

Abstract
Cyclin-dependent kinase-5 (Cdk5) was reported to downscale neurotransmission by sequestering synaptic vesicles (SVs) in the release-reluctant resting pool, but the molecular targets mediating this activity remain unknown. Synapsin I (SynI), a major SV phosphoprotein involved in the regulation of SV trafficking and neurotransmitter release, is one of the presynaptic substrates of Cdk5, which phosphorylates it in its C-terminal region at Ser549 (site 6) and Ser551 (site 7). Here we demonstrate that Cdk5 phosphorylation of SynI fine tunes the recruitment of SVs to the active recycling pool and contributes to the Cdk5-mediated homeostatic responses. Phosphorylation of SynI by Cdk5 is physiologically regulated and enhances its binding to F-actin. The effects of Cdk5 inhibition on the size and depletion kinetics of the recycling pool, as well as on SV distribution within the nerve terminal, are virtually abolished in mouse SynI knock-out (KO) neurons or in KO neurons expressing the dephosphomimetic SynI mutants at sites 6,7 or site 7 only. The observation that the single site-7 mutant phenocopies the effects of the deletion of SynI identifies this site as the central switch in mediating the synaptic effects of Cdk5 and demonstrates that SynI is necessary and sufficient for achieving the effects of the kinase on SV trafficking. The phosphorylation state of SynI by Cdk5 at site 7 is regulated during chronic modification of neuronal activity and is an essential downstream effector for the Cdk5-mediated homeostatic scaling.
Introduction
Within nerve terminals, synaptic vesicles (SVs) are organized in pools that determine synaptic performance and represent key targets for homeostatic scaling. Functional SV pools include the readily releasable pool of SVs ready to be exocytosed during stimulation, the recycling pool (RP) of SVs undergoing exo-endocytosis during long-lasting stimulations, and the resting pool (RestP) of release-reluctant SVs. The latter pool, identified in central synapses, has not yet been ascribed a clear function (Alabi and Tsien, 2012).
Synapsins (Syns) are a family of SV-associated phosphoproteins that directly contribute to the fine tuning of synapse formation, maturation, and plasticity. In adult synapses, Syns regulate the trafficking of SVs by interacting with actin and SVs, ultimately affecting the availability of SVs for release (Cesca et al., 2010; Bykhovskaia, 2011). Synapsin I (SynI), the most abundant member of the family, is an excellent substrate for several protein kinases, including protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinases (CaMK) I/IV and II, extracellular signal-regulated kinases (Erk) 1/2, Src, and cyclin-dependent kinases (Cdks) 1/5 (Jovanovic et al., 1996, 2001; Matsubara et al., 1996; Onofri et al., 2000). Phosphorylation of SynI by PKA, CaMKs, Erk, or Cdk1 decreases the binding of SynI to SVs and/or F-actin, resulting in an increased availability of SVs (Cesca et al., 2010). Conversely, SynI phosphorylation by Src favors reclustering of recycled SVs by enhancing the binding of SynI to SVs and F-actin (Messa et al., 2010).
Cdk5 is a proline-directed serine/threonine kinase highly expressed in the brain, in which it is involved in development, synaptic transmission, and plasticity (Barnett and Bibb, 2011; Lopes and Agostinho, 2011). At the presynaptic level, Cdk5 is essential for SV endocytosis, because it rephosphorylates the dephosphins (dynamin-1, amphiphysin-1, PIPKIγ, intersectin) after their dephosphorylation by the Ca2+-dependent phosphatase calcineurin during repeated cycles of exo-endocytosis (Cousin and Robinson, 2001; Tan et al., 2003; Tomizawa et al., 2003; Evergren et al., 2007). In addition to SynI, several other Cdk5 substrates were identified at the presynaptic level, such as sept5 (Taniguchi et al., 2007; Amin et al., 2008), Munc-18 (Fletcher et al., 1999; Lilja et al., 2004), CASK (Samuels et al., 2007), Pctaire-1/Cdk16 (Cheng et al., 2002; Fu et al., 2011), and N-type Ca2+ channels (Su et al., 2012; Kim and Ryan, 2013), suggesting additional presynaptic functions for this kinase. Indeed, Cdk5 was implicated recently in activity-dependent homeostatic plasticity at the presynaptic level (Kim and Ryan, 2010; Mitra et al., 2012). The kinase appears to lock SVs in the RestP, in which they are excluded from the exo-endocytotic cycle, thereby decreasing the effective size of the RP (Kim and Ryan, 2010).
Because SynI is a key regulator of SV trafficking and a Cdk5 substrate in vitro, we asked whether SynI is a downstream effector of Cdk5 in the regulation of the RP size and depletion kinetics. Furthermore, we examined whether the phosphorylation state of SynI at the Cdk5 sites is physiologically regulated and influences the molecular interactions of SynI and the spatial organization of SVs in the nerve terminal. We found that phosphorylation of SynI by Cdk5 at Ser551 is regulated in an activity-dependent manner in intact neurons and is both necessary and sufficient to explain the effects of Cdk5 on the RP/RestP partitioning and the spatial organization of SVs within the terminal.
Materials and Methods
Materials.
SynI knock-out (KO) mice were generated by homologous recombination (Chin et al., 1995) and fully backcrossed on a C57BL/6J background (Charles River) for >10 generations. Offspring of homozygous KO mice and wild-type (WT) littermates of either sex were used throughout. All experiments were performed in accordance with the guidelines established by the European Community Council (Directive 2010/63/EU of September 22, 2010) and approved by the Italian Ministry of Health.
SynI was purified from bovine brain under dephosphorylating conditions (Bähler and Greengard, 1987) and phosphorylated in vitro by purified active Cdk5/p35 (3–6 U/sample; Millipore). Phosphorylation was performed by incubation in phosphorylation buffer [40 mm 3-(N-morpholino)-propanesulfonic acid, pH 7.0, 20 mm MgCl2, 50 μm [γ-32P]ATP, and 1.5 μCi] and protease inhibitors for 30 min at 30°C at a stoichiometry of ∼0.3 mol P/mol SynI. Actin was prepared from an acetone powder of rabbit skeletal muscle and further purified by gel filtration (Bähler and Greengard, 1987). Highly purified rat SVs were obtained through controlled-pore glass chromatography and depleted of endogenous SynI by limiting dilution in 200 mm NaCl buffer (Huttner et al., 1983).
The following primary antibodies were used: monoclonal anti-SynI (catalog #106 001, Research Resource Identifiers (RRID):AB_887805; Synaptic Systems); polyclonal anti-phospho-Ser551 SynI (catalog #ab32532, RRID:AB_778198; Abcam); anti-GFP (catalog #A11122, RRID:AB_221569; Invitrogen); monoclonal anti-synaptobrevin-2 (Syb2; catalog #104 202, RRID:AB_887810; Synaptic Systems); monoclonal anti-actin and anti-synaptophysin (catalog #A4700, RRID:AB_476730 and catalog #S5768, RRID:AB_477523; Sigma-Aldrich); and monoclonal anti-Cherry antibody (catalog #632543, RRID:AB_2307319; Clontech). Amino-5-phosphonopentanoic acid (d-AP-5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), (2S)-3-[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid (CGP58845), bicuculline (BIC), tetrodotoxin (TTX), and bafilomycin A1 were from Tocris Cookson. Cell culture media were from Invitrogen. Bafilomycin was dissolved in dimethylsolfoxide (DMSO) and used at a concentration of 1 μm. R-Roscovitine (R-Rosco; Calbiochem) were dissolved in DMSO and used at a concentration of 100 μm. To exclude off-target effects, the S-Roscovitine enantiomer (S-Rosco; Calbiochem) that has little or no effects on Ca2+ channel gating properties (Buraei and Elmslie, 2008; Yarotskyy and Elmslie, 2010) was also used. Restriction enzymes were from New England Biolabs. All other chemicals were from Sigma-Aldrich.
SV binding assays.
The binding of purified SynI to Syn-depleted SVs was performed as described previously (Benfenati et al., 1989). Syn-depleted SVs (10 μg/sample) were incubated for 1 h at 0°C with increasing concentrations (30–300 nm) of either dephosphorylated or Cdk5-phosphorylated SynI in a buffer containing 220 mm glycine, 30 mm NaCl, 5 mm Tris, 4 mm HEPES, pH 7.4, 0.22 mm NaN3 10 mm EDTA, 0.2 mm PMSF, 2 μg/ml pepstatin, 1 mm NaF, 1 mm Na3VO4, and 100 μg/ml bovine serum albumin. After the incubation, SV-bound SynI was separated by high-speed centrifugation (400,000 × g for 45 min) through a 10% (w/w) sucrose cushion. Aliquots of the resuspended pellets were subjected to SDS-PAGE and immunoblotting with phospho-specific and pan-specific anti-SynI antibodies. The recovery of SVs, used to correct the amounts of bound SynI, was determined by immunoblotting with anti-synaptophysin antibodies. In some experiments, the quantification of the bound Cdk5-phosphorylated SynI was also determined in parallel by 32P-radioactivity counting. Binding isotherms were fitted using the one-ligand/one-site binding function.
Actin binding assays.
Globular actin (5 μm) was polymerized for 1 h at room temperature by the addition of 100 mm KCl/1.2 mm MgCl2 and incubated at 30°C for 2 h with 0.3 μm of either dephosphorylated or Cdk5-phosphorylated SynI. The incubation was performed in a buffer containing the following (in mm): 12.5 NaCl, 0.6 ATP, 100 KCl, 1.2 MgCl2, 1.5 2-mercaptoethanol, 6 HEPES, and 8 Tris, pH 7.4. After the incubation, samples were subjected to high-speed centrifugation (400,000 × g for 45 min at 4°C) for recovery of total F-actin (Bähler and Greengard, 1987), and the pellet and supernatant fractions were subjected to SDS-PAGE. Actin recovery was followed by densitometric scanning of Coomassie blue-stained gels. The amount of SynI bound to actin filaments was determined by immunoblotting with phospho-specific and pan-specific anti-SynI antibodies and/or by 32P-radioactivity counting.
Primary cultures of hippocampal neurons.
Pregnant mice were killed by inhalation of CO2, and 17-d-old embryos were immediately removed by cesarean section. Removal and dissection of hippocampi were described previously (Baldelli et al., 2007). Briefly, hippocampi were dissociated by enzymatic digestion in 0.125% trypsin for 20 min at 37°C and then triturated with a fire-polished Pasteur pipette. No antimitotic drugs were added to prevent glia proliferation. Neurons were plated on poly-l-lysine (0.1 mg/ml; Sigma-Aldrich)-treated 25 mm glass coverslips at a density of 60,000 cells per coverslip (low-density cultures) for immunocytochemistry, imaging, and electrophysiology experiments and on poly-l-lysine (0.01 mg/ml)-treated 35 mm plastic wells at a density of 250,000 cells per well for immunoblots.
cDNA subcloning and site-directed mutagenesis.
Green fluorescent protein-tagged rat SynIa (GFP–SynIa) was kindly donated by H.-T. Kao (Brown University, Providence, RI). mCherry-tagged SynIa was generated by substituting mCherry for GFP from the mammalian expression vector pmCherry-C1 (Clontech) using the AgeI and BglII sites. The dephosphomimetic mutants 7A-SynI, and 6,7A-SynI were generated by replacing Ser with Ala residues at the respective phosphorylation sites using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) using the following primers: 7A-SynI, forward, 5′-CCC GCC GGC CTC CCC AGC TCC ACA GCG TCA GGC A-3′; reverse, 5′-TGC CTG ACG CTG TGG AGC TGG GGA GGC CGG CGG G-3′; and 6,7A-SynI, forward, 5′-CCC GCC GGC CGC CCC AGC TCC ACA GCG TCA GGC A-3′; reverse, 5′-TGC CTG ACG CTG TGG AGC TGG GGC GGC CGG CGG G-3′. The absence of Cdk5 phosphorylation at the mutated site was verified by in vitro phosphorylation with purified Cdk5 of SynI variants expressed in COS-7 cells and immunoprecipitated.
Synaptophysin–pHluorin (SypHy) was a kind gift from Dr. C. Stevens (Salk Institute, La Jolla, CA). To facilitate subcloning, a unique SalI restriction site was created in the beginning of the SypHy sequence, and an AgeI site was added at the end of the sequence, using the following primers: SypHy1x–AgeI, forward, 5′-TAA CCG GTA TGG ACG TGG TGA ATC AGC-3′; SypHy1x-SalI, reverse, 5′-ATA GTC GAC TCA GGC GTA ATC GGG CAC ATC G-3′.
Lentivirus production and neuron transduction.
The production of vesicular stomatitis virus-pseudotyped third-generation lentiviruses was performed as described previously (De Palma and Naldini, 2002). The SypHy cDNA was inserted into the self-inactivating lentiviral vector 282.pCCLsin.PPT.hPGK.deltaNGFRpre kindly provided by Prof. L. Naldini (Telethon Institute for Gene Therapy, Milan, Italy). Briefly, the pSypHy–EGFP–N1 was linearized with the restriction enzyme XhoI. The 5′ protruding ends were blunted with the Quick Blunting kit (New England Biolabs). The SypHy cDNA was excised with the restriction enzyme XbaI. The 282.pCCLsin.PPT.hPGK.deltaNGFRpre vector was digested with the restriction enzymes SmaI and XbaI. Ligation was performed with the Quick Ligation kit (New England Biolabs). The cDNAs of WT mCherry–SynI and of its mutants were digested from and ligated into the plasmid pCCL–sin–PPT–prom–EGFP–Wpre provided by Dr. M. Amendola (Telethon Institute for Gene Therapy) using AgeI and SalI sites. Viral titers ranging from 1.0 to 5.0 × 108 transforming units/ml were obtained. Primary hippocampal neurons were infected with SypHy and either mCherry-tagged WT SynI (WT-SynI) or its phosphorylation site mutants (6,7A-SynI, 7A-SynI) at 7 d in vitro (DIV) at 10 multiplicity of infection. After 24 h, half of the medium was replaced with fresh medium. All experiments were performed between 16 and 18 DIV. For chronic activity deprivation experiments, primary neurons, infected as described above at 7 DIV, were incubated in the presence or absence of 1 μm TTX for 72 h and recorded at 18 DIV.
Live imaging experiments.
A wide-field inverted epifluorescence microscope (AF6000LX/TIRF; Leica) with a 63× (1.4 numerical aperture) oil-immersion objective and a short-arc lamp was used. The filter set for SypHy consisted of 484 ± 20 nm excitation filter, 514 nm dichroic mirror, and 525 ± 50 nm emission filter. To visualize mCherry, the filter set consisted of 575 ± 20 nm excitation filter, 604 nm dichroic mirror, and 628 ± 75 nm emission filter. Images were captured at a depth of 16-bits using an Orca-ER IEEE1394 CCD camera (Hamamatsu Photonics). For electrical field stimulation, an SIU-102 stimulator (Warner Instruments) was used. The Tyrode's solution contained 136 mm NaCl, 2.5 mm KCl, 10 mm HEPES buffered to pH 7.3, 10 mm d-glucose, 2 mm CaCl2, 1.3 mm MgCl2, 25 μm CNQX, and 50 μm d-APV to block glutamatergic transmission. For the NH4Cl challenge, a modified Tyrode's solution in which 50 mm NaCl was replaced by 50 mm NH4Cl was used.
To obtain a high percentage of cells expressing mCherry–SynI and/or SypHy and high numbers of synapses to be analyzed in each experiment, we transduced neurons with lentiviral vectors at 7 DIV. For imaging experiments, coverslips were mounted into the imaging chamber (∼100 μl volume; Quick Exchange Platform; Warner Instruments). Cells were perfused with Tyrode's solution at room temperature using laminar flow perfusion. Vacuum suction was applied to the opposite site of the chamber to stabilize the chamber volume. Action potentials (APs) were evoked using 20–40 mA pulses of 1 ms to obtain a peak-to-peak bipolar square wave current of 2 V, thereby yielding an electric field of 4 V/cm (1% duty cycle).
For SynI dispersion experiments (Chi et al., 2001), synapses were identified by a decrease in mCherry–SynI fluorescence at the start of a train of 900 APs at 10 Hz, and circular regions of interest (ROIs) of 1.7 μm diameter were manually positioned. The fluorescence changes (ΔF) of mCherry–SynI at each ROI were normalized to the starting fluorescence level (F0; average of 10 frames before the onset of stimulation) and corrected for bleaching. Time constants for dispersion and reassociation were determined by fitting the experimental points with the single-exponential decay function
 and the single-exponential rise function
and the single-exponential rise function
 respectively, where x is time, y is the specific mCherry–SynI fluorescence (ΔF/F0), a is the plateau value, and τ is the time constant of dispersion/recovery. Data were expressed as means ± SEM for the number of coverslips.
respectively, where x is time, y is the specific mCherry–SynI fluorescence (ΔF/F0), a is the plateau value, and τ is the time constant of dispersion/recovery. Data were expressed as means ± SEM for the number of coverslips.
For SypHy experiments (Burrone et al., 2006), after 10 s of baseline acquisition (F0), neurons were stimulated with a train of 1000–1200 APs at 10 Hz in the presence of 1 μm bafilomycin. At the end of the stimulation protocol, cells were perfused with 50 mm NH4Cl (Fmax). For each coverslip, a “time-lapse” dataset was acquired in the green channel (not binned, 1024 × 1024 pixels) at 2 Hz with 100 ms exposures. One image in the red channel was acquired at the beginning of each experiment to verify the relative expression of SynI at synaptic boutons. Images were analyzed using the Leica Application Suite and custom-written software scripts. Active synapses were identified by an increase >0.1 of the normalized SypHy signal in response to stimulation at 10 Hz and on the concomitant SynI expression in the red channel. To ensure unbiased analysis, custom-written software was used for automatic identification and selection of the ROIs representative of synapses in which fluorescence changes were evoked by the stimulation. ROIs were positioned, and quantitative measurements of fluorescence intensity at individual boutons were obtained by averaging pixel intensity within each ROI. Images were acquired with constant gain and exposure times across all experiments. The total increase in the fluorescence signal (ΔF) was calculated by subtracting F0. For each ROI, the ΔF was normalized to the fluorescence value obtained by alkalization of the entire vesicle pool using NH4Cl (ΔFmax). The size of the RP and the kinetics of RP depletion were calculated by fitting the experimental points with the monoexponential rise function (Eq. 2), where x is time, y is ΔF/Fmax, a is the plateau value corresponding to the RP size, and τ is the time constant of depletion. Data were expressed as means ± SEM for the number of coverslips. In all live imaging experiments, the number of synapses analyzed per coverslip ranged between 15 and 45.
Immunofluorescence and immunoblotting.
Primary hippocampal neurons were fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized with 0.1% Triton X-100 in PBS for 2 min before incubation with primary Syb2, phospho-Ser551 SynI, and SynI antibodies, followed by Alexa Fluor- conjugated 488 and 568 secondary antibodies. For experiments with acute stimulation, primary neurons were mounted in the live imaging chamber with or without electrical stimulation and lysed or fixed immediately after. For experiments with chronic modification of network activity, primary neurons of 15 DIV were incubated in the presence or absence of 1 μm TTX or 30 μm BIC for 72 h and lysed or fixed at the end of the incubation.
To allow semiquantitative comparisons of synapse immunoreactivity, experimental groups were processed under identical conditions, and samples were acquired and analyzed blindly. Synapses were semiautomatically detected using an in-house iterative algorithm based on serially decreasing thresholds similar to the previously described one (Bergsman et al., 2006) that was implemented in NIS-Elements software (Nikon). Peak fluorescence intensity for each synapse was obtained from an area of 2 × 2 pixels located on its center of mass, and an image average was generated as described previously (Orenbuch et al., 2012a). Fluorescence intensity profiles for each synapse were fitted according to the following Gaussian function:
 where xc is the center of the punctum maximum, y0 is the fluorescence offset, w2 is the variance of the Gaussian, and A its amplitude, using Origin (OriginLab). The integrated fluorescence values of single boutons were then calculated from the parameters of the Gaussian equation for a fixed length of 1.4 μm centered on the position of the fluorescence peak (Orenbuch et al., 2012b). The fluorescence values of all images acquired in each imaging session were normalized by the average value of the control group (either untreated WT neurons or KO neurons transduced with WT-SynI) that were imaged in parallel. The same number of synapses was selected in each image using a random selection routine. Image averages were then calculated to generate grand averages across images for each experimental condition.
where xc is the center of the punctum maximum, y0 is the fluorescence offset, w2 is the variance of the Gaussian, and A its amplitude, using Origin (OriginLab). The integrated fluorescence values of single boutons were then calculated from the parameters of the Gaussian equation for a fixed length of 1.4 μm centered on the position of the fluorescence peak (Orenbuch et al., 2012b). The fluorescence values of all images acquired in each imaging session were normalized by the average value of the control group (either untreated WT neurons or KO neurons transduced with WT-SynI) that were imaged in parallel. The same number of synapses was selected in each image using a random selection routine. Image averages were then calculated to generate grand averages across images for each experimental condition.
For immunoblotting, neurons were scraped in lysis buffer (20 mm HEPES, pH 7.4, 5 mm EDTA, and 2% SDS) plus protease and phosphatase inhibitors (0.2 mm PMSF, 2 μg/ml pepstatin, 1 mm NaF, and 1 mm NaVO4). After centrifugation at 16,000 × g for 5 min, samples were loaded on 10% SDS-PAGE gel, transferred to nitrocellulose, and immunoblotted with phospho-specific and pan-specific anti-SynI antibodies.
Electron microscopy.
Primary hippocampal neurons were fixed with 1.3% glutaraldehyde in 66 mm sodium cacodylate buffer, pH 7.4, postfixed in 1% OsO4, 1.5% K4Fe(CN)6, and 0.1 M sodium cacodylate, en bloc stained with 0.5% uranyl acetate, dehydrated, and embedded in Epon. Ultrathin sections were contrasted with 2% uranyl acetate and Sato's lead solution and observed with a Jeol JEM-1011 microscope at 100 kV equipped with an ORIUS SC1000 CCD camera (Gatan). Morphometric analysis of cross-section synaptic areas, active zone (AZ) lengths, total number of SVs, and docked SVs was done using NIH ImageJ. The AZ was identified based on the presence of physically docked SVs with a visible synaptic cleft, and a line passing through the AZ start and end points was calculated (see Fig. 4A). Total SVs were counted up to a maximal distance of 1400 nm from the center of the AZ segment. For the analysis of the spatial distribution of SVs, the position of each SV was located in NIH ImageJ, and the coordinates were transferred to CrimeStat (Ned Levine and Associates, National Institute of Justice, Washington, DC) in which the distance from the closest SV [mean nearest neighbor distance (MNND)] was calculated. For the evaluation of the synaptic versus perisynaptic SVs, the presynaptic region within 700 nm from the center of the AZ was defined as the synaptic area, whereas the presynaptic region extending from 700 to 1400 nm from the center of the AZ was calculated as the perisynaptic area. For the analysis of the SV distribution as a function of distance from the AZ, the minimal distance of each SV from the segment representing the AZ was calculated, and a frequency distribution graph was then built by grouping SVs in shells of increasing distance.
Patch-clamp recordings.
All patch experiments were performed on morphologically identified pyramidal neurons by the teardrop-shaped somata and the presence of an apical-like dendrite that was GABA negative in retrospective immunocytochemistry analysis. Patch electrodes, fabricated from thick borosilicate glass, were pulled and fire-polished to a final resistance of 5–7 MΩ. Whole-cell patch-clamp recordings from postsynaptic neurons were performed using an Axon Multiclamp 700B/Digidata1440A system (Molecular Devices) and an upright BX51WI microscope (Olympus) equipped with Nomarski optics. Recordings with either leak currents >100 pA or series resistance >15 MΩ were discarded. Experiments were performed at 22–24°C. The standard internal solution contained the following (in mm): 126 K gluconate/KCl, 4 NaCl, 1 MgSO4, 0.02 CaCl2, 0.1 BAPTA, 15 glucose, 5 HEPES, 3 ATP, and 0.1 GTP, pH 7.4. EPSCs were recorded in extracellular solution (Tyrode's solution) containing the following (in mm): 2 CaCl2, 140 NaCl, 1 MgCl2, 10 HEPES, 4 KCl, and 10 glucose, pH 7.3. Experiments were performed on 18 DIV neurons. Miniature EPSCs (mEPSCs) were recorded in the presence of TTX (300 nm) with d-AP-5 (50 μm), CGP58845 (5 μm), and BIC (30 μm) in the extracellular solution (Lignani et al., 2013).
Statistical analysis.
Data were analyzed by using the Student's t test or, in case of more than two experimental groups, by one-way ANOVA, followed by post hoc multiple-comparison tests using the SPSS software. Significance level was preset to p < 0.05. The distribution of experimental data was assessed using the Kolmogorov–Smirnov test.
Results
SynI is constitutively phosphorylated by Cdk5 and its phosphorylation is regulated by neuronal activity
SynI bears consensus sequences for phosphorylation by proline-directed kinases, such as Erk, Cdk1, and Cdk5. Two sites (Ser549 and Ser551; sites 6 and 7, respectively) were identified as substrates for Cdk5 in vitro (Jovanovic et al., 1996, 2001; Matsubara et al., 1996). To ascertain whether Cdk5 phosphorylates SynI in live neurons under basal conditions and whether the phosphorylation of Cdk5 sites in SynI is sensitive to Cdk5 inhibition, we used phosphorylation state-specific antibodies to phospho-site 7 of SynI (Yun et al., 2013). We first tested the specificity of the antibody using purified bovine SynI that was phosphorylated in vitro by activated Cdk5 (Cdk5/p35 complex) and found that the antibody recognized the SynI band only during incubation with the kinase (Fig. 1A). Specificity of the antibody was further assessed by immunoblotting and immunocytochemistry from KO neurons transduced with either the dephosphomimetic mutant 7A-SynI or WT-SynI used as a control. As expected, no specific signal was revealed in neurons expressing 7A-SynI, confirming the specificity of the antibody (Fig. 1B). Moreover, when GFP-tagged 7A-SynI or WT-SynI overexpressed in COS-7 cells was immunoprecipitated with anti-GFP antibodies and phosphorylated in vitro by activated Cdk5, the antibody only recognized phosphorylated WT-SynI (Fig. 1C).

Phosphorylation of SynI at site 7 by Cdk5 is constitutively high under basal conditions and is regulated by neuronal activity. A, Purified bovine SynI (1 μg) was incubated under phosphorylation conditions in the absence or presence of purified active Cdk5. Representative blot showing phospho-Ser551 (site 7; P-SynI) and total SynI immunoreactivities. B, Left column, Representative images of KO neurons transduced at 7 DIV with WT-SynI (SynI) or the Cdk5-dephosphomimetic mutant 7A-SynI and stained with the phospho-site 7 (P-SynI; green) and total (SynI; red) SynI antibodies. Scale bar, 10 μm. Right column, Phospho-SynI and total SynI immunoreactivities in lysates (15 μg protein/lane) of KO neurons transduced at 7 DIV with either WT-SynI (SynI) or the Cdk5-dephosphomimetic mutant 7A-SynI. C, GFP-tagged chimeras of either WT-SynI (SynI) or its phosphorylation mutant 7A-Syn I were overexpressed in COS-7 cells, purified by GFP immunoprecipitation (IP), phosphorylated “on bead” with purified active Cdk5, and assayed by immunoblotting with phospho-site 7-specific (P-Syn) and total SynI (SynI) antibodies. Input refers to the starting COS-7 cell lysates in which SynI is mostly dephosphorylated. The S551A mutation totally abolishes the in vitro site-7 phosphorylation by Cdk5. The experiments shown in A–C reveal the specificity of the phospho-Syn antibody toward the phosphorylated SynI site 7. D, Phospho-SynI and total SynI immunoreactivities in lysates (15 μg protein/lane) of WT hippocampal neurons (14 DIV) in the absence (Ctrl) or presence of S-Rosco (100 μm) for 30 min before harvesting. E, Phospho-SynI and total SynI immunoreactivities in lysates (15 μg protein/lane) of WT primary hippocampal neurons (14 DIV) incubated under resting conditions (Ctrl) or electrically stimulated with 1200 APs at 10 Hz before harvesting. Shown in D and E are representative blots (left) and the respective changes in the phospho-SynI/SynI ratio (right; means ± SEMs) evaluated by densitometric scanning of blots from three to four independent experiments. F, G, Left columns, Representative images of WT hippocampal neurons stained with phospho-Syn (green) and total SynI (red) antibodies under control conditions (Ctrl), after 30 min incubation with S-Rosco (100 μm; F), or after 1200 APs at 10 Hz (1200AP; G). Scale bar, 10 μm. Right columns, Mean values (left) and frequency distribution (right) of the phospho-SynI/SynI ratio in control (black bars), S-Rosco-treated (gray bars), and electrically stimulated (white bars) terminals. Data are means ± SEMs of three to four independent experiments (n = 150 terminals per experimental group per experiment). *p < 0.05, **p < 0.01, paired Student's t test. Superimposed on the frequency distribution histograms are Gaussian fits showing the shift of the phospho-SynI/SynI intensity ratio during treatments. Distributions were analyzed using the Kolgomorov–Smirnov test (p < 0.05, S-Rosco vs Ctrl).
The level of site-7 phosphorylation of SynI was constitutively high in primary hippocampal neurons (14 DIV) under basal conditions, and treatment with the Cdk5 inhibitor S-Rosco significantly reduced it (Fig. 1D). The level of site-7 phosphorylation also significantly decreased when neurons were subjected to electrical field stimulation with 1200 APs at 10 Hz as revealed by Western blot analysis (Fig. 1E), indicating that the phosphorylation state of site 7 is physiologically regulated. Accordingly, when neuronal cultures either treated with S-Rosco (Fig. 1F) or stimulated with 1200 APs at 10 Hz (Fig. 1G) were subjected to immunocytochemistry with site-7 phospho-specific antibodies, the high resting phospho-SynI/total SynI ratio of synaptic boutons was significantly decreased. The ratio exhibited a relatively high intersynaptic variability with a Gaussian distribution, which was left shifted by both treatments (Fig. 1F,G, right panels).
Cdk5 phosphorylation enhances SynI binding to actin filaments
Actin is an important and highly dynamic cytoskeletal regulator of SV trafficking at nerve terminals (for review, see Cingolani and Goda, 2008). SynI was reported to bind and bundle actin filaments and to stimulate actin monomer polymerization in a phosphorylation-dependent manner (for review, see Cesca et al., 2010). Therefore, we assessed the effect of Cdk5 phosphorylation on the interactions of SynI with actin filaments. Purified bovine SynI, phosphorylated by Cdk5 as described above, was incubated with preformed actin filaments. Interestingly, Cdk5-phosphorylated SynI bound to actin filaments more avidly than unphosphorylated SynI, with a significantly higher pellet/supernatant ratio (Fig. 2A). In this respect, the effect of Cdk5 phosphorylation of SynI resembles that of phosphorylation of Tyr301 by Src and is opposite to that exerted by PKA, Erk, or CaMKII.

Cdk5 phosphorylation enhances SynI binding to actin filaments. A, Top, Representative immunoblot of a SynI–actin binding experiment. Purified bovine SynI (500 nm) was phosphorylated by Cdk5 in vitro as shown in Figure 1A. Dephosphorylated (DP-SynI) or Cdk-phosphorylated (P-SynI) SynI was subsequently incubated with polymerized actin (5 μm) for 30 min and subjected to high-speed centrifugation. The immunoblots show total SynI and F-actin and their recovery in the pellet (bound). Bottom, The amounts of dephosphorylated (DP-SynI; black bars) or Cdk5-phosphorylated (P-SynI; gray bars) SynI bound to F-actin are reported in percentage of total SynI. Data are expressed as means ± SEMs of six independent experiments. *p < 0.05, Student's t test. Cdk5 phosphorylation of SynI significantly increased its binding to actin filaments. B, Top, Binding isotherms of dephosphorylated (DP-SynI; black symbols) and Cdk5-phosphorylated (P-SynI; white symbols) SynI to highly purified SVs that were quantitatively depleted of endogenous Syns. The mean ± SEM bound SynI values (picomoles per milligram of protein) were plotted against the respective mean ± SEM free SynI concentration (nanomolar) and fitted as described in Materials and Methods. No significant changes in either the Bmax or KD values were observed. Bottom, The binding of dephosphorylated (DP-SynI; black bars) and Cdk5-phosphorylated (P-SynI; gray bars) SynI to SVs is plotted as a percentage of the total SynI at the indicated total SynI concentrations. Data are means ± SEMs of six independent experiments. Cdk5 phosphorylation did not significantly affect the binding of SynI to SVs. C, Top row, Representative images showing punctate staining for mCherry–SynI transduced in KO hippocampal neurons at 7 DIV and analyzed at 14 DIV under resting conditions (rest), during (900AP), or after (recovery) field stimulation with 900 APs at 10 Hz. Scale bar, 5 μm. Bottom row, Representative time courses (left) of fluorescence changes in synapses before, during, and after field stimulation (red horizontal bar) in primary neurons expressing mCherry–SynI in the absence (black) or presence (gray) of R-Rosco. Dotted lines represent bleaching in the absence (black) or presence (gray) of R-Rosco. Rates of SynI dispersion during stimulation (middle) and reclustering after stimulation (right) in the absence (Ctrl, black bar) or presence (gray bar) of R-Rosco are shown as means ± SEMs of 13 and 12 coverslips, respectively. *p < 0.05, Student's t test. Cdk5 inhibition was ineffective in regulating the kinetics of activity-dependent SynI dispersion but significantly impaired the recovery phase.
Site-specific phosphorylation of SynI by distinct kinases is known to modify its interactions with SVs (for review, see Cesca et al., 2010). To directly study the role of Cdk5 phosphorylation on the binding of SynI to SVs, highly purified SynI-depleted SVs were incubated with purified bovine SynI that was phosphorylated previously in vitro by active Cdk5 or was left dephosphorylated. After separation of free and SV-bound SynI by high-speed centrifugation, the dephosphorylated and phosphorylated forms of SynI were analyzed in both total and pellet fractions by immunoblotting with total and phospho-specific SynI antibodies. The binding isotherms of dephosphorylated and Cdk5-phosphorylated SynI were similar, demonstrating that, in contrast to other kinases, Cdk5 does not modify the ability of SynI to bind SVs in vitro (Fig. 2B).
Synaptic activity induces dispersion of SynI along the axon because of its dissociation from the SV membrane, followed by SynI reclustering within presynaptic boutons as the stimulus ceases (Chi et al., 2001, 2003; Menegon et al., 2006; Orenbuch et al., 2012b). Thus, we tested whether Cdk5 phosphorylation also affects the dispersion and reclustering kinetics of mCherry–SynI transduced in hippocampal KO neurons. As reported previously, electrical field stimulation (900 APs at 10 Hz) caused a marked decrease in mCherry fluorescence intensity at nerve terminals, reflecting the dissociation of SynI from SVs and its dispersion along the axon. After returning to resting conditions, fluorescence recovered to reach ∼60% of its initial value. Interestingly, inhibition of Cdk5 activity with R-Rosco had no effect on the kinetics of dispersion, whereas it significantly impaired the reclustering rate (Fig. 2C). Consistent with the in vitro data, these results suggest that Cdk5 phosphorylation does not alter SynI affinity for SVs during activity in intact neurons, whereas it affects SynI reclustering at the end of the stimulation, likely because of the increased binding to F-actin.
Cdk5 inhibition is ineffective in regulating the recycling/RP ratio in SynI KO neurons
Cdk5 was implicated in the activity-dependent regulation of neurotransmitter release by determining the relative sizes of the RP and the RestP (Kim and Ryan, 2010). Because SynI is endogenously phosphorylated by Cdk5 in intact nerve terminals, we next investigated whether SynI acts as a molecular target for Cdk5 in the regulation of SV trafficking by comparing the effects of Cdk5 blockade on evoked SV exocytosis in WT and KO hippocampal neurons.
We used SypHy as a reporter of SV cycling (Sankaranarayanan et al., 2000; Burrone et al., 2006; Granseth et al., 2006). Experiments were performed in the presence of bafilomycin, a selective blocker of the SV proton pump, to evaluate exocytosis without the contribution of ongoing endocytosis (Sankaranarayanan and Ryan, 2001). This is important because endocytosis is strongly modulated by Cdk5 (Tan et al., 2003; Tomizawa et al., 2003; Evans and Cousin, 2007; Liang et al., 2007). At 14–17 DIV, neurons were subjected to a standard field stimulation protocol (1200 AP at 10 Hz), followed by NH4Cl alkalinization to estimate the total SV complement of the terminals (Fig. 3A,B).

Cdk5 inhibition is ineffective in regulating the RP/RestP ratio in SynI KO neurons. A, Top row, Representative images of SynI/SypHy cotransduced KO neurons analyzed at 14 DIV (7 d after transduction). SynI (red) and SypHy (green) displayed almost complete colocalization as shown in the merge panel. Bottom row, Representative images of SypHy fluorescence of an axon at rest, at the peak response to 1200 APs at 10 Hz (1200AP), and after the application of 50 mm NH4Cl. Scale bar, 5 μm. B, SypHy fluorescence traces plotted for WT (left), KO (middle), and KO + SynI (right) neurons stimulated with 1200 APs at 10 Hz in 1 μm bafilomycin in the presence (gray traces) or absence (black traces) of R-Rosco (100 μm). Traces show the mean ± SEM of the change in ΔF values (F − F0) normalized by the respective Fmax and represent the RP as a fraction of the total SV pool. Data are from 8 and 10 coverslips for WT cells, 13 and 8 coverslips for KO cells, and 12 and 10 coverslips for KO + SynI cells, in the absence or presence of R-Rosco, respectively. C, Evaluation of total dequenched fluorescence levels (Fmax; arbitrary units) of SypHy transduced in WT neurons (WT), SynI KO neurons (KO), or SynI KO neurons cotransduced with WT-SynI (KO + SynI) in the absence or presence of either R-Rosco or S-Rosco. Data (means ± SEMs) are from the same experiments shown in B plus additional nine coverslips for KO cells and nine for KO + SynI cells in the presence of S-Rosco. SynI KO neurons display a substantially reduced Fmax compared with WT neurons. Fmax in KO neurons is fully rescued by the expression of WT-SynI. Cdk5 blockade induces a reduction of Fmax in WT neurons and in KO + SynI but not in KO neurons. D, Mean ± SEM of the RP fraction of the total SV pool calculated from the exponential fit of the individual plateau values of RP depletion in the absence or presence of either R-Rosco or S-Rosco. E, The ensemble traces reported in B were normalized to the respective plateau values to visualize the kinetics of RP depletion. F, The rising phase of the individual fluorescence traces were fitted using a monoexponential function, yielding individual time constants (τ) whose mean ± SEM values are shown in the plot. Statistical analysis was performed using one-way ANOVA, followed by the Bonferroni's multiple comparison test. **p < 0.01, ***p < 0.001 versus the respective untreated control. Δp < 0.05; ΔΔΔp < 0.001 across genotype.
Previous work showed that deletion of SynI decreases the size of the total SV pool in presynaptic terminals (Li et al., 1995; Rosahl et al., 1995; Takei et al., 1995; Fassio et al., 2011). To assess the total SV pool in our system, we measured the fluorescence values of SypHy during alkalinization (Fmax). In agreement with previous reports, we observed a significant decrease in Fmax in KO neurons compared with WT neurons, an effect that was fully rescued by reintroduction of WT-SynI (Fig. 3C). Acute blockade of Cdk5 by either S- or R-Rosco for 30 min resulted in a significant decrease in the Fmax values that was observed in WT neurons or KO neurons expressing exogenous WT-SynI but not in KO neurons. Although the decrement in the Fmax values of KO neurons is likely contributed by the reported decrease in the total nerve terminal pool of SVs, the acute decrease during Cdk5 blockade suggests that Fmax may reflect changes in SV distribution within the terminal. However, the effect of roscovitine was strictly SynI dependent because it was virtually absent in KO neurons (Fig. 3B,C). Therefore, it is possible that Cdk5 not only controls the partitioning of SVs between RP and RestP (Kim and Ryan, 2010) but also affects, in an SynI-dependent manner, SV clustering and dispersion between the nerve terminal and the perisynaptic region of the axon.
To investigate the role of SynI in the regulation of the partitioning of SVs between RP and RestP, we analyzed the stimulation-induced fluorescence increase (ΔF = F − F0) as a percentage of the Fmax plateau. Under our experimental conditions, ∼40% of all SVs recycled during long-lasting stimulation in control synapses, representing the RP, whereas the remaining SVs were reluctant to be released, thus representing the RestP (Fig. 3C,D). Consistent with previously reported data (Kim and Ryan, 2010), blockade of Cdk5 activity by R-Rosco resulted in a higher percentage of SVs recruited to the RP at the expense of the RestP. When the same experimental protocol was applied to KO synapses, two interesting results were observed. First, the percentage of SVs belonging to RP was higher compared with control synapses. This is likely a compensatory effect for the reduced overall nerve terminal complement of SVs and is consistent with the higher SV dispersion and mobility reported recently in terminals lacking Syns (Fornasiero et al., 2012; Orenbuch et al., 2012a). Second, SV redistribution between the RP and RestP in KO neurons was unresponsive to Cdk5 inhibition, an effect that was fully rescued by reintroduction of WT-SynI (Fig. 3C,D).
The effects of Cdk5 inhibition on RP/RestP partitioning were reported to be associated with an acceleration of the kinetics of RP depletion, as assessed from the time course of the fluorescence increase during stimulation in the presence of bafilomycin (Kim and Ryan, 2010). Indeed, Cdk5 inhibition induced an intense acceleration of the kinetics in WT synapses but was completely ineffective in KO synapses, whose kinetics of RP depletion were much faster. Remarkably, the accelerating effect of Cdk5 blockade on the release kinetics was fully rescued by reexpression of WT-SynI in KO neurons (Fig. 3E,F).
The similar effects obtained with either roscovitine enantiomer rules out the possibility of off-target effects of the Cdk5 inhibitor on Ca2+ channels (see Materials and Methods). Together, the data suggest that SynI represents a key molecular pathway for the regulation of the RP/RestP ratio and the kinetics of SV depletion by Cdk5.
Cdk5 inhibition alters SV density and distribution in WT but not SynI KO synapses
Considering the acute effects of roscovitine on the Fmax values and the recently reported role of Syns in the spatial organization of SVs at the synapse (Fornasiero et al., 2012; Orenbuch et al., 2012a), we investigated, by electron microscopy, whether Cdk5 inhibition was able to modify the distribution of SVs within nerve terminals.
We performed ultrastructural analysis of WT and KO terminals in the absence or presence of R-Rosco and analyzed the total SV number, the degree of clustering, and their distribution between synaptic and perisynaptic areas (Fig. 4A; see Materials and Methods). Consistent with our previous observations (Fornasiero et al., 2012; Orenbuch et al., 2012a), the number of SVs in the total presynaptic area of KO terminals was significantly decreased (Fig. 4B, top), and SVs were more dispersed, as illustrated by their higher MNND (Fig. 4B, bottom). Although acute R-Rosco treatment did not significantly alter the total SV number in either genotype (Fig. 4B, top), it increased MMND and thus SV dispersion in WT, but not in KO, terminals (Fig. 4B, bottom). Analysis of SV distribution between the synaptic and the perisynaptic areas revealed that both R-Rosco treatment and SynI deletion similarly and significantly altered SV distribution by increasing the number of SV at perisynaptic sites at the expense of SVs at synaptic sites (Fig. 4C). Consistent with a direct role of SynI in mediating the Cdk5 effect, R-Rosco was virtually ineffective in altering the synaptic/perisynaptic SV distribution in KO terminals with a complete occlusion of the effects of Cdk5 inhibition and SynI deletion (Fig. 4C, bottom). These effects were investigated in more detail by analyzing the spatial distribution of SVs in 50 nm shells from the AZ. R-Rosco induced a right shift of the SV distribution histogram by decreasing the number of SVs in the proximal shells and increasing it in the distal shells, with a noticeable spread toward the perisynaptic regions (Fig. 4D, left). When the same analysis was performed in KO neurons, we observed that the distribution of SVs was already more disperse, did not exhibit a peak in the proximal shells, and was insensitive to R-Rosco treatment (Fig. 4D, right). No genotype- or treatment-dependent changes were observed in the synaptic area (WT, 0.90 ± 0.09 μm2; WT + Rosco, 0.83 ± 0.09 μm2; KO, 0.91 ± 0.01 μm2; KO + Rosco, 0.84 ± 0.09 μm2), perisynaptic area (WT, 0.29 ± 0.03 μm2; WT + Rosco, 0.30 ± 0.04 μm2; KO, 0.29 ± 0.03 μm2; KO + Rosco, 0.29 ± 0.03 μm2), and length of the AZ (WT, 0.43 ± 0.03 μm; WT + Rosco, 0.44 ± 0.03 μm; KO, 0.44 ± 0.03 μm; KO + Rosco, 0.44 ± 0.05 μm).

Cdk5 inhibition alters SV density and distribution in WT, but not in SynI KO, synapses. A, Representative electron micrographs of WT and KO hippocampal synapses (14 DIV) either untreated or treated with R-Rosco (100 μm) for 30 min. In the WT panel, a schematic representation of the morphometric procedure followed to calculate SV distribution at synapses is shown. After identification of the AZ (orange line) and tracing of the line passing through the start (C1) and the end (C2) points of AZ (dotted line), the distance of individual SVs from the AZ was calculated in the Cartesian plane. The synaptic zone (blue area) was defined as the presynaptic area between 0 and 700 nm distance from the center of the AZ segment, whereas the perisynaptic zone (green areas) was defined as the presynaptic area between 700 and 1400 nm from the AZ segment. Green spheres represent perisynaptic SVs (for additional details, see Materials and Methods). Scale bar, 200 nm. B, Morphometric analysis of the total SV number (distance from the center of the AZ < 1400 nm; top) and extent of clustering (MNND; bottom) in WT and KO synapses treated as described in A. No changes in the terminal area and the AZ length were observed across experimental groups. Statistical analysis was performed using one-way ANOVA, followed by the Bonferroni's multiple comparison test. ***p < 0.001 versus the respective untreated control. ΔΔp < 0.005, ΔΔΔp < 0.001 across genotype. C, Morphometric analysis of the SV number in synaptic (SV distance from AZ < 700 nm) and perisynaptic (700 nm < SV distance from AZ < 1400 nm) areas in WT (top) and KO (bottom) synapses treated as described in A. No changes in the synaptic and perisynaptic areas were observed across experimental groups. Statistical analysis was performed using one-way ANOVA, followed by the Bonferroni's multiple comparison test. ***p < 0.001 versus the respective untreated control. D, Distribution of SVs in WT (left) and KO (right) nerve terminals incubated for 30 min in the absence (black bars) or presence (gray bars) of R-Rosco (100 μm). The absolute number of SVs located within successive 50 nm shells from the AZ is shown. The frequency distribution of SVs as a function of the distance from the AZ was analyzed by using the Kolgomorov–Smirnov test. R-Rosco treatment significantly altered the SV distribution in WT (p = 0.001), but not in KO, synapses. The effects of R-Rosco within the respective genotype were compared by using the Student's t test within shells (*p < 0.05, ***p < 0.005). Gray squares represent perisynaptic SVs as evaluated in C. Data are means ± SEMs of 58 (WT), 60 (WT+R-Rosco), 67 (KO), and 71 (KO+R-Rosco) synapses from four independent preparations.
Phosphorylation of SynI at Ser551 is necessary and sufficient for mediating the effects of Cdk5 on the SV RP size and depletion kinetics
Of the two Cdk5 phosphorylation sites located in the SynI D-domain, Ser549 (site 6) is also targeted by Erk and Cdk1, whereas Ser551 (site 7) is a specific target for Cdk5 (Jovanovic et al., 1996, 2001; Matsubara et al., 1996). To unequivocally demonstrate a role of SynI as a Cdk5 target in the regulation of the RP/RestP pool ratio and SV depletion kinetics, we performed the same experimental protocol described above while transducing SynI KO neurons with either WT-SynI or the dephosphomimetic mutants 6,7A-SynI and 7A-SynI. Both WT-SynI and its mutants fused to mCherry were expressed to a similar extent in KO neurons, as shown by immunoblotting and measurement of mCherry fluorescence levels at SypHy-positive synapses that responded to stimulation (Fig. 5A).

Phosphorylation of SynI is necessary and sufficient for the effects of Cdk5 on the SV RP size and depletion kinetics. A, Left panels, Representative images of 14 DIV KO neurons transduced at 7 DIV with either WT-SynI (SynI) or the Cdk5-dephosphomimetic mutants 6,7A-SynI and 7A-SynI. Scale bar, 10 μm. Middle panel, Representative immunoblot with anti-Cherry (α-Cherry) and anti-SynI (α-SynI) antibodies shows comparable expression levels of WT-SynI, 6,7A-SynI, and 7A-SynI in lysates of transduced KO primary hippocampal neurons. Actin immunoreactivity (α-Actin) was used as a loading control. Right panel, Expression levels of transduced SynI variants in SypHy-positive ROIs corresponding to active synaptic boutons, as evaluated from the fluorescence intensity of the mCherry signal. WT and mutant exogenous SynI are similarly expressed and correctly targeted to nerve terminals in SynI KO neurons. B, Total dequenched fluorescence levels (Fmax; arbitrary units) of SypHy in SynI KO neurons cotransduced with WT-SynI (SynI), 6,7A-SynI (6,7A-SynI), or 7A-SynI (7A-SynI) in the absence or presence of either R-Rosco or S-Rosco. Data (means ± SEMs) are from 12 and 10 KO + SynI coverslips, 16 and 10 KO + 6,7A-SynI coverslips, and 12 and 7 KO + 7A-SynI coverslips in the absence or presence of R-Rosco, respectively. Both 6,7A-SynI and 7A-SynI neurons exhibited lower Fmax values compared with WT-SynI transduced neurons. Cdk5 blockade induced a reduction of Fmax in KO + SynI, whereas it was ineffective in KO + 6,7A-SynI and KO + 7A-SynI neurons. C, Mean ± SEM of RP over the total SV pool in the absence or presence of R-Rosco. D, Mean ± SEM time constants (τ) of RP depletion obtained from monoexponential fitting of individual curves in the absence or presence of R-Rosco. Statistical analysis was performed by using one-way ANOVA, followed by the Bonferroni's multiple comparison test. **p < 0.01, ***p < 0.001 versus the respective untreated control. Δp < 0.05, ΔΔΔp < 0.005 across SynI variants.
To assess the effect of the mutants, we compared their influence on synaptic parameters with that of WT-SynI. Specifically, we looked at Fmax, RP/RestP size, and SV depletion kinetics in the absence or presence of R-Rosco (Fig. 5B–D). Fmax values of both mutants were significantly lower than those of WT-SynI and were insensitive to Cdk5 inhibition by R-Rosco (Fig. 5B). Both SynI mutants significantly increased the RP/RestP partitioning, and the 7A-SynI mutant significantly accelerated the kinetics of SV depletion with respect to WT-SynI. Moreover, the effects of acute Cdk5 inhibition on both parameters were fully occluded by both mutations (Fig. 5C,D). The similar efficacy of the site-6/site-7 double mutation and of the single site-7 mutation, which is specific for Cdk5, suggests a preferential involvement of the latter in mediating the effects of the kinase.
Together, the data show that mutation of phosphorylation site 7 in SynI phenocopies the effect of Cdk5 inhibition on the redistribution of SVs between the RP and RestP and that the dephosphomimetic mutation of this very site is sufficient to abolish the effects of Cdk5 inhibition on SV exocytosis. Therefore, phosphorylation of SynI site 7 is both necessary and sufficient for the action of Cdk5 in controlling nerve terminal homeostasis.
Mutation of Cdk5 phosphorylation sites of SynI mimics the effects of Cdk5 inhibition on SV density and distribution
We next verified whether the expression of the Cdk5 dephophomimetic mutants of SynI in KO neurons was also able to affect the distribution of SVs at the ultrastructural level and to mimic the effects of Cdk5 inhibition (Fig. 6A). Similar to roscovitine (Fig. 4B), expression of either dephosphomimetic mutant of SynI did not affect the number of SVs in the total presynaptic area (Fig. 6A,B). However, either mutant significantly altered the distribution of SVs by increasing SV dispersion (MNND; Fig. 6B) and the number of SVs at perisynaptic sites at the expense of SVs at synaptic sites (Fig. 6C). Consistently, either mutant induced a right shift of the SV distribution histogram with fewer SVs in the proximal shells and more SVs in the distal shells compared with WT-SynI (Fig. 6D). No mutant-dependent changes with respect to WT-SynI were observed in the synaptic area (WT-SynI, 0.79 ± 0.07 μm2; 6,7A-SynI, 0.80 ± 0.05 μm2; 7A-SynI, 0.81 ± 0.06 μm2), perisynaptic area (WT-SynI, 0.28 ± 0.03 μm2; 6,7A-SynI, 0.34 ± 0.03 μm2; 7A-SynI, 0.28 ± 0.02 μm2), and length of the AZ (WT-SynI, 0.52 ± 0.05 μm; 6,7A-SynI, 0.45 ± 0.04 μm; 7A-SynI, 0.50 ± 0.05 μm). The data indicate that phosphorylation of the Cdk5 sites in SynI does not affect the total SV number, as deletion of SynI does. Rather, the inability of Cdk5 to phosphorylate SynI alters SV distribution by increasing SV dispersion and consequently perisynaptic SVs. These effects of dephosphomimetic mutants on SV distribution were similar to those caused by the complete absence of SynI or by the acute inhibition of Cdk5 by roscovitine in WT neurons.

Mutation of Cdk5 phosphorylation sites of SynI mimics the effects of Cdk5 inhibition on SV density and distribution. A, Representative electron micrographs of KO neurons (14 DIV) transduced at 7 DIV with WT-SynI (SynI), 6,7A-SynI, or 7A-SynI. Scale bar, 200 nm. B, Morphometric analysis of the total SV number (left) and MNND (right) in KO synapses of neurons transduced as described in A. No changes in the terminal area or the AZ length were observed across experimental groups. Data are means ± SEMs of 100 (SynI, black bars), 93 (6,7A-SynI, dark gray bars), and 119 (7A-SynI, light gray bars) KO synapses from two independent preparations. Data were analyzed by one-way ANOVA, followed by the Dunnett's multiple comparison test. ΔΔΔp < 0.001 versus SynI. C, Morphometric analysis of the SV number in synaptic and perisynaptic areas in the KO neurons described in B. No changes in the synaptic and perisynaptic areas were observed across experimental groups. Data are analyzed by one-way ANOVA, followed by the Dunnett's multiple comparison test. ΔΔΔp < 0.001 versus SynI. D, Distribution of SVs in KO + SynI and KO + 6,7A-SynI (left) or KO + 7A-SynI (right) nerve terminals. The absolute number of SVs (means ± SEMs) located within successive 50 nm shells from the AZ is shown. Gray squares represent perisynaptic SVs as evaluated in C. The frequency distribution of SVs as a function of the distance from the AZ was analyzed by the Kolgomorov–Smirnov test. Either SynI mutant significantly right shifted the SV distribution with respect to WT-SynI (p = 0.0027, 6,7A-SynI vs SynI; p = 0.003, 7A-SynI vs SynI). Within shells, the effect of either mutant was compared with that of WT-SynI by using the Student's t test (**p < 0.01).
Quantitative immunofluorescence analysis of the endogenous SV marker Syb2 (Takamori et al., 2006) was also performed to analyze modifications in clustering of SVs at synaptic sites during roscovitine treatment (Fig. 7A,B) and expression of the 6,7A-SynI mutant (Fig. 7C,D). Immunostaining of primary hippocampal neurons was analyzed by examining both the peak fluorescence at the center of each synaptic bouton, a measure that represents the maximal density of SVs within the terminal, and the integrated fluorescence, a measure that reflects the total SV population in the synaptic area (see Materials and Methods). Under basal conditions, both the normalized Syb2 peak intensity in synaptic puncta and the normalized integrated fluorescence of KO neurons were significantly lower than the respective values of WT neurons, a finding in accordance with the decreased absolute SV density and the higher SV dispersion demonstrated in the absence of SynI (Figs. 4, 7A,B; Fornasiero et al., 2012). Examination of the spatial distribution of Syb2 fluorescence revealed that synaptic puncta in KO neurons were more diffuse with respect to the compact and intense appearance of WT terminals. Consistent with the predictions based on the SypHy Fmax values and the ultrastructural data, the acute treatment with roscovitine significantly decreased both the peak and the integrated Syb2 fluorescence intensity of WT synaptic puncta to approximately the same value observed in KO terminals (Fig. 7B). In the latter terminals, the effect of acute Cdk5 inhibition was completely occluded. These observations support the idea that either SynI deletion or Cdk5 inhibition destabilizes SV clusters, allowing redistribution of SVs that disperse into the perisynaptic area. Interestingly, when primary KO neurons were transduced with the 6,7A-SynI mutant, significant decreases in both peak and integrated Syb2 fluorescence intensity were observed with respect to WT-SynI transduced neurons, mimicking the effects of either roscovitine treatment or SynI deletion on SV distribution (Fig. 7C,D).

Quantitative immunofluorescence analysis with the endogenous SV marker Syb2. A, Immunostaining for Syb2 in WT and KO neurons that were treated with either vehicle (Ctrl) or R-Rosco (100 μm) for 30 min. The Syb2 fluorescence intensity at synaptic boutons is lower in both untreated KO neurons and WT neurons treated with R-Rosco with respect to untreated WT neurons, confirming SV dispersion. Scale bar, 5 μm. B, Normalized peak fluorescence intensity and integrated fluorescence at synaptic puncta of WT and KO neurons incubated in the absence or presence of R-Rosco. Notice that, in untreated KO neurons and in both WT and KO neurons treated with R-Rosco, the fluorescence is less concentrated at synaptic boutons. Data are means ± SEMs of 42 (WT), 45 (WT + R-Rosco), 42 (KO), and 41 (KO + R-Rosco) images from three independent preparations. Statistical analysis was performed using two-way ANOVA, followed by the Bonferroni's multiple comparison test. *p < 0.05, **p < 0.01 versus the respective untreated control. ΔΔp < 0.01, ΔΔΔp < 0.001 across genotype. C, Immunostaining for Syb2 in KO neurons that were transduced with mCherry chimeras of either WT-SynI (left) or 6,7A-SynI mutant (right). The bottom row displays the merge of the green (Syb2) and red (mCherry) channels showing that the two proteins colocalize at synaptic boutons. The Syb2 fluorescence intensity at synaptic boutons is lower in KO neurons transduced with 6,7A-SynI with respect to WT-SynI. Scale bar, 2 μm. D, Normalized peak fluorescence intensity and integrated fluorescence at synaptic puncta of neurons transduced with WT-SynI (SynI) or 6,7A-SynI. Expression of the dephosphomimetic mutant decreases Syb2 fluorescence at synaptic boutons. Data are means ± SEMs of 15 (WT-SynI) and 12 (6,7A-SynI) images from three independent preparations. Statistical analysis was performed using the Student's t test. *p < 0.05, **p < 0.01.
Together with the ultrastructural data and SypHy Fmax determinations, these results confirm our previous observations in SynI/II/III KO mice (Fornasiero et al., 2012; Orenbuch et al., 2012a) and directly implicate SynI in the control of SV clustering/dispersion within nerve terminals. The observations that the effect of roscovitine on SV distribution is occluded in KO terminals (Figs. 4, 7B) and that the effect of Cdk5 inhibition is mimicked by the expression of the Cdk5 phosphorylation mutant indicate SynI as the main Cdk5 effector on SV distribution.
SynI phosphorylation by Cdk5 at site 7 is changed during chronic modifications of electrical activity in mature hippocampal synapses
A pivotal role of Cdk5 in synaptic scaling was reported recently in several experimental models (Kim and Ryan, 2010; Mitra et al., 2012; Peng et al., 2013). Chronic silencing of neuronal activity by TTX was reported to strongly downregulate the expression of Cdk5 at the presynaptic level (Kim and Ryan, 2010). If SynI is the main target for the Cdk5 homeostatic action on SV trafficking, then the phosphorylation state of SynI site 7 should change during chronic modifications of electrical activity that also alter SV partitioning into pools. Thus, we treated WT primary hippocampal neurons with either TTX or BIC for 72 h and analyzed the phosphorylation state of SynI at site 7 by immunoblotting and immunocytochemistry.
In total culture extracts, long-lasting blockade of activity by TTX significantly reduced SynI phosphorylation, whereas the opposite effect was observed after chronic increase of network activity by BIC (Fig. 8A). Analysis of the phospho-SynI/SynI ratio at single nerve terminals by double immunocytochemistry fully confirmed the results on total extracts (Fig. 8B). A clear-cut left shift in the frequency distribution of the phospho-SynI/total SynI ratio, reminiscent of that obtained during acute Cdk5 inhibition with roscovitine, was observed after chronic activity blockade with TTX, whereas an opposite right shift was brought about by sustained hyperexcitability induced by chronic GABAA receptor blockade with BIC (Fig. 8B). Thus, the homeostatic responses to sustained activity changes are associated with specific changes in the Cdk5 phosphorylation of SynI that can, in turn, modify SV trafficking.

SynI phosphorylation at site 7 by Cdk5 in mature hippocampal synapses is regulated by homeostatic synaptic scaling. A, Left, Representative blot showing Ser551 phosphorylated (P-SynI) and total (SynI) SynI immunoreactivity in lysates from primary WT neuronal cultures that were left untreated (Ctrl) or treated with either TTX (1 μm) or BIC (30 μm) for 72 h. Right, Phospho-SynI/SynI ratios calculated by densitometric scanning of the blots. Data are means ± SEMs of three independent experiments run in duplicate. *p < 0.05, **p < 0.01, one-way ANOVA, followed by Dunnett's multiple comparison test. B, Top panels, Representative images of hippocampal synapses labeled with phospho-Syn (green) and total SynI (red) antibodies under control conditions (Ctrl) or after incubation with either TTX or BIC for 72 h. Scale bar, 10 μm. Bottom panels, Mean values (left) and frequency distribution (right) of the phospho-SynI/SynI ratio in terminals of control (black bars; same data from Fig. 1F), TTX-treated (white bars), or BIC-treated (gray bars) neurons. Data are means ± SEMs of four independent experiments (n = 150 terminals per experimental group per experiment). Chronic TTX treatment significantly decreased the extent of Cdk5 phosphorylation of SynI and induced a left shift in the distribution profile of the phospho-SynI/SynI ratio compared with untreated terminals. In contrast, chronic BIC treatment significantly increased the degree of SynI phosphorylation by Cdk5 and caused a right shift in the distribution profile. *p < 0.05, **p < 0.01, one-way ANOVA, followed by Dunnett's multiple comparison test. Frequency distributions were analyzed using the Kolgomorov–Smirnov test (p = 0.011, TTX vs control; p = 0.003, BIC vs control).
The Cdk5-dependent homeostatic scaling of presynaptic activity in response to chronic silencing of neuronal activity occurs through phosphorylation of SynI at Ser551
Because the Cdk5-linked homeostatic scaling of neuronal activity during chronic TTX is characterized by an increased size of the releasable pool of SVs and an increased proportion of presynaptically active synapses (Kim and Ryan, 2010), we addressed the question as to whether phosphorylation of SynI site 7 is also essential in mediating this homeostatic response. Thus, we expressed either WT-SynI or its site-7 dephosphomimetic mutant in KO neurons that were subjected to long-lasting (72 h) treatment with vehicle or TTX and analyzed the RP/RestP ratio, the kinetics of RP depletion, and the frequency and amplitude of mEPSCs, as a standard readout for homeostatic synaptic scaling. Under control conditions, the dephosphomimetic mutant, in addition to increasing the RP/RestP ratio and accelerating RP depletion (Figs. 5C,D, 9A,B), significantly increased the frequency of mEPSCs in the absence of changes in their amplitude (Fig. 9C–E).

Mutation of phosphorylation site 7 in SynI mimics and occludes the effects of chronic silencing of neural activity on SV trafficking and mEPSC frequency. A, Ensemble average traces of SypHy fluorescence plotted in KO synapses transduced with either WT-SynI (SynI; left) or 7A-SynI (right). Neurons, incubated for 72 h in the absence (black traces) or presence (gray traces) of 1 μm TTX, were stimulated with 1200 APs at 10 Hz in 1 μm bafilomycin. Solid traces show the average ± SEM time course of the changes in ΔF values (F − F0) normalized by the respective Fmax and represent the RP as a fraction of the total SV pool. Data are from seven, four, six, and seven coverslips for WT-SynI, WT-SynI + TTX, SynI-7A, and SynI-7A + TTX groups, respectively. B, The mean ± SEM fraction of the RP fraction of the total SV pool, calculated from the individual plateau values (left), and the means ± SEMs of the individual time constants (τ) of RP depletion, calculated from the monoexponential fitting of the individual fluorescence traces (right), are shown for WT-SynI- or 7A-SynI-expressing neurons incubated for 72 h in the absence (black columns) or presence (gray columns) of TTX. Statistical analysis was performed using one-way ANOVA, followed by the Bonferroni's multiple comparison test. *p < 0.05, ***p < 0.001 versus the respective untreated control. Δp < 0.05, ΔΔΔp < 0.001 across genotype. C, mEPSCs were recorded in KO synapses transduced with either WT-SynI (SynI; left) or 7A-SynI (right) incubated for 72 h in the absence (black traces) or presence (gray traces) of 1 μm TTX. D, Cumulative probability of the interevent intervals (left) and amplitude (right) of mEPSCs recorded in KO synapses transduced with either WT-SynI (top) or 7A-SynI (bottom) and incubated in the presence (gray line) or absence (black line) of TTX. E, The frequency (left) and amplitude (right) of mEPSCs in transduced synapses are reported as means ± SEMs for the four experimental groups described above. Cumulative curves and mean values were obtained from 3 min recordings from n = 15, 13, 12, and 12 neurons for WT-SynI, WT-SynI + TTX, SynI-7A, and SynI-7A + TTX groups, respectively. Statistical analysis was performed using two-way ANOVA, followed by the Bonferroni's multiple comparison test. *p < 0.05 versus respective untreated control. ΔΔΔp < 0.001 across genotype.
Chronic silencing of neuronal activity by TTX generated the expected homeostatic responses consisting of increased RP/RestP ratio (Fig. 9B, left), accelerated kinetics of RP depletion (Fig. 9B, right), and increased frequency and amplitude of mEPSCs (Fig. 9C–E). However, when the homeostatic responses were assessed in neurons expressing the site-7 dephosphomimetic mutant, no significant differences in all parameters were observed with respect to either chronic TTX alone or the SynI mutant in the absence of TTX, except for mEPSC amplitude whose response was not affected by the SynI mutation (Fig. 9E).
The data show that the SynI mutant fully mimics the presynaptic effects of the strong Cdk5 downregulation occurring during chronic blockade of activity (Kim and Ryan, 2010), and its expression under chronic TTX completely occludes the presynaptic scaling response. This suggests that the downregulation of the Cdk5 pathway impinging on SynI phosphorylation is a main contributor of the homeostatic response at the presynaptic level. The preserved quantal size in neurons expressing the SynI mutant and the lack of modulation of the homeostatic increase in mEPSC amplitude evoked by chronic TTX are consistent with a purely presynaptic site of action of SynI phosphorylation by Cdk5.
Discussion
Cdk5 plays numerous regulatory roles in synaptogenesis, synaptic transmission, and plasticity (Hawasli and Bibb, 2007; Barnett and Bibb, 2011; Guan et al., 2011). By phosphorylating key substrates at both presynaptic and postsynaptic levels, Cdk5 is a key regulator of synaptic transmission that acts in a concerted manner with the phosphatase calcineurin. At the presynaptic level, in addition to the well established regulation of SV endocytosis (Cousin and Robinson, 2001; Tan et al., 2003; Tomizawa et al., 2003; Evergren et al., 2007), an important role of Cdk5 in the modulation of presynaptic voltage-gated Ca2+channels and SV trafficking was demonstrated recently. Although the functional impact on N-type Ca2+ channels and presynaptic Ca2+ influx is still controversial (Su et al., 2012; Kim and Ryan, 2013), it was shown that Cdk5 downregulates the RP by sequestering SVs in the RestP, an effect that is counteracted by calcineurin (Kim and Ryan, 2010; Marra et al., 2012). SynI is a good candidate for mediating these effects because of its function in the regulation of SV trafficking. Although SynI was identified as a Cdk5 substrate in the nineties (Jovanovic et al., 1996, 2001; Matsubara et al., 1996), no data were provided so far about the potential function of Cdk5 phosphorylation of SynI in presynaptic physiology. Only recently, phosphorylation of SynI by Cdk5 was reported to play a role in neurite outgrowth and SynI recruitment to nascent synapses (Easley-Neal et al., 2013; Yun et al., 2013).
Here, we demonstrate that SynI phosphorylation by Cdk5 is physiologically regulated and that SynI is a necessary and sufficient mediator of Cdk5 actions on SV trafficking at the presynaptic level. In fact, the effects of R-Rosco or S-Rosco on RP/RestP ratio and kinetics of RP depletion were virtually abolished in both KO neurons and KO neurons expressing the nonphosphorylatable mutant 6,7A-SynI or 7A-SynI. This evidence rules out potential off-target effects of roscovitine, whose R (but not S) enantiomer was reported to affect Ca2+ conductances, and of chronic SynI deletion, because reexpression of SynI rescues both the phenotype and the roscovitine effect. The observation that single-point mutations at the Cdk5 phosphorylation sites totally abolish the roscovitine effect unequivocally implicates SynI as the downstream effector of Cdk5 on SV trafficking. Moreover, the ability of the single site mutant 7A-SynI to phenocopy the effects of the absence of SynI identifies site 7, a specific Cdk5 phosphorylation site, as the central switch for these effects.
What is the mechanism by which SynI phosphorylation sequesters recycling SVs to the release-reluctant RestP? We found that, although the SV binding of SynI is unaffected by Cdk5 phosphorylation, its association with F-actin is increased. Nerve terminal actin is a dynamic regulator of SV trafficking and represents a scaffolding element for SV cycling, turnover, and mobility (Cingolani and Goda, 2008). Although actin does not participate in determining the RP size, inhibition of actin remodeling disrupts the spatial segregation of recycling SVs (Marra et al., 2012; Ratnayaka et al., 2012). Although actin binding by SynI is negatively regulated by PKA, CaMK, and Erk phosphorylation (Cesca et al., 2010), phosphorylation by Src (Tyr301; Messa et al., 2010) and Cdk5 (Ser551; this study) were found to enhance the interactions with F-actin. The latter effect can also explain the decreased rate of SynI reclustering induced by Cdk5 inhibition. It was reported recently that release-reluctant SVs can be rapidly converted into fast-releasing/recycling SVs in an actin-dependent manner (Lee et al., 2012). Thus, the positive modulation of SynI–actin interactions provides a model in which decreased phosphorylation of SynI at the Cdk5 sites by either calcineurin or homeostatic downregulation of Cdk5 expression (Kim and Ryan, 2010) disrupts the interaction with actin filaments and releases SV clusters from the RestP to the RP. Although it is not possible to exclude that the SynI phosphorylation mutants interfere with the recently reported effects of Cdk5 on Ca2+ influx (Su et al., 2012; Kim and Ryan, 2013), this possibility is unlikely in view of the lack of detectable association of SynI with either N- or P/Q-type Ca2+ channels (Medrihan et al., 2013). SV trafficking in mature neurons critically depends also on other Syn isoforms. Putative consensus sites for Cdk5 phosphorylation also exist in SynIII but not in SynII (Candiani et al., 2010). Although SynIII is downregulated in mature neurons, we found recently that both the RP size and total synaptic SVs are decreased in parallel in SynII KO neurons, with no significant changes in the RP/RestP ratio (Corradi et al., 2014). Thus, SynI appears to represent the main, if not the only, mechanism responsible for the action of Cdk5 on the SV partition between releasable and nonreleasable pools in mature neurons.
In addition to the increase in the RP/RestP ratio, acute Cdk5 inhibition induced unclustering and dispersion of SVs from synaptic to perisynaptic areas, an effect that was shared by SynI deletion (Fornasiero et al., 2012; this study). Notably, effects on SV dispersion similar to those elicited by acute roscovitine were observed by expressing the dephosphomimetic SynI mutants, with both conditions leaving the absolute number of SVs unaffected. This phenomenon was assessed by analyzing SV counts in the nerve terminal and perisynaptic areas with electron microscopy and was confirmed by the quantification of the fluorescence of the endogenous SV marker Syb2 and the exogenously expressed SV marker SypHy at synaptic boutons. Indeed, a decreased SV clustering at synaptic sites both in the absence of SynI during acute roscovitine treatment or after expression of 6,7A-SynI was observed by both quantitative Syb2 immunocytochemistry and SypHy Fmax evaluation. The previously observed decreases in Fmax in SynI and SynII KO neurons were attributed to an impaired SV density at synaptic sites (Fassio et al., 2011; Corradi et al., 2014). However, because Fmax is related to the expression level and SV targeting of the engineered SypHy at synaptic sites, a decrease in its level can be also ascribed to an augmented SV dispersion. In all experiments, the effects of acute roscovitine on Fmax values, Syb2 fluorescence and ultrastructural SV distribution were fully occluded when the treatment was performed in KO neurons or in KO neurons expressing dephosphomimetic SynI mutants, consistent with a common origin of these effects.
The dephosphomimetic 7A-SynI mutant increased the frequency of spontaneous miniature exocytotic events. The source of SVs undergoing spontaneous fusion is still unclear, and conflicting evidence exists for an origin from either the RestP (Sara et al., 2005; Fredj and Burrone, 2009; Ramirez et al., 2012) or the releasable pools of SVs (Lemke and Klingauf, 2005; Groemer and Klingauf, 2007; Hua et al., 2010). The increase in mEPSC frequency observed during expression of Cdk5 phosphorylation mutants of SynI that parallels the increase in the RP/RestP ratio supports the latter hypothesis and is compatible with the increased SV availability and recruitment of silent synapses reported during inhibition of the Cdk5 pathway (Kim and Ryan, 2010).
Recent evidence strongly indicates that Cdk5 is a master regulator of synaptic homeostasis and participates in the scaling of synaptic strength to compensate the effects of sustained hypoactivity or hyperactivity (Seeburg et al., 2008; Mitra et al., 2012; Peng et al., 2013). Interestingly, chronic silencing of firing activity strongly downregulates nerve terminal Cdk5, producing a homeostatic roscovitine-like effect that increases recruitment of SVs to the RP and activates previously silent synapses, thus enhancing the signaling capability of nerve terminals in response to activity deprivation (Kim and Ryan, 2010). We found that SynI is part of this homeostatic mechanism and responds to heightened or decreased activity with opposite changes in the phosphorylation state of site 7. In this respect, chronic blockade of activity, associated with decreased expression of Cdk5, inhibits SynI phosphorylation at site 7, resulting in the upscaling of synaptic strength and RP size, a condition mimicked by expression of the Cdk5-dephosphomimetic mutants of SynI under basal conditions. Moreover, the occlusion between the effects of chronic TTX and SynI site-7 mutation on the RP/RestP ratio, the dynamics of RP depletion, and mEPSC frequency indicates that the homeostatic regulation of presynaptic activity attributable to Cdk5 occurs through, and is reflected by, changes in the phosphorylation state of SynI.
The effects of Cdk5 at the nerve terminal level are counterbalanced by Ca2+-dependent dephosphorylation by calcineurin (Kim and Ryan, 2010, 2013; Ratnayaka et al., 2012). Therefore, this mechanism provides an efficient push–pull system for the fine tuning of the exocytotic “power” at individual synapses from silence to maximal performance, by shifting the SV complement between releasable and nonreleasable compartments. We demonstrated that SynI is the main Cdk5 effector in this mechanism and that it mediates the effects of Cdk5 based on the phosphorylation state of Ser551 during both normal activity and homeostatic adaptations. The identification of SynI as the main effector protein of the Cdk5-linked pathway that impinges on SV dynamics will contribute to a more complete description of the homeostatic regulatory mechanisms governing SV trafficking and synaptic strength.
Footnotes
This study was supported by research grants from the Italian Ministry of University and Research (Research Programs of Relevant National Interest; F.B. and F.V.), the Italian Ministry of Health Progetto Giovani (A.F.), the San Paolo Company, Torino (A.F., F.B., and F.V.), Citizens United for Research in Epilepsy Innovator Award (F.B.), Israel Science Foundation Grant 1427/12 (D.G.), the Pierfranco and Luisa Mariani Foundation, the Jerome Lejeune Foundation (A.F.), and Telethon Italy Grant GGP13033 (F.B. and F.V.). We thank Drs. Hung-Teh Kao (Brown University, Providence, RI) and Paul Greengard (Rockefeller University, New York, NY) for providing us with the SynI KO mice, Drs. Luigi Naldini and Mario Amendola (Telethon Institute for Gene Therapy, Milan, Italy) for invaluable help in lentiviral production protocols, and Drs. Marina Nanni, Roberta Pelizzoli, and Tatiana Tkatch (Italian Institute of Technology, Genoa, Italy) for valuable help with cell cultures and lentivirus production.
The authors declare no conflicts of interest.
- Correspondence should be addressed to Dr. Anna Fassio, Department of Experimental Medicine, Section of Human Physiology, University of Genoa, Viale Benedetto XV, 3, I-16132 Genoa, Italy. anna.fassio{at}unige.it





{kind=link}
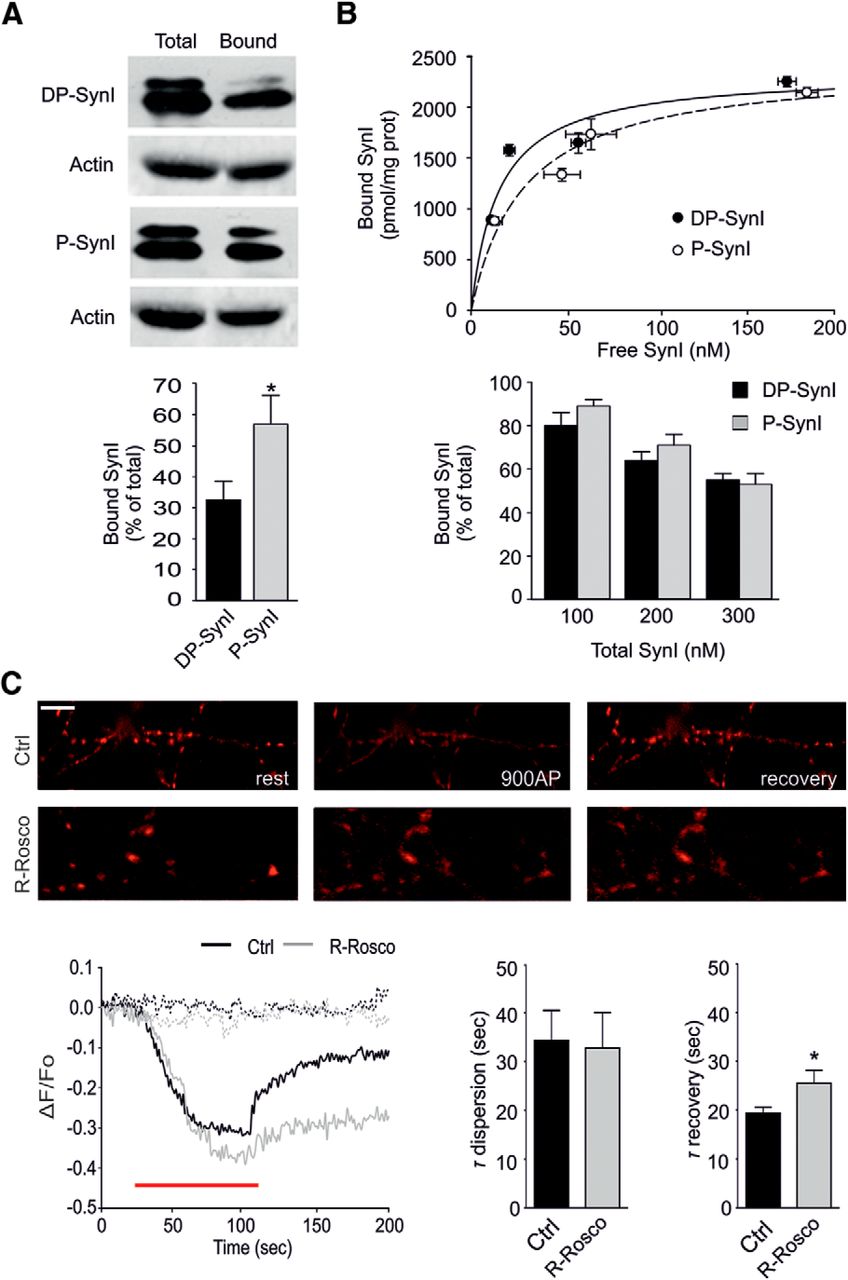{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}