and reserve pool unloading during immediate unloading were unaffected by the simultaneous silencing of both AP-1γ and AP-3δ (Fig. 8C,D). Similarly, RRP replenishment after 30 min was unaffected (Fig. 8E). Importantly, the inhibition of reserve pool replenishment in the simultaneous absence of both AP-1γ and AP-3δ was not significantly different to the inhibition observed by their single knockdown (Fig. 8E, compared to Figs. 4G and 5G, reserve pool size as percentage of empty vector; shRNA AP-1b, 68.6 ± 1.0; AP-3b, 63.5 ± 6.4; AP-1b plus AP-3b, 61.7 ± 9.7; not significant one-way ANOVA). In addition, silencing of either AP-1γ or AP-3δ expression did not produce any additional inhibition of reserve pool replenishment to that already observed with BFA (Fig. 9). Since the knockdown of AP-1γ and AP-3δ has no additive effect on reserve pool replenishment, we conclude that they control the same SV budding pathway and thus generate the same SV population from bulk endosomes.

Simultaneous silencing of AP-1γ or AP-3δ inhibits reserve pool replenishment to the same extent as individual silencing. Cultures were cotransfected with either shRNA against AP-1γ and AP-3δ (AP-1b, AP-3b) or a double concentration of empty shRNA vector. A, After 72 h, cultures were loaded with FM1-43 (10 μm) using an 800 action potential (80 Hz) train. Dye was washed away immediately and after 2 min the RRP (30 Hz, 2 s) and reserve pool (RP, three trains of 40 Hz, 10 s) were sequentially unloaded (Immediate Unload). The same unloading stimuli were delivered after a 30 min rest period (Second Unload). B, Representative images are shown for neurons cotransfected with AP-1b and AP-3b shRNA vector (left, mCer, mCerulean), FM1-43 loading in the same field of view (middle, FM1-43), and a merged image of transfected neuron (red) and FM1-43 loading (green). Scale bar, 10 μm. C, D Representative traces of dye unloading during both the immediate and second unloads for cells transfected (gray) with either a double concentration of empty vector (C) or cotransfection with AP-1b and AP-3b (D). Graphs also display unloading from untransfected cells (control, black) from the same field of view. Both Immediate Unload and Second Unload traces are normalized to the total recycling pool. E, F, Bar graphs display mean values of the size of the RRP and RP of the Second Unload (E) and the Total Second Unload (F). All values were first normalized to the total immediate recycling pool and then to the equivalent unload of untransfected nerve terminals in the same experiment (n = 3 independent experiments; all ± SEM, **p < 0.01, one-way ANOVA and Student's t test for E and F respectively).

Silencing of either AP-1γ or AP-3δ does not increase the inhibition of reserve pool replenishment observed with BFA. Cultures were transfected with shRNA that was either empty or expressing oligonucleotides against AP-1γ (AP-1b) or AP-3δ (AP-3b). A, After 72 h, cultures were loaded with FM1-43 (10 μm) using an 800 action potential (80 Hz) train. Dye was washed away immediately and after 2 min the RRP (30 Hz, 2 s) and reserve pool (RP, 3 trains of 40 Hz, 10 s) were sequentially unloaded (Immediate Unload). The same unloading stimuli were delivered after a 30 min rest period (Second Unload). Brefeldin A (10 μg/ml) was exposed to cultures before the Immediate Unload and was present thereafter. B, Representative traces of dye unloading during both the immediate and second unloads for cells transfected with either empty vector plus BFA (black), AP-1b plus BFA (red), or AP-3b plus BFA (blue). Both Immediate Unload and Second Unload traces are normalized to their respective recycling pool. C, Bar graphs display mean values of the size of the RRP and RP of the second unload. All values were first normalized to the total immediate recycling pool and then to the equivalent unload of untransfected nerve terminals in the same experiment (n = 3 independent experiments; all ± SEM, one-way ANOVA).
Discussion
We have shown an essential requirement for both AP-1 and AP-3 complexes in the generation of SVs from bulk endosomes, which translates into a specific replenishment of the reserve SV pool. Evidence for this is as follows: (1) BFA inhibits generation of SVs from HRP-labeled bulk endosomes; (2) BFA and shRNA knockdown of either AP-1 or AP-3 inhibits the replenishment of the reserve pool from bulk endosomes; and (3) AP-1 and AP-3 play no role at the plasma membrane in either ADBE or CME.
Previous biochemical studies in neuroendocrine cells have shown clear evidence that the AP-3 complex is essential for vesicle generation from an endosome compartment (Faundez et al., 1998; Blumstein et al., 2001). Furthermore, studies in central neurons have shown that AP-3 is required in concert with the BLOC-1 complex for the traffic of SV protein cargo from cell bodies to nerve terminals (Larimore et al., 2011). The presynaptic localization of both AP-1 and AP-3 complexes (Glyvuk et al., 2010; Newell-Litwa et al., 2010), coupled to the fact that BFA only affects SV recycling during elevated neuronal activity (Voglmaier et al., 2006; Kim and Ryan, 2009), suggested that either or both could be required for SV generation from bulk endosomes. To determine this, we designed assays to specifically monitor SV generation from bulk endosomes as opposed to classical early endosomes or other endosomal compartments. The strategy involved loading bulk endosomes with a reporter molecule (FM1-43 or HRP), depleting nerve terminals of SVs that were generated by CME, and then following the generation of new SVs from bulk endosomes (Cheung et al., 2010). This approach permits a pharmacological intervention (such as BFA application) once ADBE is complete, allowing separation of effects on bulk endosome budding from any potential effects on plasma membrane endocytosis. It also allows a molecular dissection of the replenishment of specific SV pools by separate SV endocytosis modes via use of either overexpression or shRNA silencing vectors, providing these maneuvers have no direct effects on SV exocytosis (Cheung et al., 2010).
A measurable reserve pool of SVs can be generated even after pharmacological or molecular ablation of AP-1 and AP-3 function, which raises questions regarding the origin of these SVs. The most likely scenario is that a few SVs are still being generated from bulk endosomes due to incomplete knockdown of AP-1 and AP-3 or incomplete inhibition by BFA. Another potential possibility is that a different adaptor protein may substitute for AP-1 or AP-3, as seen when AP-2 expression is silenced during CME (Kim and Ryan, 2009). It is unlikely that these remaining reserve pool SVs are generated at the plasma membrane by CME, since reserve pool replenishment can be ablated by addition of the dynamin antagonist dynasore after dye loading is complete (G. Cheung, unpublished observations). Thus, the SVs that replenish the reserve pool are generated by a dynamin-dependent event that occurs after stimulation is complete, suggesting they are derived from bulk endosomes.
Different AP complexes generate SVs from different compartments
Different APs concentrate protein cargo during vesicle budding at different membrane compartments (Robinson, 2004). For example, AP-1 has a defined role in budding vesicles from the trans-Golgi network (Ghosh et al., 2003). In addition to this classical role, AP-1 has been implicated in vesicle budding from recycling endosomes to either the cell surface or the Golgi complex (Pagano et al., 2004), whereas AP-3 is present on early endosomes and aids delivery of cargo to late endosomes and lysosomes (Newell-Litwa et al., 2007). AP-3 is also essential for the generation of vesicles from endosomes in neuroendocrine cells and for sorting SV cargo for transport to nerve terminals (Blumstein et al., 2001; Larimore et al., 2011). In contrast, AP-2 is required for CME at the plasma membrane (Robinson, 2004). Recent studies using shRNA silencing of AP-2 in primary hippocampal cultures confirmed that it plays a role in SV endocytosis during mild stimulation, whereas BFA treatment or knockdown of AP-1γ had no effect (Kim and Ryan, 2009). Our studies agree with these findings, showing that retrieval of SVs by CME or dextran internalization by ADBE was not affected by knockdown of either AP-1γ or AP-3δ, highlighting the lack of a role for either AP-1 or AP-3 at the nerve terminal plasma membrane.
We have shown an essential role for both AP-1 and AP-3 in the generation of SVs from bulk endosomes. This key shared molecular function explains previous observations in a number of neuronal systems. For example, BFA inhibited a slow route of SV protein cargo traffic during high intensity, but not low intensity, stimulation in primary hippocampal neurons (Voglmaier et al., 2006). Also, deletion of the AP-3μB subunit reduced SV numbers in inhibitory nerve terminals (Nakatsu et al., 2004), which typically have higher activity rates (Bartos et al., 2007). Mocha mice (which are null for AP-3δ and have little or no other AP-3 subunits) have SVs of irregular size (Newell-Litwa et al., 2010). They also display defects in asynchronous neurotransmitter release and a higher mEPSC frequency (Scheuber et al., 2006). Mice with a genetic ablation of AP-1σB have a marked reduction of SVs, a phenotype that is exacerbated by stimulation (Glyvuk et al., 2010). Finally, cultured neurons from AP-1σB-null mice displayed a stimulation-dependent increase in endosome number compared to wild-type and a robust defect in the replenishment of the recycling pool (Glyvuk et al., 2010). An arrest of SV generation from bulk endosomes specifically during ADBE explains these prior observations and provides a direct molecular locus for these AP complexes in the SV life cycle.
Dual essential requirement for AP-1 and AP-3 in SV generation from bulk endosomes
We demonstrate a shared requirement for both AP-1 and AP-3 in SV reserve pool replenishment from bulk endosomes. This suggests that: (1) AP-1 and AP-3 are not functionally redundant; and (2) they both have key and specific roles in the generation of SVs from bulk endosomes. In different cellular systems AP-1 and AP-3 have displayed varying degrees of functional overlap. For example, they have partially redundant functions in sorting cargo to melanosomes (Theos et al., 2005; Delevoye et al., 2009; Lakkaraju et al., 2009), where AP-1 and AP-3 form distinct vesicles with different melanosome cargo in in vitro budding assays (Chapuy et al., 2008). However, this does not seem to occur at the bulk endosome, since we have shown that both AP complexes are essential for the generation of the same SV pool. This is not due to neurons exclusively expressing either AP-1 or AP-3, since we have shown by immunofluorescence that almost all neurons in culture express both AP-1γ and AP-3δ (92.3 ± 2.2% AP-1γ; 88.1 ± 1.4% AP-3δ).
Why are AP-1 and AP-3 both essential for the production of SVs from bulk endosomes? One possibility is that these complexes work in a sequential manner to generate SVs. In this model a precursor SV buds from a bulk endosome (using for example AP-1) and fuses with a classical early sorting endosome. A mature SV may then bud from the early endosome (using AP-3) and replenish the reserve pool (Fig. 10). There is no current morphological evidence to suggest that this occurs; however, recent work from a number of laboratories have suggested that movement of SV membrane through classical early endosomes may contribute toward SV recycling in central nerve terminals (Hoopmann et al., 2010; Uytterhoeven et al., 2011). We are currently testing this hypothesis by examining the potential contributions from classical early endosomes to SV recycling during different stimulation intensities.

Potential shared roles for AP-1 and AP-3 complexes in ADBE. The dual requirement for AP-1 and AP-3 in ADBE can be explained by a sequential requirement for AP-1 and AP-3. After formation of the bulk endosome, an immature SV is formed in an AP-1-dependent manner. This immature SV delivers SV cargo to a classical early endosome, and then a mature SV (with a full complement of cargo) buds from this compartment using AP-3.
The shared essential roles for both AP-1 and AP-3 in SV budding from bulk endosomes highlight a key conceptual divergence between ADBE and CME. During CME, SV protein cargo is concentrated by AP-2 and potentially other non-classical adaptors at the plasma membrane (Jung and Haucke, 2007; Kelly and Owen, 2011). However, the essential requirement for AP-1 and AP-3 at bulk endosomes suggests that little or no sorting of SV protein cargo occurs at the plasma membrane during ADBE. This makes conceptual sense, since the major physiological role of ADBE is to rapidly correct for large changes in plasma membrane surface area, rather than create a fully functional SV. Other forms of fluid phase uptake actively exclude specific plasma membrane proteins (Mercanti et al., 2006), suggesting that ADBE may use a similar mechanism, allowing the rate-limiting step of SV cargo clustering to be performed once inside the neuron. Thus, during ADBE, SV cargo selection and processing may occur at the bulk endosome rather than at the plasma membrane.
In summary, we have demonstrated a molecular locus of action for AP-1 and AP-3 in the SV life cycle in central nerve terminals. This essential molecular requirement for SV generation from bulk endosomes will provide a novel point of intervention to determine how ADBE controls neurotransmission in both neuronal physiology and pathology.
Footnotes
This work was supported by a grant from the Wellcome Trust (Ref. 084277). The anti-delta SA4 antibody was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. Dr. Alan Prescott and Mr. John James (both University of Dundee) are thanked for excellent technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Mike Cousin, Membrane Biology Group, Centre for Integrative Physiology, George Square, University of Edinburgh, EH8 9XD, Scotland, UK. M.Cousin{at}ed.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}