

Abstract
Formation of the CNS white matter is developmentally tightly regulated, but the molecules and mechanisms of myelination control in the postnatal CNS are poorly understood. Here, we show that myelin growth is controlled by fibroblast growth factor (FGF) signaling, originally identified as a proliferative signal for oligodendrocyte precursor cells (OPCs) in vitro. We created two lines of mice lacking both FGF receptor 1 (Fgfr1) and Fgfr2 in oligodendrocyte-lineage cells but found that in these mice OPC proliferation and differentiation were unaffected. In addition, axonal ensheathment and the initiation of myelination were on time. However, the rapid growth of CNS myelin, normally occurring in the second postnatal week, was strongly inhibited. Throughout adulthood, the myelin sheath remained disproportionately thin relative to the axon caliber. In adult mice, mutant oligodendrocytes were normal in number, whereas the transcription of major myelin genes was reduced. This FGF receptor-mediated stimulation of mature oligodendrocytes could also be modeled in vitro, demonstrating that enhanced expansion of oligodendroglial processes requires signaling by extracellular signal regulated kinase-1 and -2 (Erk1/2), downstream mediators of mitogen-activated protein kinase (MAPK). In vivo, Erk1/2-MAPK activity was reduced in the hypomyelinated CNS of Fgfr1/Fgfr2 mutant mice. These studies reveal a previously unrecognized function of FGF receptor signaling in oligodendrocytes that contributes to the regulation of myelin sheath thickness and that uncouples the initiation of ensheathment from the later phase of continued myelin growth.
Introduction
A key question in cellular neuroscience is how oligodendrocytes and Schwann cells are instructed to myelinate axons and what molecular mechanisms control myelin thickness to allow long-range connections to operate with millisecond precision. In the peripheral nervous system (PNS), the neuronal growth factor neuregulin-1 type III (NRG1) has emerged as a “master-regulator” of Schwann cell development and myelination (Nave and Salzer, 2006). Oligodendrocytes differ from Schwann cells in myelination control (Chan et al., 2004), and the role of NRG1/ErbB signaling for myelinating CNS axons has been questioned (Roy et al., 2007; Brinkmann et al., 2008; Taveggia et al., 2008). Earlier evidence that CNS myelination is controlled by insulin-like growth factor 1 (IGF-1) remains indirect because it is unclear whether the reported effects on myelination were on oligodendrocyte precursor cells (OPCs) and oligodendrocytes or on myelination per se (Carson et al., 1993; Joseph D'Ercole and Ye, 2008), suggesting the involvement of another growth factor in this process.
Fibroblast growth factors (FGFs) are a family of growth factors that, like neuregulins, serve diverse functions in proliferation, differentiation, and organogenesis, and FGF receptors (Fgfr1–4) such as ErbB receptors are receptor tyrosine kinases that activate phosphoinositol 3 kinase/Akt/mammalian target of rapamycin (mTOR) and Ras/Raf/Mek/Erk (extracellular signal regulated kinase) pathways (Mason, 2007; Turner and Grose, 2010). Neurons and astrocytes produce several FGFs, prominently FGF1 and FGF2, which are expressed concomitantly with active myelination and are also identified in axons (Elde et al., 1991; Riva and Mocchetti, 1991; Gómez-Pinilla et al., 1992; Matsuyama et al., 1992; Nakamura et al., 1999; Becker-Catania et al., 2011; Ratzka et al., 2011). Oligodendrocyte-lineage cells express FGF receptors in a developmentally regulated manner (Bansal et al., 1996; Fortin et al., 2005). Fgfr1 is expressed in both OPCs and differentiated oligodendrocytes, and Fgfr3 only in OPCs, suggesting at least one role of FGFs in early oligodendrocyte development. Indeed, in vitro studies have shown that FGF2 stimulates the proliferation of isolated OPCs and thereby inhibits their differentiation (McKinnon et al., 1990; Bansal, 2002). Recently, we showed that Fgfr1 and Fgfr2 signaling plays a role in the specification of OPCs from the embryonic ventral forebrain, similar to sonic hedgehog (Furusho et al., 2011). Interestingly, Fgfr2 is specifically expressed in differentiated oligodendrocytes, becomes enriched in lipid raft microdomains, and is preferentially associated with paranodal myelin (Bryant et al., 2009) (our unpublished observations). In vitro, FGF receptor activation of differentiated oligodendrocytes stimulates process outgrowth (Fortin et al., 2005), raising the possibility that FGF receptor signaling has another distinct function in myelination by mature oligodendrocytes.
Given the overlap of Fgfr1 and Fgfr2 expression in mature oligodendrocytes (Fortin et al., 2005), single gene deletions are unlikely to reveal the hypothetical FGF receptor function in myelination (Kaga et al., 2006). We, therefore, generated and analyzed two lines of mice lacking both Fgfr1 and Fgfr2 following Cre recombination selectively in either CNP+ or Olig1+ OPCs and oligodendrocytes. While Fgfr1/Fgfr2-independent signaling suffices to allow oligodendrocyte differentiation, axonal ensheathment, and the initiation of myelination, Fgfr1/Fgfr2-deficient oligodendrocytes are unable to fully express myelin protein genes and to increase myelin thickness to normal levels during active myelination. This hypomyelination is associated with reduced activation of Erk1/2-mitogen-activated protein kinase (MAPK) signaling, which can be modeled in vitro by inhibiting FGF/FGFR/Erk1/2 signaling in process-bearing oligodendrocytes. We suggest that FGF receptors are “late-stage” regulators of CNS myelination.
Materials and Methods
Generation of Fgfr1/Fgfr2 double conditional knock-out mice.
Conditional double knock-out mice of either sex, referred to as Fgfr1−/−;Fgfr2−/−, were generated by mating Fgfr1flox/flox;Fgfr2flox/flox mice with CNPCre/+ mice (Lappe-Siefke et al., 2003) to produce progeny with disrupted Fgfr1 and Fgfr2 genes in CNP-expressing oligodendrocyte-lineage cells and Schwann cells (Kaga et al., 2006; Furusho et al., 2009; Wang et al., 2009). The efficiency and specificity of Fgfr1 and Fgfr2 recombination was confirmed by PCR, immunoblotting, in situ hybridization, and analysis of CNPCre/+;Rosa-26-YFP reporter mice, as described previously (Kaga et al., 2006; Furusho et al., 2009; Wang et al., 2009; and data not shown). To confirm key findings from the Fgfr1−/−;Fgfr2−/−;CNP-Cre line, especially those related to OPC proliferation and differentiation, we also generated a second line of conditional Fgfr1−/−;Fgfr2−/− double knock-out mice by mating Fgfr1flox/flox;Fgfr2flox/flox with Olig1cre/+ mice (Lu et al., 2002), to produce progeny in which disruption of Fgfr1 and Fgfr2 genes occurs in Olig1-expressing OPCs even earlier in the lineage than in the CNP-Cre line. Littermates of the genotypes Fgfr1flox/flox;Fgfr2flox/flox;CNP+/+ and Fgfr1flox/flox;Fgfr2flox/flox;Olig1+/+ were used as “controls.”
The mutant mice live a normal lifespan, and Fgfr1flox/flox;Fgfr2flox/flox;CNPCre/+ mice perform normally on rotarod test (Furusho et al., 2009). However, these mice showed prolongation of auditory brainstem response peak latency, indicative of slower nerve conduction (Wang et al., 2009).
Immunolabeling.
As described previously (Kaga et al., 2006; Furusho et al., 2011), matched transverse cryostat sections (15 μm) from the cervical regions of the spinal cords (C5–C8) of mutants and littermate controls of either sex were immunolabeled for myelin basic protein (MBP; 1:3000; a gift from E. Barbarese, University of Connecticut Medical School, Farmington, CT), or double immunostained for 5-bromo-2′-deoxyuridine (BrdU; 1:50; Becton-Dickinson) and Olig2 (1:100; Immuno-Biological Laboratories), or platelet-derived growth factor receptor α (PDGFRa; 1:200; a gift from W. B. Stallcup, Sanford-Burnham Medical Research Institute, La Jolla, CA). Spinal cord cultures were immunolabeled, as described previously (Furusho et al., 2011), with oligodendrocyte-lineage markers O4 (1:25) (Bansal et al., 1992), CNP (1:100; Sternberger Monoclonal), and HPC7 [1:25 (HPC7 labels immature oligodendrocytes similar to anti-galactocerebroside)] (Baas and Barnstable, 1998).
Cell culture.
Dissociated cultures of spinal cord were prepared individually from newborn control and mutant pups of either sex from the same litter and grown in defined media, as described previously for embryonic brains (Furusho et al., 2011). Briefly, the cervical and thoracic portions of the spinal cords were dissected, and meninges were removed. The spinal cords were chopped with a blade, transferred to 1.5 ml tubes, trypsinized (0.025%) for 30 min at 37°C, and gently triturated and plated at a density of 50,000/cm2 on poly-d-lysine-coated plastic dishes in 4% FCS/DMEM. After 17 h, the medium was changed to Bottenstein/Sato-defined media (DMEM with 100 μg/ml transferrin, 5.2 ng/ml sodium selenium, 5 μg/ml insulin, 8.8 μg/ml putrescine, 6 ng/ml progesterone, 10 IU/ml penicillin, and 100 μg/ml streptomycin; all from Sigma) plus 0.5% FCS.
Mouse OPCs from neonatal control and mutant forebrains were isolated by immunopanning with O4 antibody and were grown, as described previously, before harvest for quantitative real-time (qRT)-PCR (Dugas et al., 2006).
Rat OPCs were purified as described previously (Fortin et al., 2005). Briefly, OPCs were obtained from mixed primary cultures from neonatal rat telencephalon (P1–P2) of either sex by overnight shaking, followed by differential adhesion and complement lysis with anti-galactocerebroside. This process resulted in the enrichment of OPCs by the removal of terminally differentiated oligodendrocytes, astrocytes, and macrophages. Cells were plated in 5% FCS/DMEM in tissue culture plates coated with poly-d,l-ornithine (Sigma; 50 μg/ml). Following cell attachment for 2–3 h, the medium was changed to serum-free, defined medium mN2 [DMEM with d-glucose (4.5 mg/L), human transferrin (50 μg/ml), bovine pancreatic insulin (5 μg/ml), 3,3,5-triiodo-l-thyronine (15 nm), sodium selenium (30 nm), d-biotin (10 nm), hydrocortisone (10 nm), sodium pyruvate (0.11 mg/ml), penicillin-streptomycin (10 IU/ml and 100 μg/ml, respectively)], 0.1% BSA (all from Sigma), and 10 ng/ml PDGF for 1–2 d to expand the OPCs. To obtain differentiated mature oligodendrocytes, PDGF was removed and OPCs were allowed to differentiate into mature oligodendrocytes by growth in serum-free mN2 media for 4 d, followed by 2 d in cytosine arabinoside (2 μm) to eliminate the few remaining undifferentiated proliferating OPCs. After removal of the cytosine arabinoside, mature oligodendrocytes were grown for an additional 2 d in the absence or presence of FGF2 (10 ng/ml) with or without the Mek inhibitor U0126, at a dose of 12.5 or 25 μm (which gave the same effects), or PD98059, at a dose of 50 μm (Calbiochem). For immunoblot analysis for Erk1/2, oligodendrocyte cultures were grown as described in 60 mm dishes at high density and treated with FGF2 and inhibitors as indicated in the experiments. Inhibitors were added to the cultures 1 h before the addition of FGF2.
Oligodendrocyte process growth analysis was performed using low-density oligodendrocyte cultures to provide enough space for the full expansion of their process network and to clearly visualize individual oligodendrocytes. Measurement of the size of oligodendrocytes was performed using National Institutes of Health image analysis. Although very few isolated rat oligodendrocytes make membrane sheaths per se, they do make an extensive network of processes and are mostly circular, with the cell body in the middle. We measured the diameter (in micrometers) of the whole oligodendrocyte (including cell body and processes) in vertical and horizontal planes, and the mean diameter value was used to calculate the area of the oligodendrocyte. Approximately, 100 mature oligodendrocytes from at least three wells each were measured. Because the size of the cell body of the oligodendrocytes did not change with any treatment, the increase in the total size of the oligodendrocytes reflects an increase in the lengths of their processes. Two to three independent experiments were performed.
Electron microscopy.
Electron microscopy (EM) and morphometric analysis on spinal cords and optic nerves was performed as described previously (Marcus et al., 2006). Briefly, mice of either sex were transcardially perfused with 0.1 m Millonig's buffer containing 5% glutaraldehyde and 4% paraformaldehyde and postfixed for 2 weeks. Tissue was harvested, processed, and sectioned for standard electron microscopic analysis and imaged using a JEOL JEM 1200EX transmission electron microscope.
In situ hybridization.
Matched transverse cryostat sections (15 μm) from cervical regions of the spinal cords (C5–C8) and sagittal sections of the whole brain of mutants and littermate controls of either sex were analyzed by in situ hybridization, as previously described (Kaga et al., 2006; Furusho et al., 2011) using riboprobes specific for proteolipid protein (PLP) mRNA (a gift from W. B. Macklin, University of Colorado School of Medicine, Aurora, CO), MBP mRNA (M. Qiu, University of Louisville, KY), or PDGFRa mRNA (a gift from W. D. Richardson, University College London, UK). Briefly, after incubation in 1 μg/ml proteinase K at 37°C for 30 min, sections were hybridized overnight at 65°C with a digoxigenin-labeled antisense cRNA probe and washed in 50% formamide, 2× SSC and 1% SDS at 65°C for 2–3 h, followed by rinses in 2× SSC, 0.2× SSC at room temperature, and 0.1× SSC at 60°C. After blocking in 1% Tween 20 and 1% normal goat serum (1 h), sections were incubated (2 h) in an alkaline phosphatase-conjugated antidigoxigenin antibody (1:5000; Roche Diagnostics). Color was developed with 4-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate, and nuclei were counterstained with Hoechst blue dye 3342 (1 μg/ml; Sigma).
Immunoblotting.
Immunoblotting was performed as described previously (Fortin et al., 2005). Briefly, equal amounts of total proteins were loaded by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and immunolabeled for phosphorylated-Erk1/2 (1:1000; Sigma), pan-Erk1/2 (1:5000; Promega), PLP (1:1000; M. Lees), oligodendrocyte-specific protein (OSP; 1:1000; Invitrogen), MBP (1:10,000; a gift from E. Barbarese, University of Connecticut Medical School, Farmington, CT), N-myc downstream-regulated gene1 (NDRG1; 1:2000; a gift from U. Suter, Swiss Federal Institute of Technology, Zurich, Switzerland), glial fibrillary acidic protein (GFAP; I:10,000; Dako), or Neurofilament M (NF-M; 1:5000; Millipore).
Quantitative real-time PCR.
qRT-PCR was performed from 1 μg of total RNA, reverse transcribed to cDNA, using iQ SYBR Green Supermix (Bio-Rad), according to the manufacturer's instructions. Primers used were as follows: PLP, 5′-GTATAGGCAGTCTCTGCGCTGAT-3′ and 5′-AAGGGCAGCAATCATGAAGG-3′; and MBP, 5′-TACCTGGCCACAGCAAvGTAC-3′ and 5′-GTCACAATGTTCTTGAAG-3′. Quantification of PCR products was performed using the 2-ΔΔCt method. Quantities of mRNA were normalized to the housekeeping gene glycerol phosphate dehydrogenase.
Results
Myelin sheath thickness is reduced in Fgfr1−/−;Fgfr2−/− mutants
To determine the differentiation of CNP+ oligodendrocytes that lack Fgfr1 and Fgfr2 in vivo, spinal cord sections from conditional double-mutant mice were immunolabeled for MBP (Fig. 1A). The initiation of myelination, as judged by MBP expression at age postnatal day 4 (P4), was normal in these double mutants. However, the comparison to littermate controls at age P30 revealed significantly less MBP-positive myelin, and the overall white matter area was smaller. The levels of several myelin-specific proteins (PLP, MBP, OSP, and NDRG1) were examined in spinal cord homogenates (age P15) and showed a significant reduction in the mutants (Fig. 1B), further suggesting that myelin synthesis in the double-mutant mice is perturbed.

Myelin sheath thickness is reduced in Fgfr1−/−;Fgfr2−/− mutants. A, Cervical spinal cord sections from P4 and P30 Fgfr1−/−;Fgfr2−/− (CNP-Cre line) mutants and controls were immunolabeled for MBP (green) and neurofilament-M (red). Aa–Ad, At P4 (Aa, Ab) initiation of myelination appeared normal, but by P30 (Ac, Ad), the white matter area was significantly reduced (area of lateral-ventral columns: controls, 3.9 ± 0.3 mm2; mutants, 2.6 ± 0.2 mm2; p < 0.05, N = 3–8). High-magnification images (inset) show less MBP+ myelin staining around axons in P30 spinal cords. Scale bars: (in Aa, Ab) Aa–Ad, 200 μm; (in Ad) Ac, Ad, insets, 10 μm. B, Quantification of myelin proteins, PLP, MBP, OSP, and NDRG1, by immunoblotting in P15 spinal cords shows a significant reduction of these proteins in the mutants. β-Actin was used as a loading control. Error bars indicate SEM (N = 3). *p < 0.05. C, EM images of ventral spinal cords at P15, P30, and 10 months (10M) from controls and Fgfr1−/−;Fgfr2−/− (CNP-Cre line) show that at P15 axons are similarly wrapped by thin myelin sheaths in both controls and mutants but at P30 myelin thickness appears reduced in the mutants compared with controls. This difference becomes even more pronounced at 10M. Scale bar, 1 μm. D, Scatter plots of g-ratios of individual fibers in relation to respective axon diameters quantified at P15, P30, 5M, and 10M from Fgfr1−/−;Fgfr2−/− mutants (red circles) and littermate controls (gray circles) confirms that the mutants have thinner myelin (higher g-ratios) than controls at all the ages examined. E, Average g-ratio values when plotted as a function of age show that myelin thickness continues to increase in the controls during adulthood but is arrested in the mutants at approximately the level attained by P15, becoming significantly different from controls from P30 onward. Approximately 90–200 axons from two to four mice of each age and genotype were analyzed; *p < 0.01. F, Percentage of myelinated and unmyelinated axons, counted at P15, P30, and 10M, showed no difference between the control and the mutants. Approximately 300–600 axons from six EM images magnified 5000× were counted at each time point from at least two mice per genotype.
Next, we compared the myelination of the spinal cord in Fgfr1−/−;Fgfr2−/− mutants (CNP-Cre line) and littermate controls at ages P15, P30, 5 months, and 10 months by EM. Surprisingly, the thickness of myelin was almost similar in mutants and controls (P15), but while myelin thickness increased in control mice with age, CNS myelination appeared to completely stall in double-mutant mice (Fig. 1C). Morphometric quantification of myelin thickness by g-ratio analysis (ratio of axon diameter to myelinated fiber diameter, expressed as scatter plots) confirmed a relative reduction of myelin thickness in the mutants (Fig. 1D) at all ages examined, from P30 to 10 months. Indeed, consistent with previous studies (Blakemore, 1974; Peters et al., 2001; Shepherd et al., 2012), when average g-ratios were plotted as a function of age (Fig. 1E), the myelin thickness in the spinal cords of normal mice continued to increase (with decreasing g-ratios) up to at least 10 months of age. In contrast, the double mutants remained “arrested” in development, with the same g-ratios that had been reached after 2 weeks. The numbers of myelinated and unmyelinated axons were similar in mutants and controls (Fig. 1F).
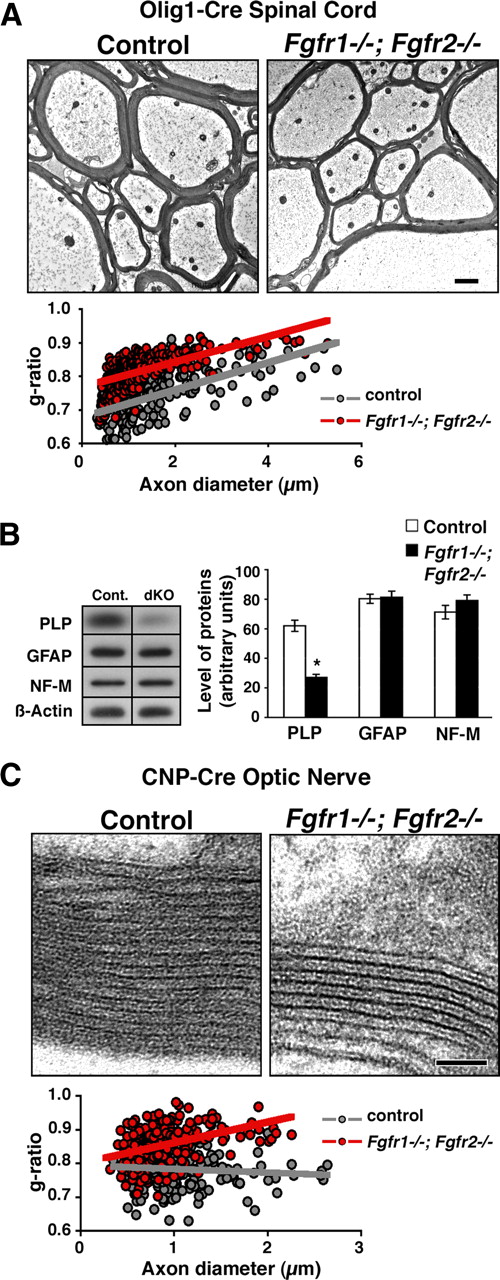
To confirm our findings above, we generated and analyzed a second line of conditional knock-out mice in which Fgfr1/Fgfr2 were ablated by the Olig1-Cre driver. EM analysis of the spinal cords of these mutants showed that, as for the CNP-Cre line, myelin sheath was thinner in the mutant mice than in controls (Fig. 2A). Quantification of myelin thickness by g-ratio analysis confirmed that axons of all diameters were wrapped by significantly thinner myelin [compare average g-ratios in controls (0.74 ± 0.07) with those in mutants (0.82 ± 0.05); p = 4.7 × 10−40]. The numbers of total axons that were myelinated in the spinal cords of the Olig1-Cre Fgfr1/Fgfr2 mutant mice were also similar to those in controls (myelinated axons: controls, 89%; mutants, 86%). No obvious ultrastructural abnormalities were noted in the spinal cords of these mutants compared with controls. Further, immunoblot analysis of spinal cord homogenates from these mice showed a reduction in the levels of myelin protein (PLP) but not of the astrocytic (GFAP) or neuronal (NF-M) proteins, indicating that there was no obvious effect on either the neuronal or astrocytic compartment attributed to Fgfr1/Fgfr2 ablation in the Olig1-Cre line (Fig. 2B).

Reduction in myelin sheath thickness occurs in different regions of the CNS and in a different Cre-driver line of Fgfr1−/−;Fgfr2−/− conditional mutants. A, EM analysis of ventral cervical spinal cord sections from 7-month-old Fgfr1−/−;Fgfr2−/− (Olig1-Cre line) shows that myelin thickness is reduced in the mutants compared with controls. Quantification of myelin thickness shown as scatter plots of g-ratios of individual fibers in relation to respective axon diameters from Fgfr1−/−;Fgfr2−/− mutants (red circles) and littermate controls (gray circles) confirms that the mutants have thinner myelin than controls have. Scale bar, 1 μm. Percentages of total axons that were myelinated did not show a significant difference between controls and mutants (myelinated axons: controls, 89%; mutants, 86%). For each genotype, 500–700 axons were analyzed. B, Quantification of myelin proteins (PLP), astrocytic protein (GFAP), and axonal protein (NF-M) by immunoblotting of P15 spinal cords from control (cont.) and Fgfr1−/−;Fgfr2−/− mutant mice (Olig1-Cre line) (dKO) shows a significant reduction in the levels of PLP but not GFAP or NF-M. β-Actin was used as a loading control. Error bars indicate SEM (N = 4–5). *p < 0.05. C, High-magnification EM images of optic nerves from 10-month-old controls and Fgfr1−/−;Fgfr2−/− mice (CNP-Cre line) shows fewer myelin wraps in the mutants than in controls. Note that the ultrastructure and periodicity of compact myelin sheaths in the mutants are indistinguishable from those of the control. Scatter plots of g-ratios of individual fibers in relation to respective axon diameter quantified from Fgfr1−/−;Fgfr2−/− mutants (red circles) and littermate controls (gray circles) confirm that the mutants have thinner myelin (higher g-ratios) than controls. Percentages of total axons that were myelinated, counted in the optic nerve did not show any difference between the control and mutants (myelinated axons: controls, 98%; mutants, 97%). Approximately 400–500 axons from six EM images magnified 5000× were counted from two mice per genotype. Scale bar, 0.05 μm.
To examine whether ablation of FGF receptor signaling has a general effect on myelin thickness in different regions of the CNS or is specific to the spinal cord, we analyzed myelin thickness in the optic nerves of 10-month-old Fgfr1/Fgfr2 (CNP-Cre line) mutant mice by EM analysis (Fig. 2C). As seen in the spinal cord, myelin thickness was also reduced in the optic nerves of the mutants compared with controls. Quantification of myelin thickness by g-ratio analysis (expressed as scatter plots) confirmed the reduction of myelin thickness in the mutants (average g-ratios: controls, 0.78 ± 0.05; mutants, 0.85 ± 0.06; p = 7.4 × 10−39). Given that the ultrastructure and the periodicity of myelin sheets in the mutants were unaffected (Fig. 2C), we conclude that the overall reduction of myelin thickness in the mutants results from a decrease in the number of wraps.
Disruption of Fgfr1/Fgfr2 signaling does not affect OPC proliferation or oligodendrocyte differentiation in vivo or in cultures initiated from mutant spinal cords
The hypomyelination in the Fgfr1−/−;Fgfr2−/− mutants could arise from early defects in OPC proliferation, survival, and/or differentiation, resulting in fewer oligodendrocytes. Therefore, to examine the effects of Fgfr1/Fgfr2 disruption on OPC proliferation and differentiation, we analyzed not only the CNP-Cre line but also the Olig1-Cre driver line of mutant mice in which Fgfr1/Fgfr2 were deleted from OPCs at a stage even earlier than in CNP-Cre line. The complete deletion of Fgfr1 in OPCs was confirmed by qRT-PCR analysis of isolated OPCs from neonatal forebrains of these mutants, and deletion of Fgfr2 was demonstrated by immunoblotting, showing reduced expression of Fgfr2 protein in the spinal cords of mutant mice (Fig. 3C). To examine the proliferation of OPCs, we double-labeled control and mutant spinal cord sections with BrdU and markers of OPCs (PDGFRa or Olig2) at P0 (Olig1-Cre line) (Fig. 3A,B) and at P4 and P9 (CNP-Cre line) (Fig. 3D,E). Quantification of the numbers of double-labeled cells showed that the proliferation of OPCs was not affected in the mutants compared with controls at any of the ages examined from either of the two lines of mutant mice. Further, there was no reduction in the total numbers of OPCs as determined by the quantification of PDGFRa mRNA+ cells at P0 (Fig. 3Ac,Ad,B) or Olig2+ cells at P9 (Fig. 3Dc,Dd,F).

Disruption of Fgfr1/Fgfr2 signaling does not reduce the proliferation or the numbers of OPCs in the Fgfr1−/−;Fgfr2−/− mutants. Mice were injected intraperitoneally with BrdU (100 mg/kg body weight) 1 h before harvest. A–F, To identify proliferating cells, transverse sections of cervical spinal cords from Fgfr1−/−;Fgfr2−/− mutants and littermate control from P0 Olig1-Cre line (A, B) or P4 and P9 CNP-Cre line (D–F) were double labeled for BrdU (red) and PDGFRa or Olig2 (green). Insets, arrowheads, Examples of double-labeled cells. B, E, Total numbers of BrdU;PDGFRa or BrdU;Olig2 double-labeled cells were counted in the entire sections and expressed as percentages of PDGFRa or Olig2-positive cells. Quantification of the total numbers of OPCs, identified by in situ hybridization for PDGFRa mRNA at P0 in the Olig1-Cre line (Ac, Ad, B) or by immunostaining for Olig2 at P9 in the CNP-Cre line (F), shows no differences in the numbers of OPCs between controls (cont.) and mutant mice. Three sections each from three to five mice of each group were analyzed. Error bars indicate SEM (N = 3–5). Scale bars, A, D: 200 μm; insets, 100 μm. C, qRT-PCR for the expression of Fgfr1 mRNA in OPCs isolated from the forebrains of neonatal Fgfr1−/−;Fgfr2−/− mutants and littermate controls from two separate litters of the Olig1-Cre line shows the complete loss of Fgfr1 from mutant OPCs, as expected. Immunoblotting for Fgfr2 protein in the P15 spinal cords from the Olig1-Cre line of Fgfr1/Fgfr2 mutants (dKO) shows reduced expression of Fgfr2 in the mutants compared with controls.
We next examined the differentiation of OPCs into oligodendrocytes by PLP in situ hybridization and CC1 immunostaining (data not shown), as markers of differentiated oligodendrocytes, in the spinal cords of Fgfr1−/−;Fgfr2−/− mutants and littermate controls from the Olig1-Cre line at P0 and P9 (Fig. 4A,B) and from CNP-Cre line at P4 and P9 (data not shown) (Fig. 4B). There were no differences in the total numbers of differentiated oligodendrocytes in the white matter at any of these ages in either of the two lines. OPC differentiation into oligodendrocytes was also analyzed in dissociated cultures initiated from spinal cords of newborn mutant and control mice from the CNP-Cre line (Fig. 4C,D) and the Olig1-Cre line (data not shown). The percentage of oligodendrocyte-lineage cells (O4+) that differentiated into oligodendrocytes at 4 d (HCP7+) and 7 d (CNP+) in culture (DIC) did not differ between the control and mutants.

Disruption of Fgfr1/Fgfr2 signaling does not affect the extent and timing of oligodendrocyte differentiation in vivo or in cultures initiated of mutant spinal cords. A, B, Transverse sections of cervical spinal cord at P0 and P9 from control and mutants from Olig1-Cre line were analyzed by in situ hybridization for PLP mRNA expression. All PLP mRNA+ oligodendrocytes in the whole section were counted. No statistically significant differences were observed in the total numbers of PLP mRNA+ oligodendrocytes between control and mutant mice at any of the ages examined. Three sections each from three to seven mice of each group were analyzed. Error bars indicate SEM (N = 3–7). Scale bar, 200 μm. C, D, Dissociated cultures initiated from spinal cords of P0 Fgfr1−/−;Fgfr2−/− mutants and littermate controls from the CNP-Cre line were double immunolabeled with O4 and markers of differentiated oligodendrocytes (HPC7 at 4 d and CNP at 7 d). The percentage of total oligodendrocyte-lineage cells (O4+) that differentiated into oligodendrocytes in mutants was comparable to that of controls at both days. Individual cultures initiated from each pup from two separate litters were analyzed. Three representative wells from each group were counted. Error bars indicate SEM (N = 3). Scale bar, 100 μm.
We conclude from these in vivo and in vitro studies, performed in two independent lines of mice, that the loss of Fgfr1/Fgfr2 signaling does not adversely affect OPC proliferation or survival or the extent and timing of oligodendrocyte differentiation.
Levels of PLP and MBP mRNA, but not numbers of oligodendrocytes, are reduced in the adult Fgfr1−/−;Fgfr2−/− mutant mice
We next examined the numbers of differentiated oligodendrocytes as a function of age from P9 to 8 months in the spinal cords of Fgfr1−/−;Fgfr2−/− mutants (CNP-Cre line) and littermate controls by PLP in situ hybridization (Fig. 4A,B). As seen for earlier postnatal ages (above), the total numbers of differentiated oligodendrocytes present in the mutant spinal cords did not differ from those in controls during adulthood. Interestingly, the intensity of the PLP mRNA signal per oligodendrocyte appeared markedly reduced in the mutants compared with the controls. This difference was most pronounced at P15. It has been shown earlier that during normal myelination, PLP mRNA levels are upregulated during the active phase of myelination and then are reduced as myelination slows down in older animals (Jordan et al., 1989). Consistent with this, in the controls we observed that PLP mRNA intensity per oligodendrocyte first increased, reaching a peak at P15, and then reduced with age as expected. In contrast, mutant oligodendrocytes showed only a very modest increase in intensity at P15 that remained low throughout (Fig. 5A,B). Similarly, MBP mRNA intensity showed an even more dramatic difference between control and mutant mice in the CNP-Cre line (Fig. 5C). To confirm these findings in a second line of Fgfr1/Fgfr2 conditional knock-out mice and to investigate whether ablation of FGF receptors also affected other regions of the CNS, we analyzed the expression of MBP mRNA in the spinal cord, cerebellum, and corpus callosum of the Olig1-Cre driver line of Fgfr1/Fgfr2 mutants (Fig. 5E,F). In all these cases, the intensity of MBP mRNA was found to be reduced in the mutants compared with controls. Quantification of mRNA levels by qRT-PCR confirmed that the expression of PLP and MBP transcripts was significantly reduced in the P15 and 1-month-old mutant spinal cords of both CNP-Cre (Fig. 5D) and Olig1-Cre (Fig. 5G) lines.

The levels of PLP and MBP mRNA, but not the numbers of oligodendrocytes, are reduced in the adult Fgfr1−/−;Fgfr2−/− mutant mice. A, B, Transverse sections of cervical spinal cord at P9, P15, 1 month (1M), 2M, and 8M were analyzed by in situ hybridization for PLP mRNA. All PLP mRNA+ oligodendrocytes in the whole white matter regions of the spinal cords were counted per section. No statistically significant differences were observed in the total numbers of PLP mRNA+ oligodendrocytes in control and mutant mice at any of the ages examined (B). However, the normal increase in PLP mRNA signal intensity per oligodendrocyte that occurs in controls at P15 did not occur in the mutants (A). C, E, Similarly, MBP mRNA signal intensity was lower in the mutants than controls in the spinal cords of both the CNP-Cre (C) and Olig1-Cre (E) lines. F, Reduced expression of MBP mRNA signal intensity in the cerebellum and corpus callosum (CC) of 2-month-old Olig-1-Cre line of Fgfr1−/−;Fgfr2−/− mice shows that these regions of the CNS were also affected in the mutants. Three sections each from three to four mice of each group were analyzed. Error bars indicate SEM (N = 3–4). Scale bars: A, C, E, 200 μm; F, 500 μm. D, G, qRT-PCR analysis of PLP and MBP mRNA levels in spinal cords of P15 and 1-month-old control and mutant mice from CNP-Cre (D) or Olig1-Cre (G) lines shows reduced PLP and MBP mRNA levels in the mutants compared with the controls. Error bars indicate SEM (N = 3–6). *p < 0.05; **p < 0.01.
We conclude that a probable cause for reduced myelin thickness in Fgfr1−/−;Fgfr2−/− mutants is not a reduction in the numbers of mature oligodendrocytes but rather a reduction in the ability of each oligodendrocyte to upregulate myelin genes and to produce sufficient myelin wraps during active myelination.
Erk1/2-MAPKs are likely downstream mediators of FGF receptor signaling in the regulation of myelin thickness
Several signaling molecules have been implicated in controlling myelin sheath thickness, most prominently the activation of Akt/mTOR (Narayanan et al., 2009). However, Erk1/2-MAPKs are also strongly activated downstream of FGF receptors and several other growth factor receptors. Immunoblotting for phosphorylated Erk1/2-MAPKs in spinal cord homogenates showed a significant decrease in phospho-Erk1/2-MAPKs levels in the mutant mice compared with controls (Fig. 6A), suggesting that the Erk1/2-MAPK pathway may be involved in the regulation of myelin thickness through Fgfr1/Fgfr2 signaling in oligodendrocytes.

Erk1/2 MAPKs are one of the likely downstream mediators of FGF receptor signaling in the regulation of myelin thickness. A, Spinal cord homogenates of P15 Fgfr1−/−;Fgfr2−/− mutants and littermate controls were analyzed by immunoblotting for phosphorylated Erk1/2 MAPKs and pan-Erk MAPK expression. Quantification of the band intensity shows a significant reduction in the levels of phospho-Erk1/2 activation in the mutants. Pan-Erk was used as a loading control and did not show any change. Seven to eight mutants and littermate controls from two separate litters of CNP-Cre line were analyzed. Error bars indicate SEM (N = 7–8). *p < 0.05. B, Mature rat oligodendrocytes grown in defined media were treated with FGF2 (10 ng/ml) for 2 d in the presence or absence of U0126 or PD98059 (inhibitors of Mek, upstream of Erk1/2) and double labeled for O4 and MBP. Whole diameter (in micrometers, cell body plus processes) of ∼100 mature oligodendrocytes was measured by NIH image analysis in each group. Total area of the oligodendrocyte, (OL) calculated from the measured diameter and plotted as arbitrary units, shows a significant increase in the relative size of oligodendrocyte processes in the presence of FGF2 and a complete attenuation of this increase by inactivation of Erk1/2. Representative images of mature oligodendrocytes from control, FGF2-treated, and FGF2+U0126-treated cultures immunostained with O4 are shown. Scale bar, 50 μm. Error bars indicate SEM (N = 3). *p < 0.05. Two to three independent experiments were performed. C, Immunoblot analysis of parallel cultures of mature oligodendrocytes shows that exposure to FGF2 (15 min) increased the phosphorylation of Erk1/2. Pretreatment of cultures with U0126 or PD98059 efficiently attenuates FGF2-mediated Erk1/2 phosphorylation at doses that inhibit oligodendrocyte process extension. Error bars indicate SEM (N = 5–6). *p < 0.05.
We next sought to establish a link between Erk1/2-MAPK activation downstream of FGF receptors and the extension of oligodendrocyte processes. Myelin membrane sheets in vivo are formed by the extension of oligodendrocyte processes. Therefore, oligodendrocyte process outgrowth in vitro is thought to mimic many of the morphological changes that occur during axon ensheathment by oligodendrocytes in vivo. We have previously demonstrated that mature oligodendrocytes with dense process network when exposed to FGF2 or FGF9 increase their process size further (Fortin et al., 2005) and that a specific inhibitor of FGF receptors (PD173074) attenuated this FGF2-mediated process outgrowth and Erk1/2-MAPK activation (Bansal et al., 2003; Fortin et al., 2005). To examine whether the FGF2-mediated increase in oligodendrocyte process elongation occurs in parallel to an increase in the phosphorylation of Erk1/2-MAPKs and whether the inactivation of Erk1/2-MAPKs inhibited both these effects, we prepared cultures of mature oligodendrocytes and treated them with FGF2 in the absence or presence of two specific inhibitors of Mek (upstream of Erks) (Fig. 6B,C) as described in Materials and Methods. One set of cultures was immunolabeled with O4 and MBP (Fig. 6B), and the other set was immunoblotted for phosphorylated-Erk1/2-MAPK (Fig. 6C). Quantification of the whole oligodendrocyte size (cell body plus processes) showed that FGF2 treatment increased the oligodendrocyte process length (Fig. 6B) and also increased Erk1/2-MAPK phosphorylation (Fig. 6C). Both these effects were completely abolished in the presence of the inhibitors (Fig. 6B,C), suggesting a correlation between the two FGF-mediated effects on mature oligodendrocytes. These in vitro data, together with the observation of reduced Erk1/2-MAPK activation in the Fgfr1/Fgfr2 mutant mice (Fig. 6A), suggests that activation of the Erk1/2-MAPK pathway by FGF receptors in oligodendrocytes is likely to play an important role in the regulation of myelin thickness.
Discussion
The molecular signals in the CNS that regulate myelination and the fine-tuning of myelin sheath thickness are poorly understood. In rodents, the peak of myelin synthesis lies in the second and third postnatal weeks, but myelination continues thereafter and in humans is known to progress during the first decades of life. We have shown in the present study that oligodendrocytes require FGF receptor signaling to assemble the normal number of myelin membrane wraps around axons, and we provide data to suggest that Erk1/2-MAPK activation in oligodendrocytes is a likely FGF receptor downstream mechanism. The failure of Fgfr1−/−;Fgfr2−/− double-mutant mice to reach full myelin sheath thickness is associated with the reduced peak levels of myelin gene expression, without affecting cell numbers or the proliferation and differentiation of OPCs. These findings strongly suggest that the two FGF receptors are key regulators at late stages of CNS myelination.
In Fgfr1−/−;Fgfr2−/− double-mutant mice, myelination is not affected in the PNS (Furusho et al., 2009), where ErbB2/ErbB3 signaling is essential for Schwann cell survival, differentiation, and myelination (Garratt et al., 2000; Michailov et al., 2004; Taveggia et al., 2005). In contrast to the PNS, NRG1/ErbB signaling to oligodendrocytes is dispensable for survival and CNS myelination (Brinkmann et al., 2008), and other reports agree that CNS myelination is possible despite perturbed NRG1/ErbB signaling (Roy et al., 2007; Taveggia et al., 2008) and that myelin thickness is normal in the spinal cords of these mutants (Taveggia et al., 2008). This left open the question of whether one or more other growth factors are responsible for stimulating oligodendrocytes and myelination in trans. Alternatively, cell-autonomous differentiation of oligodendrocytes is sufficient to trigger myelination (Rosenberg et al., 2007, 2008; Barres, 2008) and is sufficient to achieve normal myelin thickness. Our findings clearly involve FGFs in this late function, independent of any previous role of FGFs in oligodendrocyte differentiation.
Previously, other growth factors such as IGF-1, brain-derived neurotrophic factor (BDNF), and NT3 have been implicated in normal myelination (Cellerino et al., 1997; Kahn et al., 1999; Ye et al., 2002; Zeger et al., 2007; Joseph D'Ercole and Ye, 2008; Vondran et al., 2010). However, in IGF-1 receptor, NT3, TrkC, and BDNF knock-out mice, these effects are associated with and presumably caused by earlier defects in the oligodendrocyte lineage, that is, OPC proliferation and differentiation. Moreover, in conventional BDNF−/− (or BDNF+/−), NT-3−/−, and TrkC−/−-null mutant mice, the developmental effects were not specific to the oligodendroglial lineage and thus were somewhat difficult to interpret.
Our findings in oligodendrocyte-specific mutants suggest a direct role of FGF receptor signaling in myelination. In the absence of Fgfr1/Fgfr2, oligodendrocytes are normally differentiated, ensheathed axons, and even make a few compact wraps of myelin. However, the oligodendrocytes then stall, and myelin sheath thickness remains largely unaltered between day 15 and 1 year, leading to a disproportionate relationship between myelin thickness and axon diameter (Friede and Bischhausen, 1982). Thus, Fgfr1/Fgfr2-mediated regulation of myelin growth can be genetically uncoupled as a late aspect of oligodendrocyte differentiation, coinciding with a late temporal upregulation of FGFs and FGF receptors in the postnatal CNS. An ∼15-fold increase in the levels of FGF1 and a twofold to fourfold increase in the levels of FGF2, FGF22, and FGF3 transcripts (Ratzka et al., 2011) and of FGF2 protein (Riva and Mocchetti, 1991) were reported in the adult spinal cord relative to their levels at birth. At the receptor level, Fgfr2 expression begins late in oligodendrocyte-lineage cells, and Fgfr1 is upregulated upon oligodendrocyte terminal differentiation, concomitant with the expression of major myelin protein genes (Fortin et al., 2005). Thus, combined Fgfr1/Fgfr2 signaling in oligodendrocytes drives the most active phase of myelination and radial growth of the sheath. We note that the activation of FGF receptors in cultured oligodendrocytes likewise increases process outgrowth (Fortin et al., 2005).
What would be the source of FGFs that regulate myelin sheath thickness? FGFs are produced by both astrocytes and neurons. Interestingly, active FGF1 protein was recently identified in purified axolemma from adult myelinated axons (Becker-Catania et al., 2011), and FGF1, FGF2, and FGF9 immunoreactivity has also been detected in axonal fibers (Elde et al., 1991; Matsuyama et al., 1992; Todo et al., 1998). Thus, axonally derived or astrocyte-derived FGFs, associated with the axon surface, bound to extracellular heparan sulfate proteoglycans (Ford-Perriss et al., 2002), could serve as axonal signaling molecules. The receptor Fgfr2 has been detected in purified myelin membranes and in lipid raft microdomains, and is enriched at the paranodes (Fortin et al., 2005; Bryant et al., 2009; data not shown), suggesting that local axonal signals could be received and transmitted to the oligodendroglial compartment. We note that astrocytes also express FGFs (Gómez-Pinilla et al., 1992; Nakamura et al., 1999), which adds a further layer of complexity to the fine-tuning of myelin growth in the mammalian CNS.
Several known signaling pathways operate downstream of FGF receptor tyrosine kinases, including P13K/Akt/mTOR, PLC-γ, and the Ras/Raf/Mek/Erk1/2 pathway (Mason, 2007). In Fgfr1/Fgfr2 double mutants, the reduction in myelin sheath thickness was accompanied by a reduction in the level of activated Erk1/2-MAPKs; and in vitro, inhibition of Erk1/2-MAPKs in differentiated oligodendrocytes was accompanied by a failure to increase FGF2-mediated process outgrowth. This suggests that Erk1/2-MAPKs are involved in the extension of the myelin sheath, which is consistent with the suggested role of Erks in oligodendroglial process elongation in vitro (Stariha et al., 1997; Younes-Rapozo et al., 2009). However, because FGF2 can also stimulate Akt phosphorylation in mature oligodendrocytes (Bryant et al., 2009) and because constitutive activation of Akt in transgenic mice increases myelin thickness through the Akt/mTOR pathway (Narayanan et al., 2009), FGF receptors are likely activating both pathways. Consistent with this model, we find that inhibition of mTOR by rapamycin prevented high-level expression of major myelin proteins and cytoskeletal alterations in cultured oligodendrocyte, perturbing the elaboration of typical “myelin-like” membranes (Guardiola-Diaz et al., 2012).
An interesting aspect of FGF receptor function is the ability to synergistically interact with other cell surface receptors. It is thus possible that oligodendrocytes respond to FGFs in synergy with other myelinogenic signals, thereby comprising complex axonal cues. For example, axonal electrical activity promotes oligodendrocyte differentiation and myelination (Piaton et al., 2010). One proposed mechanism is the activation of adenosine receptors on oligodendrocyte-lineage cells (Stevens et al., 2002). Recently, a direct physical interaction between adenosine A2A receptor and FGF receptors has been demonstrated, leading to a synergistic stimulation of the Erk1/2-MAPK pathway and increased process extension in neurons (Flajolet et al., 2008). In addition, the activation of integrins and cell adhesion molecules (CAMs), such as NCAM, L1, and N-cadherin, can lead to the activation of FGF receptors with clear biological responses (Rusnati et al., 1997; Toledo et al., 2005; Palser et al., 2009). Direct binding and coclustering of FGF receptors with CAMs and the colocalization of FGF receptors with integrins and focal adhesion kinase have been shown in focal adhesions (Plopper et al., 1995). Given the known involvement of CAMs and integrins in axon–glia interactions and CNS myelination (Piaton et al., 2010), it is possible that oligodendroglial FGF receptors “cotransmit” these axon-derived signals to enhance myelination.
Although not all FGF family members are mitogens for OPCs in culture (Fortin et al., 2005; Prendaj et al., 2010), OPCs proliferate extensively when exposed to FGF2 and are then unable to differentiate, leading to the notion that FGF signaling in vitro is inhibitory for OPC differentiation (McKinnon et al., 1990; Bansal, 2002; Fortin et al., 2005). However, the present findings from two independent lines of Fgfr1/Fgfr2 mutant mice that exhibit normal OPC proliferation and differentiation suggest that the in vivo function of FGF signaling is more complex. The role of Fgfr3 on OPCs is less clear, because activation of Fgfr1 (but not Fgfr3) leads to proliferation in vitro (Fortin et al., 2005), and Fgfr3−/− mice (e.g., Fgfr1−/−;Fgfr2−/− mice) are normal with respect to OPC expansion (Oh et al., 2003). Given the dramatic reduction in OPC numbers in the spinal cord of PDGF-A knock-out mice (Fruttiger et al., 1999), we suggest PDGF receptor signaling, rather than FGF receptor signaling, is the more likely mechanism regulating OPC expansion in vivo.
In conclusion, we have identified Fgfr1 and Fgfr2 activation on oligodendrocytes as an important late-stage regulatory mechanism in the control of myelin growth and myelin sheath thickness in the CNS, perhaps serving a function similar to that of ErbB2 and ErbB3 signaling in the postnatal PNS. Obvious differences exist as to the source of NRG1 type III and FGF polypeptides, which are axon resident and paracrine signaling molecules, respectively. Based on our in vivo and in vitro data, we further suggest that Erk1/2-MAPKs are critical downstream signals of oligodendroglial FGF receptors that are likely to play an important role in the regulation of myelin thickness. In human multiple sclerosis patients, remyelination of lesions is often inefficient, leading to myelin sheaths that are thinner than normal, similar to the myelin observed here in the Fgfr1−/−;Fgfr2−/− mutants. Therefore, understanding the molecular mechanism that regulates normal myelin sheath thickness clearly has implications for human demyelinating disorders.
Footnotes
This work was supported by NIH Grant NS38878, and in part by National Multiple Sclerosis Society Grant RG 4087-A-3 and NIH Grant NS41078 (R.B.). A portion of the microscopy was performed at the Virginia Commonwealth University Department of Anatomy and Neurobiology Microscopy Facility, which was supported, in part, by funding from National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke Center Core Grant 5P30NS047463–02. We thank Dr. D. M. Ornitz (Washington University School of Medicine, St. Louis, MO) for providing the Fgfr1 and Fgfr2 floxed mice and Dr. D. H. Rowitch (University of California, San Francisco, San Francisco, CA) for Olig1-Cre mice. We thank M. Bryant, G. Wark, and S. Ahmad for technical assistance.
- Correspondence should be addressed to Rashmi Bansal, Department of Neuroscience, University of Connecticut Medical School, 263 Farmington Avenue, Farmington, CT 06030-3401. bansal{at}neuron.uchc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}