

Abstract
The glutamate transporter excitatory amino acid transporter 3 (EAAT3) is polarized to the apical surface in epithelial cells and localized to the dendritic compartment in hippocampal neurons, where it is clustered adjacent to postsynaptic sites. In this study, we analyzed the sequences in EAAT3 that are responsible for its polarized localization in Madin–Darby canine kidney (MDCK) cells and neurons. Confocal microscopy and cell surface biotinylation assays demonstrated that deletion of the EAAT3 C terminus or replacement of the C terminus of EAAT3 with the analogous region in EAAT1 eliminated apical localization in MDCK cells. The C terminus of EAAT3 was sufficient to redirect the basolateral-preferring EAAT1 and the nonpolarized EAAT2 to the apical surface. Using alanine substitution mutants, we identified a short peptide motif in the cytoplasmic C-terminal region of EAAT3 that directs its apical localization in MDCK cells. Mutation of this sequence also impairs dendritic targeting of EAAT3 in hippocampal neurons but does not interfere with the clustering of EAAT3 on dendritic spines and filopodia. These data provide the first evidence that an identical cytoplasmic motif can direct apical targeting in epithelia and somatodendritic targeting in neurons. Moreover, our results demonstrate that the two fundamental features of the localization of EAAT3 in neurons, its restriction to the somatodendritic domain and its clustering near postsynaptic sites, are mediated by distinct molecular mechanisms.
- dendritic sorting signal
- apical sorting signal
- glutamate transporter
- polarized trafficking
- EAAT3
- excitatory amino acid carrier
The actions of glutamate, the major excitatory neurotransmitter in the mammalian CNS, are terminated by sodium-dependent glutamate transporters located on the plasma membrane of neurons and glia. Glutamate transporters constitute a distinct gene family of which five human glutamate transporters [excitatory amino acid transporters 1–5 (EAAT1–5)] have been identified (Arriza et al., 1994, 1997; Fairman et al., 1995). The rodent homologues of EAAT1 (glutamate/aspartate transporter) and EAAT2 (glutamate transporter-1) are primarily expressed in glia (Rothstein et al., 1994; Lehre et al., 1995), whereas EAAT3 (EAAC1) and EAAT4 are expressed predominantly in neurons (Furuta et al., 1997; Dehnes et al., 1998). EAAT3 is widely expressed in the CNS as well as in epithelial cells in the kidney and gut. Surprisingly, the carrier is not found on the axons or presynaptic terminals, which release glutamate. Instead, EAAT3 and EAAT4 are targeted to the somatodendritic domain and concentrated near postsynaptic sites (Coco et al., 1997; Furuta et al., 1997; Conti et al., 1998; Dehnes et al., 1998; He et al., 2000) where they are thought to regulate synaptic signaling by limiting the diffusion of glutamate to extrasynaptic receptors (Brasnjo and Otis, 2001).
The Madin–Darby canine kidney (MDCK) cell line has been used extensively as a simple system to study the sorting of polarized membrane proteins (Scannevin et al., 1996; Kryl et al., 1999; Poyatos et al., 2000; Martinez-Maza et al., 2001; McCarthy et al., 2001). In MDCK cells, apical and basolateral proteins are segregated from each other as they exit the trans-Golgi network and then subsequently delivered to the appropriate surface. In the case of basolateral proteins, sorting depends on short motifs present in the cytoplasmic tails of the proteins. Apical sorting is less well understood. Some proposed apical sorting mechanisms include glycosylphosphatidylinositol linkage, raft association, and glycosylation (Winckler and Mellman, 1999). More recently, evidence has emerged that cytoplasmic regions of proteins may also contain information important for apical sorting. For instance, a 39-amino acid sequence in the cytoplasmic C terminus of rhodopsin directs its apical localization in MDCK cells (Chuang and Sung, 1998). Similarly, a 32-amino acid sequence at the C terminus of GABA transporter 3 (GAT-3) confers apical localization (Muth et al., 1998). This region contains a motif that may bind to postsynaptic density-95/Drosophiladisc large protein/zona occludens protein 1 domains (Muth et al., 1998), but it is not known whether this mediates its targeting. Discrete, well defined sorting motifs have yet to be defined in these, or other, apically targeted proteins.
Many proteins that are dendritically localized in neurons are concentrated on the basolateral surface of MDCK cells, and in some cases, the same motifs mediate sorting in both cell types. The endogenous localization of EAAT3 represents an exception to this idea: EAAT3 is present at the apical membrane in kidney cells and on the soma and dendrites of neurons (Coco et al., 1997; Shayakul et al., 1997). If the sorting of EAAT3 depends on its association with lipid rafts or its glycosylation, as observed for some apical proteins, this would be the first instance in which such signals mediated dendritic targeting in neurons. It would be equally unusual to find a cytoplasmic, dendritic targeting motif that mediated apical sorting in MDCK cells. Alternatively, it may be that different motifs mediate sorting in the two cell types. To elucidate the motifs involved in the sorting of EAAT3 and to resolve this paradox, we have investigated the sorting of EAAT3 in MDCK cells and in hippocampal neurons. By expressing a series of mutated proteins, we have identified a novel 11-amino acid motif in the cytoplasmic tail of EAAT3 that mediates its apical sorting in MDCK cells and its somatodendritic localization in hippocampal neurons.
MATERIALS AND METHODS
Cell culture
MDCK cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 89 U/ml penicillin, and 89 μg/ml streptomycin. Media for stable MDCK cell lines were further supplemented with 500 μg/ml geneticin (G418) (Invitrogen). The cells were plated at high density onto 12 or 24 mm Transwell-COL filters (Costar, Cambridge, MA) and grown to confluency before use.
Primary hippocampal cultures were prepared from embryonic day 18 rats as described previously (Banker and Goslin, 1998). Neurons were plated at 300,000 cells per 6 cm dish.
Mutagenesis
Various PCR-based mutagenesis protocols were used to create the mutant constructs. All the mutant constructs were sequenced to confirm that no unintended mutations were introduced. All constructs were subcloned into the pEGFPC vector (Clontech, Palo Alto, CA).
To construct the EAAT1/EAAT3 C-terminal chimeras (E1CT3 and E3CT1), silent mutations at E501L502 and L470 of EAAT1 and EAAT3 cDNAs, respectively, were introduced by using the three-primer PCR mutagenesis technique (Seal and Amara, 1998) to create the SacI restriction site. The C termini of EAAT1 and EAAT3 were exchanged at the SacI restriction site to produce the resultant E1CT3 and E3CT1 constructs. EAAT3 C-terminal deletion constructs were made with the last amino acid of the mutant being the following: E469,P484, S497, and V510 for E3Δ470–524, E3Δ485–524, E3Δ498–524, and E3Δ511–524, respectively. The constructs containing C-terminal regions of EAAT3 fused to EAAT2 were created by attaching EAAT3 C-terminal cDNA fragments to full-length EAAT2 cDNA separated by a linker (containing the sequence GCC GGA TCT GCC). The amino acid residues of EAAT3 contained in the EAAT3 C-terminal fragments are denoted in parentheses. For instance, E2–3(469–524) contains the amino acids E469–F524 of EAAT3 fused to full-length EAAT2. The QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) was used to create the triple or double alanine substitution mutants. The template was green fluorescent protein (GFP)-E2–3(498–524), and primers were created with the least nucleotide changes to the alanine codon.
Transfection
Stable transfection into MDCK cells. MDCK cells (at 50–80% confluency) in each well of a six-well plate were transfected with a mixture containing 2 μg of DNA and 40 μg of LipofectAMINE (Invitrogen) in serum-free DMEM. After 5 hr, 10% fetal bovine serum, 89 U/ml penicillin, and 89 μg/ml streptomycin were added to the mixture. Twenty-four hours after the start of transfection, the DNA-LipofectAMINE-DMEM mixture was replaced with fresh DMEM containing serum, penicillin, streptomycin, and 500 μg/ml G418 (G418 media). Forty-eight to 72 hr after the start of transfection, three wells containing the greatest number of fluorescent cells were replated at low density and placed in G418 media for 2–3 weeks to select for stably transfected cell lines. G418-resistant cell lines were screened for detectable GFP fluorescence, and cell lines with higher GFP fluorescence levels were chosen for use in this study. For each construct, results were verified with at least two independently derived stable cell lines.
Transient transfection into MDCK cells. For each construct to be transfected, 1.2 μg of DNA was combined with 2.4 μg of LipofectAMINE 2000 (Invitrogen) in serum-free DMEM and incubated at room temperature for 30 min. The mixture was then combined with MDCK cells in media and plated at high density onto a 12 mm Transwell-COL filter. The bottom of the transwell filter was filled with media. Twenty-four hours after the start of transfection, the wells of transfected cells were replaced with fresh media. The cells were allowed to grow to confluency for a further 48–72 hr before use.
Transient transfection of dissociated hippocampal neuronal cultures. Constructs were introduced into cultures at 8–10 din vitro using Effectene-mediated transfection (Qiagen, Valencia, CA). Briefly, for each construct, 1 μg of DNA was mixed with 8 μl of enhancer and then 10 μl of Effectene according to the manufacturer's instructions. After addition of the DNA–Effectene complexes to the media, the cells were incubated for 2 or 9 d at 37°C. The cells were then fixed in 4% paraformaldehyde and 4% sucrose and permeabilized in 0.25% Triton X-100, and coverslips were mounted on slides using elvanol. GFP fluorescence was used to measure cell surface distribution. Expression of the constructs for a minimum of 2 d allowed for accumulation at the cell surface and minimized the contribution of intracellular fluorescence. Under these conditions, the GFP signal appears enriched at the surface when examined at high magnification. Moreover, expression of the EAAT constructs had reached a steady state at these times.
Immunohistochemistry and confocal microscopic imaging
All steps were performed at room temperature. The cells were initially rinsed twice with PBS (containing 100 μmCaCl2 and 1 mmMgCl2) before they were fixed with 4% paraformaldehyde (with 50 mm HEPES in PBS, pH 7.4) for 20 min. Then the cells were rinsed twice with PBS and incubated with a blocking and permeabilization solution containing 5% horse serum, 1% BSA, and 0.2% Triton X-100 in PBS for 30 min. Subsequently, staining for E-cadherin was performed with the monoclonal antibody anti-uvomorulin clone DECMA-1 (Sigma, St. Louis, MO). The cells were incubated in the primary antibody for 2–3 hr. The cells were washed three times for 3 min with PBS and probed with rhodamine red-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA). The cells were washed three times for 3 min with PBS before the filter containing the cells was excised from its support and mounted onto a slide with ProLong antifade reagent (Molecular Probes, Eugene, OR). Images were constructed by measuring the fluorescence signal using confocal microscopy (MRC 1024 system; Bio-Rad, Hercules, CA) with excitation lines of 488 and 568 nm for the GFP and rhodamine signals, respectively. The z-series was collected by initially focusing on the middle section of the cells (position 0) and scanning 8 μm above to 8 μm below this plane at 0.1 μm intervals.
Cell surface biotinylation assay
Cell surface expression of the EAAT constructs was assayed with modifications of the method described previously (Daniels and Amara, 1998). All steps before the Western blot analysis were performed at 4°C unless otherwise specified. Briefly, the cells were washed once quickly with room temperature PBS and then three times for 10 min with cold PBS. The cells were then incubated with 2 mg/ml sulfosuccinimidyl 2-(biotinamido)ethyl-1,3-dithiopropionate (sulfo-NHS-SS-biotin) (Pierce, Rockford, IL) in biotinylation buffer (in mm: 2 CaCl2, 150 NaCl, and 10 triethanolamine, pH 7.5) for 40 min placed either on the top or bottom of the transwell filter to assay for apical or basolateral expression of the transporters, respectively. The side not incubated with sulfo-NHS-SS biotin was incubated with biotinylation buffer only. The reaction was quenched by incubation with 100 mm glycine in PBS for 20 min. The cells were washed with PBS and then lysed with lysis buffer [1% Triton X-100, 150 mm NaCl, 5 mm EDTA, and 50 mm Tris, pH 7.5, containing a protease inhibitor mixture (1×; Roche Molecular Biochemicals, Indianapolis, IN)], and the cell lysate was collected after centrifugation at 14,000 × g for 10 min. The cell lysate was incubated with Ultralink immobilized NeutrAvidin beads (Pierce) for 2–3 hr. The beads were separated from the supernatant by centrifugation at 5000 × g for 15 min. The beads contained cell surface proteins, whereas the supernatant contained cytosolic proteins. The beads were washed three times with lysis buffer, twice with high-salt wash buffer (0.1% Triton X-100, 500 mm NaCl, 5 mm EDTA, and 50 mm Tris, pH 7.5), and once with no-salt wash buffer (50 mm Tris, pH 7.5). Subsequently, the proteins on the beads were released by incubation with SDS loading buffer containing 200 mm DTT. The proteins were separated on 8% polyacrylamide gels and then transferred onto Immobilon-P blots (Millipore, Bedford, MA). The blots were probed for the expression of EAATs with either polyclonal antibodies to the C terminus of the EAATs or a monoclonal antibody to GFP (JL-8; Clontech) and visualized with horseradish peroxidase-conjugated secondary antibody and chemiluminescent reagent (PerkinElmer, Beverly, MA). Blots were exposed to film to obtain nonsaturated exposures, which could be used for densitometric quantitation of chemiluminescent signals. Films were scanned using a Duoscan F40 (Agfa, Ridgefield Park, NJ), and band intensities in each lane (same area size in each lane) were background-subtracted and quantitated by using the software program TINA (Fuji Medical Systems, Stamford, CT). Statistical significance was determined with the Newman–Keuls multiple-comparisons test for independent groups using WINKS evaluation software (Texasoft, Cedar Hill, TX).
Imaging and quantitation of GFP fluorescence in hippocampal neurons
Neurons were chosen for analysis only if they were sufficiently separated from other transfected cells to ensure that all labeled processes arose from the cell pictured. Images were acquired on a Leica (Nussloch, Germany) DMIRBE microscope linked to a Princeton Instruments Micromax chilled CCD camera controlled by Metamorph imaging software (Universal Imaging, Downingtown, PA). For cells at 11 d in vitro, low-magnification images were acquired using a 16×, 0.5 numerical aperture (NA) Plan Fluotar objective, and high-magnification images for quantification were acquired using a 63×, 1.32 NA Plan Apo objective. Cells at 19 d in vitro were imaged on the same microscope at 63× by acquiring a through-focal series of images at 0.2 μm intervals (∼30 planes). Stacks of these images were subject to constrained-iterative deconvolution (15 iterations) using Deltavision SoftWoRx 2.5 software (Applied Precision,Issaquah, WA) based on the point spread function determined for our microscope. High-magnification images of 19 d in vitrocells are displayed as single planes of the resulting deconvolved stacks.
Images for quantification were corrected for background fluorescence and uneven field illumination. The average fluorescence intensity in axons and dendrites was obtained by drawing a series of ∼10 one-pixel-wide lines down the center of processes on corrected high-magnification digital images. For each cell, the fluorescence for each line in the axons and dendrites was averaged to obtain the final values for axon and dendrite intensity. These averages were used to calculate the axon/dendrite ratio. Theoretically, one would expect a uniformly distributed protein to have an axon/dendrite ratio of 1. In practice, the axon/dendrite ratio for nonpolarized proteins averages between 0.5 and 1. This is probably because of a greater contribution of out-of-focus fluorescence in the thicker dendrites compared with the relatively thin axons. Statistical significance of differences between groups was determined by performing the Newman–Keuls multiple-comparisons test for independent groups using WINKS evaluation software.
RESULTS
Localization of wild-type EAATs in MDCK cells
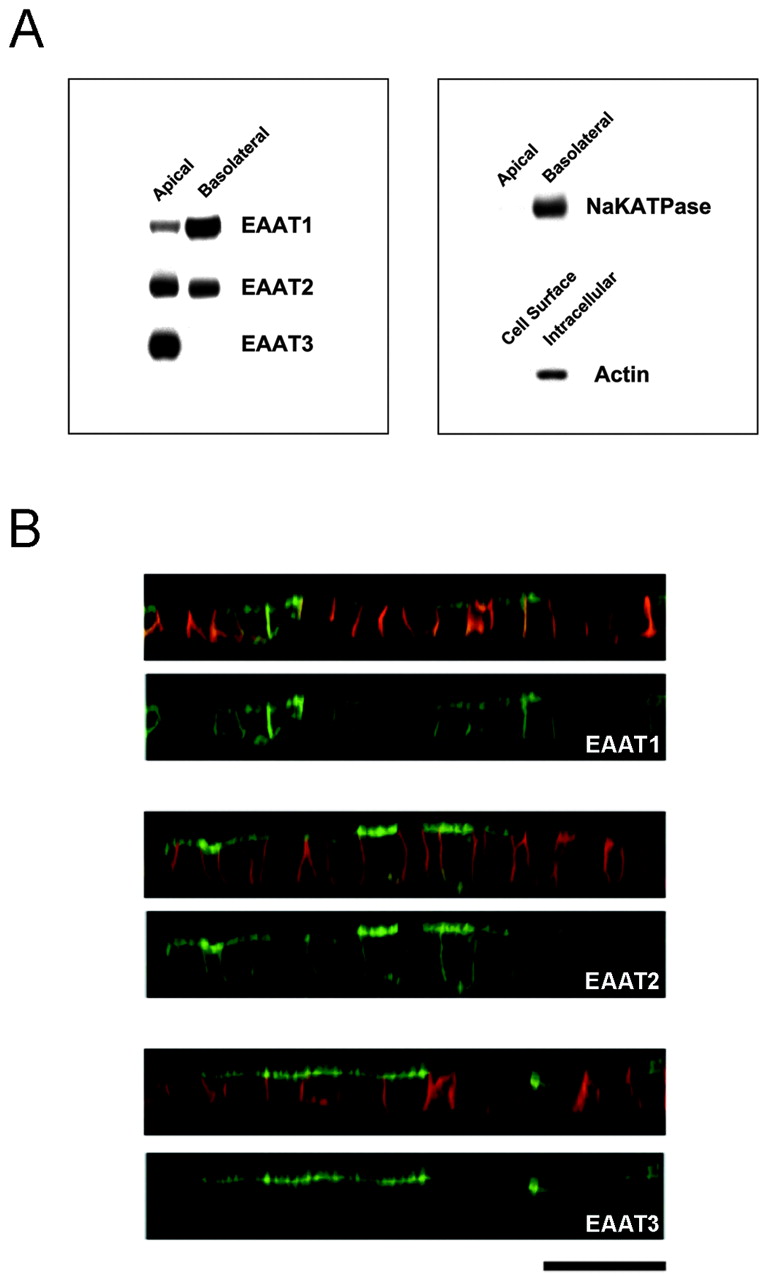
We stably expressed EAAT1–5 tagged on the N terminus with GFP in polarized MDCK cells and examined their localization using confocal microscopy and cell surface biotinylation. EAAT3 was extremely polarized to the apical surface, whereas EAAT1 and EAAT2 were expressed at both the apical and basolateral surfaces (Figs. 1,2B) with EAAT1 more basolateral-preferring than EAAT2 (22 ± 8% apical versus 54 ± 1% apical; Table 1). It is important to note that our stably transfected, GFP-tagged EAAT3 retained the normal apical localization of endogenous EAAT3 in kidney epithelial cells (Shayakul et al., 1997). EAAT4 and EAAT5 were expressed at lower levels than the other EAATs, but they appeared to be present at both cell surfaces (data not shown). Therefore, EAAT3 is the only glutamate transporter that is completely polarized in MDCK cells.

Localization of wild-type EAAT1–3 in MDCK cells. MDCK cells were stably transfected with EAAT1–3 that was tagged with GFP at the N terminus and grown on transwell filters. A, Representative Western blots from a cell surface biotinylation assay. This assay assessed the stable expression level of each EAAT at each cell surface by the application of biotin at the apical or basolateral surfaces. There is no signal when biotin was not included in the assay. The control blots show that Na+/K+-ATPase is detected at the basolateral but not the apical cell surface fraction, and actin is only detected in the intracellular fraction. B, Representative confocal images of EAAT1–3 in a vertical section (z-series) illustrating the localization of GFP-EAAT (green) and a basolateral marker, E-cadherin (red). The top panel of each construct is a composite image illustrating both GFP and basolateral marker signals, and the bottom panel contains only the GFP signal. In the vertical section, a fluorescence signal at the apical surface appears as a horizontal line at the topof the cell, whereas a signal at the basolateral surface appears asvertical lines at the sides of the cell. Occasionally, there is fluorescence attributable to the underlying filter, which appears as a horizontal line at thebases of all the cells. This fluorescence signal is not scored as basolateral localization. All EAATs are expressed at both surfaces, except EAAT3, which is restricted to the apical surface. Eachblot and image is representative of at least two additional experiments. Scale bar, 20 μm.

Schematic representation and localization of the EAAT3 mutants. A, Topology of EAAT1 (Seal et al., 2000) and regions that are exchanged with EAAT3. The C terminus (orange), the large second extracellular loop (purple), and the N-terminal region (blue) are shown. Numbers denote transmembrane domains. OUT, Extracellular; IN, intracellular. B, Schematic diagram of the EAAT1/3 C-terminal chimeras, EAAT3 deletions, and EAAT3 C terminus attached to the end of full-length EAAT2. Regions containing sequences from EAAT1, EAAT2, and EAAT3 are denoted by yellow,red, and green, respectively. The localization of the stably transfected mutants was determined by both confocal microscopic imaging and a cell surface biotinylation assay. The presence or absence of a signal at a domain is denoted by + or −, respectively.
Quantitative analysis of the cell surface expression of stably transfected EAAT constructs in MDCK cells
The C-terminal motif directs apical localization of EAAT3 in MDCK cells
To identify the region of EAAT3 important for its apical localization, we made chimeras between EAAT1 and EAAT3, exchanging the three large less conserved regions: the N terminus, the large second extracellular loop (which contains consensus N-linked glycosylation sites), and the C terminus (Fig. 2A). Replacing the C terminus of EAAT3 with the C terminus of EAAT1 (E3CT1) abolished apical targeting (Figs. 2B, 3; Table 1). Conversely, replacing the C terminus of EAAT1 with the analogous C-terminal region of EAAT3 (E1CT3) conferred specific apical localization, changing from 22 ± 8% apical localization of wild-type EAAT1 to 97 ± 1% apical localization of E1CT3 (Table 1). Replacing the large second extracellular loop or the N-terminal region of EAAT3 with the corresponding regions of EAAT1 did not affect the polarization of EAAT3 (although the chimera including the N-terminal region of EAAT1 showed little cell surface expression). Replacing these domains of EAAT1 with the corresponding regions of EAAT3 did not cause EAAT1 to become apically localized. These substitutions indicated that the cytoplasmic C terminus of EAAT3 contains information important for its apical localization.
To further test the role of the EAAT3 C terminus, we constructed addition and deletion mutants. We first tested whether this small region of EAAT3 could redirect a nonpolarized protein to the apical domain. The addition of the C terminus of EAAT3 to full-length EAAT2 [E2–3(469–524)] changed its localization from nonpolarized to apically polarized as judged by cell surface biotinylation assays (Fig.3A, Table 1) and confocal microscopy (Fig. 3B). The C terminus of EAAT3 similarly redirected the taurine transporter to the apical surface (data not shown). Conversely, deleting the C terminus of EAAT3 (E3Δ470–524) resulted in a nonpolarized localization (54 ± 3% apical); this mutant could be detected at both the basolateral and apical surfaces (Figs. 2B, 3). Together, the results from stably transfected chimeric, addition, and deletion mutants in MDCK cells demonstrate that the C terminus of EAAT3 is necessary for its apical sorting and sufficient to confer apical localization on a nonpolarized protein.

The cytoplasmic C terminus of EAAT3 is important in its apical localization. Representative Western blot analysis (A) and confocal vertical (z-series) images (B) of MDCK cells stably transfected with EAAT1–3 C-terminal chimeras or truncation mutant constructs are shown. The presence of the C terminus of EAAT3 in the mutant proteins confers stable expression predominantly at the apical surface, whereas its absence results in a more nonpolarized localization. Results are representative of at least two additional experiments. Scale bar, 20 μm.
Next, we sought to narrow down the region within the C terminus of EAAT3 that is important for its apical localization. First, we made successively smaller truncations of the C terminus of EAAT3. All the truncated EAAT3 mutants have reduced apical localization (Fig.2B, Table 1). Subsequent analyses (see next paragraph) showed that all of the truncations eliminated all or part of the sequence required for apical sorting. We also used a complementary approach of adding successively smaller regions of the EAAT3 C terminus to the end of full-length EAAT2. This strategy revealed the importance of the region between the last 27 and 14 amino acids of EAAT3. The mutant E2–3(498–524) showed an apical localization, whereas E2–3(511–524) had a significantly less polarized distribution (Figs.2B, 4; Table 1). Similar results were found when the same regions of the C terminus of EAAT3 were fused to the full-length taurine transporter (data not shown). These results show that amino acids between D498 and D511of the C terminus of EAAT3 contain an apical localization signal.

A 14-amino acid sequence in the C terminus of EAAT3 contains the signal for apical localization. Representative Western blots (A) and confocal images (B) of MDCK cells stably transfected with constructs containing full-length EAAT2 fused to various regions of the EAAT3 C terminus are shown. The mutant proteins containing at least the most distal 27-amino acid sequence of the EAAT3 C terminus attached to EAAT2 [E2–3(498–524)] shows an apical localization, whereas the mutant with a smaller distal region (14-amino acid sequence) of the C terminus of EAAT3 [E2–3(511–524)] has a more nonpolarized localization. These results suggest that the region in EAAT3 between 27 and 14 amino acids from the C-terminal end contains the sorting motif. Results are representative of at least two additional experiments. Scale bar, 20 μm.
We used two additional, complementary methods to pinpoint residues within the 14 amino acids of the EAAT3 C terminus responsible for apical targeting. For these experiments, constructs were expressed transiently, and localization was assessed by confocal microscopy. First, we examined constructs of EAAT2 fused to short segments from the C terminus of EAAT3 (Fig. 5A; see supplemental data on-line at www.jneurosci.org). Constructs containing residues D498–V510were apically polarized, whereas constructs containing residues upstream of V504 were not. This indicates that residues N505–V510(NGGFAV) are crucial for apical localization. Second, we made sequential triple or double alanine substitutions in the EAAT3 region of the mutant consisting of EAAT2 attached to amino acids D498–F524 of EAAT3 [E2–3(498–524)] (Fig. 5). The substitution with three alanine residues between amino acids V504 and A509 disrupted apical localization; substitution of amino acids K501–Y503had a smaller effect (40% of transfected cells exhibited a polarized distribution). Alanine substitutions N-terminal to amino acid K501 or C-terminal to amino acid A509 did not disrupt apical localization. Next, we examined double alanine substitutions between amino acids V504 and D511. Substitution of the residues V504N505 and F508A509resulted in loss of apical polarity. Substitution of residues V510D511slightly disrupted the polarized distribution (70% of the transfected cells still showed apical localization). Replacing residues G506G507 with alanine did not alter apical localization of the mutant protein, but this may be because alanine and glycine are similar in structure. Substituting bulkier amino acid residues at positions 506 and 507 may have an impact on the polarity of EAAT3. The substitution of A509 with serine (the rat homolog of EAAT3 has a serine at this position) did not alter the apical localization of the mutant construct (data not shown). Thus, the residues V504N505 and F508A509within the motif are critical in apical sorting, whereas the neighboring residues may influence the specific localization motif. The apical localization sequence motif is KSYVNGGFAVD (amino acids K501–D511).

Delineation of the residues in the sorting motif for apical localization of EAAT3. A, Representative confocal images of MDCK cells transiently transfected with constructs that contained full-length EAAT2 fused to either short regions of the EAAT3 C terminus or the most distal 27-amino acid sequence of EAAT3 [E2–3(498–524)] with triple or double alanine substitution mutations. Both E2–3(498–510) andE2–3(485–497) contained a 12-amino acid sequence from the C terminus of EAAT3, but only E2–3(498–510) was apically localized. B, Sequences of the constructs containing sequential triple or double alanine substitution of E2–3(498–524) and localization of the mutants. Regions of EAAT2 and 3 are represented in red and green, respectively, and the residues that are substituted with alanines are denoted in black. Substitution of residues with alanine of the following sequence disrupts the apical localization of the parent construct: KSYVNGGFAVD (the critical residues are gray). Results are representative of at least three additional experiments in duplicate. Scale bar, 20 μm.
Quantitative analysis of the polarity of EAAT constructs in hippocampal neurons
The C-terminal sorting motif targets EAAT3 to the somatodendritic region in neurons
Next, we tested whether the apical targeting motif in EAAT3 also directs the localization of EAAT3 in neurons. We transfected both wild-type and mutated EAATs tagged with GFP or yellow fluorescent protein (YFP) into polarized neurons in primary hippocampal cultures. We quantified the average fluorescence in axons and dendrites for each cell and used the ratio of axon to dendrite fluorescence as a measure of polarity. The wild-type EAAT3-YFP was extremely polarized to dendrites (Figs. 6A,7B), with an average axon/dendrite ratio of 0.06 ± 0.02 (Table 2), a value comparable with that of other dendritically polarized proteins such as transferrin and low-density lipoprotein receptors (G. Banker, unpublished observations). In older neurons (19 d in vitro), which are highly innervated, EAAT3 was present in clusters on the dendritic surface and on spines and filopodia (Fig. 7C). Some of these clusters lie adjacent to synaptophysin-positive presynaptic specializations (data not shown), consistent with the postsynaptic localization of EAAT3 that has been reported previously. Thus, in all respects, transfected GFP- or YFP-tagged EAAT3 showed the same polarized distribution in neurons as for endogenous EAAT3 (Coco et al., 1997). When live cells were imaged, transport of EAAT3 carriers was readily observed within the dendrites, but no carriers were visible in the axon (data not shown). Thus in neurons, the polarization of EAAT3 in the dendritic membrane reflects its selective delivery to dendrites, not its selective retention in the dendritic membrane.

Distribution of wild-type and mutant EAATs in polarized hippocampal neurons. Hippocampal cultures were transiently transfected with GFP- or YFP-tagged EAATs, and expression of the constructs was assessed in 11-d-old hippocampal neurons. The EAAT3-YFP signal (A) is completely restricted to dendrites, whereas cotransfected soluble cyan fluorescent protein (CFP; B) fills the entire cell, including its axon. Mutant E3(504–509AAAAAA) with a six-alanine replacement in the apical sorting domain is present in both axons and dendrites (C), as is EAAT2(D). Arrows indicate axons;arrowheads indicate dendrites. Scale bar, 100 μm.

Higher-magnification images of wild-type and mutant EAATs in polarized hippocampal neurons. EAAT3-YFP is enriched on the surface of dendrites but absent from axons (B). Coexpressed soluble cyan fluorescent protein (CFP) fills the entire volume of the axon and dendrites (A). The mutant EAAT3 [E3(504–509AAAAAA)] and EAAT2 are present in both dendrites and axons (D, E). In older neurons (transfected at 10 d in vitro and imaged 9 d later), both wild-type and mutant EAAT3 are frequently present in clusters on spines and filopodia in the dendrites, and mutant EAAT3 is present in axons. Dendrites (arrowheads) are thicker, taper as they extend away from the cell body, and have filopodia and developing spines present all along them. Axons (arrows) are thinner, maintain a relatively uniform diameter, and lack spines. Scale bars: A, B, D, E, 10 μm; C, E, F, 2 μm.
In contrast, two EAAT3 C-terminal truncation mutants lacking the apical-targeting motif (E3Δ470–524 and E3Δ485–524) were significantly less polarized (average axon/dendrite ratios, 0.28 ± 0.03 and 0.29 ± 0.05, respectively). Moreover, the substitution of residues 504–509 within the sorting motif of EAAT3 with alanines [E3(504–509AAAAAA)] disrupted somatodendritic polarity to the same extent as deleting the entire cytoplasmic tail (average axon/dendrite ratio, 0.26 ± 0.02). In all cases, mutant EAAT3s extended throughout the entire axonal arbor, a result not observed with the wild-type protein. However, disruption of the sorting motif in EAAT3 did not result in a complete loss of polarity (compare with EAAT2, whose axon/dendrite ratio was 0.68). Therefore, other regions of EAAT3 may also contain dendritic targeting information. Although such mutations disrupted the polarization of EAAT3, they did not prevent its clustering on dendritic spines (Fig. 7F). These results suggest that the apical sorting signal is not required for the synaptic clustering of EAAT3. Rather, these results show that the apical sorting motif in EAAT3 is responsible for its dendritic targeting in neurons.
We next investigated whether adding the EAAT3 sorting motif to a uniformly distributed protein was sufficient to cause its redistribution to the dendrites. When EAAT2 was expressed in hippocampal neurons, it was approximately equally distributed in axons and dendrites, exhibiting an average axon/dendrite ratio of 0.68 ± 0.09 (Figs. 6D, 7D; Table 2), a value comparable with that found for nonpolarized proteins such as NgCAM-related cell adhesion molecule and CD8 (B. Sampo and G. Banker, unpublished observations). To test whether the EAAT3 sorting motif could redirect EAAT2 to the dendrite, we fused the last 27 amino acids of EAAT3 (containing the sorting motif) to full-length EAAT2 [E2–3(498–524)]. E2–3(498–524) was significantly more dendritic than wild-type EAAT2 (axon/dendrite ratio, 0.31 ± 0.04) but was less polarized than wild-type EAAT3. A similar mutant, which differed only in that alanines were substituted for residues 504–506 within the sorting motif [E2–3(498–524:504–506AAA)], was as nonpolarized as wild-type EAAT2 (axon/dendrite ratio, 0.82 ± 0.10). This indicates that the EAAT3 sorting motif has some ability to redirect a uniform protein in neurons but is not sufficient to completely redirect EAAT2, as it is in MDCK cells. It may be that EAAT2 itself contains its own sorting information that influences its localization in neurons and thus competes with the EAAT3 motif.
Together, our results clearly show that EAAT3 contains a sorting motif in its cytoplasmic tail. This motif, KSYVNGGFAVD, directs the apical localization of EAAT3 in MDCK cells and is required for its dendritic localization in hippocampal neurons.
DISCUSSION
In this study, we identified a novel sorting motif in the cytoplasmic C-terminal region of EAAT3 (KSYVNGGFAVD) that directs its specific localization to the apical domain of MDCK cells and the somatodendritic region of hippocampal neurons. This is the first well defined motif known to be required for apical sorting in MDCK cells and dendritic sorting in hippocampal neurons and capable of redirecting the localization of nonpolarized proteins to these domains. A database search did not reveal any other known membrane proteins that contain the motif VNGGFA, but other epithelial sorting motifs shared by different proteins have proved difficult to identify solely on the basis of the amino acid sequence.
The EAAT3 sorting motif is one of several different motifs found in the cytoplasmic tails of neuronal proteins that target them to dendrites (Jareb and Banker, 1998; Poyatos et al., 2000; Ruberti and Dotti, 2000). It is, however, one of the first instances of a defined cytoplasmic motif that mediates apical sorting in MDCK cells. Traditionally, it has been thought that basolateral and dendritic sorting shared many common features, as did apical and axonal sorting (Dotti and Simons, 1990). Over the past few years, it has become clear that apical proteins are not always segregated to the axonal domain in neurons. For example, other amino acid transporters, such as GAT-3 and glycine transporter 2, are sorted to the apical domain in MDCK cells but are uniformly distributed in neurons (Ahn et al., 1996; Poyatos et al., 2000). Our results, however, represent the first instance in which a well- defined dendritic targeting motif mediates apical targeting in epithelia.
The EAAT3 sorting motif likely directs dendritic and apical targeting by interacting with other proteins involved in targeting. Proteins that bind to the EAAT3 sorting motif have not yet been identified. Recently, the protein glutamate transporter EAAC1-associated protein (GTRAP)3–18 has been shown to interact with the C terminus of the rat homolog of EAAT3 (Lin et al., 2001). However, increasing the expression of GTRAP3–18 in cells decreases the affinity of the transporter for its substrate but does not alter the cell surface expression of the transporter. Thus, it is not likely that GTRAP3–18 is involved in the sorting of EAAT3 to the appropriate cell surface.
The mechanism by which the EAAT3 sorting motif may direct apical and dendritic targeting could involve any of the steps in membrane protein targeting. It is thought that sorting motifs, particularly those that reside in the cytoplasmic domain, are recognized by cytoplasmic adaptor proteins (APs). These adaptor proteins concentrate the membrane proteins within discrete domains and link them with coat proteins. The coat proteins then induce vesicle budding, hence leading to the sorting of distinct membrane proteins into unique transport carriers (Gu et al., 2001). For example, specific subunits of two adaptor complexes, AP-1 and AP-4, have been shown to be necessary for the basolateral sorting of some proteins containing C-terminal sorting signals (Folsch et al., 1999; Simmen et al., 2002). It is possible that neurons and epithelia express a common adaptor that recognizes the EAAT3 motif and sorts EAAT3 protein into a specific population of carrier vesicles but that the fate of these carriers differs in neurons and epithelia. Because there are profound differences in the organization of microtubules in neurons and epithelia, it is possible that differences in the microtubule-based transport of EAAT3 carriers contribute to their apparently divergent localization in neurons and epithelia. In MDCK cells, microtubules are oriented with their minus ends directed toward the apical surface and their plus ends directed toward the basolateral domain (Bacallao et al., 1989). In neurons, microtubules in the axons are oriented with the plus end directed away from the cell body, whereas microtubules in the dendrites have a mixed orientation (Baas et al., 1988). Thus a minus end-directed motor might be expected to move carrier vesicles to the apical domain in MDCK cells and to the dendritic domain in neurons, as proposed by Goldstein and Yang (2000).
There is already evidence for the involvement of the minus end motor dynein in the apical sorting of rhodopsin, an example of an apical protein whose sorting is directed by a cytoplasmic motif (Chuang and Sung, 1998; Tai et al., 2001). The 39-amino acid tail of rhodopsin interacts with the dynein light chain TcTex-1 but not another light chain dynein, RP3 (Tai et al., 1999). RP3 and TcTex-1 can compete for binding to the dynein complex, and the overexpression of RP3 in MDCK cells leads to a decrease in the expression of TcTex-1 as well as the nonpolarized localization of rhodopsin (Tai et al., 2001). Thus, the apical targeting of rhodopsin in MDCK cells is dependent on TcTex-1-mediated dynein function. It would be of interest to determine whether the sorting motif in EAAT3 interacts with components of dynein or other minus end-directed motor proteins, such as kinesin family member C2 (Goldstein and Yang, 2000; Yang et al., 2001).
The clustering of EAAT3-GFP on dendritic spines and filopodia also deserves comment. In the brain, EAAT3 has a “perisynaptic” location, lying just beyond the active zone where postsynaptic glutamate receptors are most highly concentrated. This location suggests that neuronal glutamate transporters are poised to regulate the activity of extrasynaptic glutamate receptors by controlling the diffusion of glutamate away from the synapse, as has been demonstrated recently at parallel fiber synapses in the cerebellum (Brasnjo and Otis, 2001). These results indicate that the correct localization of glutamate transporters is critical to their role in regulating neurotransmission. The EAAT3 clusters we observed, which might be difficult to detect by immunostaining of intact tissue, bear a striking resemblance to postsynaptic receptor clusters seen on cultured hippocampal neurons (Allison et al., 2000; Naisbitt et al., 2000). The clustering of EAAT3 is not affected by deletion or mutation of the dendritic targeting signal, suggesting that it is mediated by a different molecular mechanism. It would be of interest to know whether interaction with membrane scaffolding proteins contributes to the localization of postsynaptic transporters such as EAAT3.
Footnotes
This work was supported by Howard Hughes Medical Institute and National Institutes of Health Grants NS 33273 (S.G.A.), NS17112 (G.B.), and MH 66179 (G.B.). We thank Dr. D. M. Fass for critical review of this manuscript and B. Smoody for preparing the neuronal cell cultures.
Correspondence should be addressed to Dr. Susan G. Amara, Howard Hughes Medical Institute and Vollum Institute, Oregon Health and Science University, L-474, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: amaras{at}ohsu.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}