

Abstract
Cortical glutamate and substantia nigra dopamine (DA) afferents converge onto the dendritic spines of medium spiny neurons (MSNs) in the striatum where they act to modulate motor and cognitive functions. The released DA spills over from its synapse and is thought to regulate glutamatergic input by acting on distal DA receptors located on corticostriatal axon terminals. By monitoring evoked DA release directly using fast-scan cyclic voltammetry, we report a reciprocal modulation by glutamate spillover on evoked striatal DA release, induced by either glutamate uptake blockade or high-frequency stimulation of corticostriatal tracts. We demonstrate that this modulation is attributable to the activation of group I metabotropic glutamate receptors. Thus, under conditions in which glutamate escapes the confines of its synapse, it can elicit the presynaptic suppression of dopaminergic neurotransmission.
Introduction
High-affinity uptake by excitatory amino acid transporters (EAATs) restricts the diffusion of synaptically released glutamate beyond the confines of its synapse (Diamond and Jahr, 1997). Glutamate spillover, induced by pharmacological inhibition of EAATs, modifies synaptic transmission in the cerebellum and hippocampus (Mitchell and Silver, 2000; Semyanov and Kullmann, 2000; Turecek and Trussell, 2000; Brasnjo and Otis, 2001; Diamond, 2001). Whereas direct synaptic contacts between nigrostriatal dopaminergic and corticostriatal glutamatergic axon terminals have not been observed in the striatum, their boutons closely converge onto the dendritic spines of medium spiny neurons (MSNs) (Nirenberg et al., 1996). Dopamine (DA) spillover from the nigrostriatal synapse not only activates receptors on MSNs several micrometers away (Garris et al., 1994; Gonon, 1997; Gonon et al., 2000) but may also activate presynaptic DA receptors on neighboring glutamatergic corticostriatal afferents (Wang and Pickel, 2002). This presynaptic action has been reported in some studies to regulate the release of glutamate in the striatum (Cepeda et al., 2001; Tang et al., 2001). A role for glutamate spillover and a reciprocal presynaptic modulation of nigrostriatal DA transmission, however, has not been determined.
In the present study, we used EAATs blockers or high-frequency stimulation (HFS) of corticostriatal tracts to enhance the synaptic overflow of glutamate and monitor its effect on evoked DA release in the mouse striatal slice with fast-scan cyclic voltammetry. The results showed that glutamate spillover inhibits evoked DA release by acting on metabotropic glutamate 1 receptors (mGluR1).
Materials and Methods
Animals and striatal slice preparation. Two- to 5-month-old male C57BL/6 mice and mGluR5-deficient mice (B6,129-Gprcletm1Rod) were obtained from The Jackson Laboratory (Bar Harbor, ME). The use of the animals followed the National Institutes of Health guidelines and was approved by the Institutional Animal Care and Use Committee of Columbia University.
Mice were decapitated without anesthesia. Coronal striatal brain slices at 300 μm (bregma +1.54 to +0.62 mm) (Franklin and Paxinos, 1997) were prepared on a vibratome. Sagittal striatal slices, also at 300 μm, were used for the HFS experiments. The slices were allowed to recover for 1.5 hr at room temperature in a holding chamber containing oxygenated artificial CSF (ASCF) and then placed in a recording chamber superfused (1 ml/min) with ACSF (in mm: 125 NaCl, 2.5 KCl, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 0.3 KH2PO4, and 10 glucose) at 36°C.
d,l-Threo-β-hydroxyaspartate (THA), d,l-threo-β-benzyloxyaspartate (TBOA), S-α-methyl-4-carboxyphenylglycine (MCPG), 1-aminoindan-1,5-dicarboxylic acid (AIDA), 2-methyl-6-(phenylethynyl)pyridine (MPEP), (S)-3,5-dihydroxyphenylglycine (DHPG), 2R,4R-4-amino pyrrolidine-2,4-dicarboxylate[(2 R,4 R)-APDC], l-2-amino-4-phosphonobutyric acid (l-AP-4), (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), d(-)-2-amino-5-phosphonopentanoic acid (d-AP-5), CGP52432, 8-cyclopentyl-1,3-dipro-pylxanthine (DPCPX), 4-(2-[7-amino-2-(2-furyl)[1,2,4] triazolo[2,3-a][1,3,5]triazin-5-ylamino] ethyl)phenol (ZM241385), AM281, WIN55, 212-2, 2-aminoethoxydiphenylorane (2APB), and cyclopiazonic acid (CPA) were obtained from Tocris Cookson (Balwin, MO). Dihydrokainate (DHK), GABAzine (SR95531), atropine, mecamylamine, U73122, and chelerythrine chloride (Che) were from Sigma (St. Louis, MO). Xestospongin C (XeC) was from Calbiochem (San Diego, CA). All drugs at the concentration used did not alter the electrode sensitivity. The drugs were diluted from frozen aliquots immediately before use and were applied through bath application. Ultrapure water (Nanopure; Barnstead, Dubuque, IA) was used in all solutions.
Electrochemical recordings. Electrochemical recordings and electrical stimulation were adapted from Schmitz et al. (2001). Briefly, disk carbon fiber electrodes of 5 μm diameter with a freshly cut surface were placed into the dorsal striatum ∼50 μm below the exposed surface. For cyclic voltammetry (CV), a triangular voltage wave (-400 to +900 mV at 280 V/sec vs Ag/AgCl) was applied to the electrode every 100 msec. For amperometry, a constant voltage of +400 mV was applied via an Axopatch 200B (Axon Instruments, Foster City, CA). Current was recorded with an Axopatch 200B amplifier with a low-pass Bessel filter setting at 10 kHz, digitized at 25 kHz (ITC-18 board; InstruTech, Great Neck, NY). Triangular wave generation and data acquisition were controlled by a personal computer running a locally written (Dr. E. Mosharov, Columbia University, New York, NY) IGOR program (WaveMetrics, Lake Oswego, OR). Striatal slices were electrically stimulated by an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel) using a bipolar stimulating electrode placed at a distance of ∼100 μm from the recording electrode. Background-subtracted cyclic voltammograms served for electrode calibration and to identify the released substance. The current was converted to concentration based on a calibration in ACSF after the experiment. In some cases, after using CV to confirm the identity of DA, we switched to amperometry to increase temporal and detection sensitivity.
The slices were equilibrated in the recording chamber for 20 min before experiments were conducted. The slices were stimulated every 2 min by a single-pulse stimulus in the dorsal striatum, and experiments were generally initiated after five stimuli to confirm a consistent and robust response.
Data analysis and statistics. The drugs used in this study mainly affected the amplitude of the evoked DA response and usually attained a maximal effect within 10 min. Therefore, the change of DA amplitude was determined by comparing DA release before and after 10 min application of the agents, unless otherwise specified. This is expressed as a percentage of the average amplitude of the two postdrug responses divided by the average amplitude of the two predrug responses.
Experimental values in the text and in the figures are mean ± SEM. The two-tailed Student's t test was used for paired data, and one-way ANOVA followed by Dunnett's test was used for group data (GraphPad Software, San Diego, CA). The difference was considered significant at levels of p < 0.05.
Results
Inhibition of evoked DA release by EAAT blockers
To investigate whether the overflow of endogenous glutamate modulates evoked DA release, we applied a single-pulse electrical stimulus every 2 min in the dorsal striatum of acutely prepared corticostriatal slices to elicit the release of both DA and glutamate from nigrostriatal and corticostriatal afferents. DA release was monitored by CV (Schmitz et al., 2001) throughout the protocol. Under control conditions, there was a slight rundown during the 20 min experiment duration (5 ± 5%; n = 4). Bath application of the EAAT blocker THA (100 μm) (Arriza et al., 1994) suppressed evoked DA release by 4 min and reached a maximal inhibition of 33% of control levels at 10 min (Fig. 1a). A concentration-response curve for THA revealed an EC50 of ∼60 μm (Fig. 1c), and 300 μm THA suppressed evoked DA release by 34 ± 3.7% (n = 4) (Fig. 1b,c). Similarly, the THA analog TBOA (50 μm) a nonspecific EAAT blocker but one of the most potent nonsubstrate inhibitors of EAAT2 (Shimamoto et al., 1998), elicited a similar reduction of evoked DA release (44 ± 7.6%; n = 7; not statistically different from THA) (Fig. 1d).

Inhibition of evoked DA release by EAAT blockers. a, Inhibition of evoked DA release by 100 μm THA. Voltammetric responses obtained in a striatal slice during seven sequential stimuli at 2 min intervals. THA (100 μm) was added to the perfusion medium immediately after the second stimulus. b, The time course of the THA (300 μm) effect on evoked DA release (n = 8, THA; n = 4, control, i.e., perfusion medium; *p < 0.05 compared with respective control values by Student's t test). c, Dose-response curve of THA (n = 4). d, Inhibition of evoked DA release by different EAAT blockers. THA, 300 μm; TBOA, 50 μm; DHK, 300 μm (n = 4-7).
The five high-affinity sodium-dependent EAA transporters that have been cloned from human tissue are termed EAAT1 to EAAT5 (Arriza et al., 1994; Fairman et al., 1995). Three homologous transporter proteins have been identified in rodents: GLAST [for glutamate/aspartate transporter (EAAT1)], GLT1 [for glutamate transporter-1 (EAAT2)], and EAAC1 (EAAT3) (Pines et al., 1992; Storck et al., 1992). In the rat striatum, GLT1 and GLAST are expressed in astrocytes, and EAAC1 is expressed in neurons (Rothstein et al., 1994; Lehre et al., 1995). To estimate the relative contribution of glial and neuronal glutamate transporters to the DA depression, we compared the effects of THA and TBOA with the effect of DHK, a specific uptake inhibitor for GLT1 subtype (Rothstein et al., 1994). DHK (300 μm) elicited a DA depression (45 ± 7.1%; n = 5) similar to TBOA. Therefore, it seems that the DA depression by glutamate overflow was attributable mainly to the suppression of the glial glutamate transporter GLT1.
The effects of both TBOA and DHK on inhibition of evoked DA release fully recovered after a 20 min washout, whereas there was a partial recovery from the effect of THA (data not shown).
Inhibition of evoked DA release by THA was attributable to activation of mGluRs
The depression of stimulated DA release by THA was not mediated by ionotropic glutamate receptors (iGluRs), because the combination of the NMDA receptor antagonist d-AP-5 (50 μm) and the AMPA/kainate receptor antagonist CNQX (10 μm) did not occlude the THA effect (n = 5). In contrast, the broad-spectrum mGluR antagonist MCPG (300 μm) potently blocked the THA-induced inhibition of evoked DA release (n = 4) (Fig. 2). Neither d-AP-5 plus CNQX nor MCPG alone had any significant effect on evoked DA release (data not shown).

Inhibition of evoked DA release by THA was attributable to activation of mGluRs. The combination of the NMDA receptor antagonist d-AP-5 (50 μm) and the AMPA/kainate receptor antagonist CNQX (10 μm) did not occlude the response to THA (300 μm), whereas the broad-spectrum mGluR antagonist MCPG (300 μm) blocked THA inhibition of evoked DA release (n = 4-5; **p < 0.01 compared with THA, by one-way ANOVA followed by Dunnett's test).
THA reduced evoked DA release by activating group I mGluRs
To date, eight members of the mGluRs family have been identified. These are subdivided into three groups on the basis of amino acid sequence homology, signal transduction mechanisms, and pharmacological profiles (Conn and Pin, 1997). We used specific ligands to identify which mGluRs mediated the THA effect on evoked DA release. The selective group I antagonist AIDA (300 μm) blocked the depression of DA release by THA (n = 4) (Fig. 3a). Consistent with an action mediated by group I mGluRs, the group I mGluR agonist DHPG produced a dose-dependent reduction of DA release that was similar in duration and magnitude to THA, reaching a maximal inhibition of 40% of control levels. The depression recovered after a 30 min washout. No significant inhibition of stimulated DA release was observed with either the group II mGluR agonist (2R,4R)-APDC (20 or 50 μm) or the group III mGluR agonist l-AP-4 (100 or 300 μm) (Fig. 3b). AIDA alone did not enhance DA release (-14.3 ± 3.7% of control; n = 4), indicating that there was no tonic activation of mGluRs on the dopaminergic transmission in the preparation.

THA reduced evoked DA release by activating mGluR1. a, The group I mGluR antagonist AIDA (300 μm) blocked the inhibition by THA (300 μm) on evoked DA release (n = 4; **p < 0.01 compared with THA by Student's t test), whereas AIDA itself had no effect. b, The group I mGluR agonist DHPG (100 and 200 μm) and not those for group II (APDC, 20 and 50 μm) and group III mGluR (l-AP-4, 100 and 300 μm) inhibited evoked DA release (n = 4-10; **p < 0.01 compared with control by one-way ANOVA followed by Dunnett's test). c, The mGluR5 agonist CHPG (500 μm) had no effect on evoked DA release. The mGluR5 antagonist MPEP (20 μm), as well as lack of the receptor in mGluR5-deficient mice, did not block the inhibition by DHPG on evoked DA release (n = 4-6; **p < 0.01 compared with control by one-way ANOVA followed by Dunnett's test).
Group I mGlu receptors comprise mGluR1 and mGluR5. To determine which subtype was involved, we used both pharmacological tools and mGluR5-deficient mice. The mGluR5-specific agonist CHPG (500 μm) had no effect on evoked DA release (-3.6 ± 5.7%; n = 4). Consistently, the mGluR5-specific antagonist MPEP (20 μm) itself had no effect on evoked DA release (-0.3 ± 0.3%; n = 4) and did not block the effect of DHPG (-38.2 ± 3%; n = 6). Furthermore, mGluR5-deficient mice exposed to DHPG exhibited similar DA depression (-39.3 ± 2.7%; n = 4) (Fig. 3c). We therefore conclude that the inhibition of evoked DA release was attributable to the activation of mGluR1.
DHPG-induced depression of evoked DA release was mediated by mobilization of internal Ca2+ stores
Group I mGluRs are phospholipase C (PLC) coupled receptors (Pin and Duvoisin, 1995) that stimulate phosphoinositide (PI) hydrolysis and diacylglycerol (DAG) generation. To gain insight into the mechanisms involved in the depression of DA release by mGluR1 activation, we first measured evoked DA release after exposing slices to U73122 (10 μm for 20 min), a specific antagonist of PLC. U73122 itself had no effect (-6.5 ± 2.1%; n = 6) but completely blocked the effect of DHPG (-4.7 ± 6.0%; n = 7) (Fig. 4). Inhibition of protein kinase C (PKC) by Che (20 μm), which is activated downstream of PI hydrolysis, did not abolish DHPG-induced depression (-42 ± 2%; n = 4).
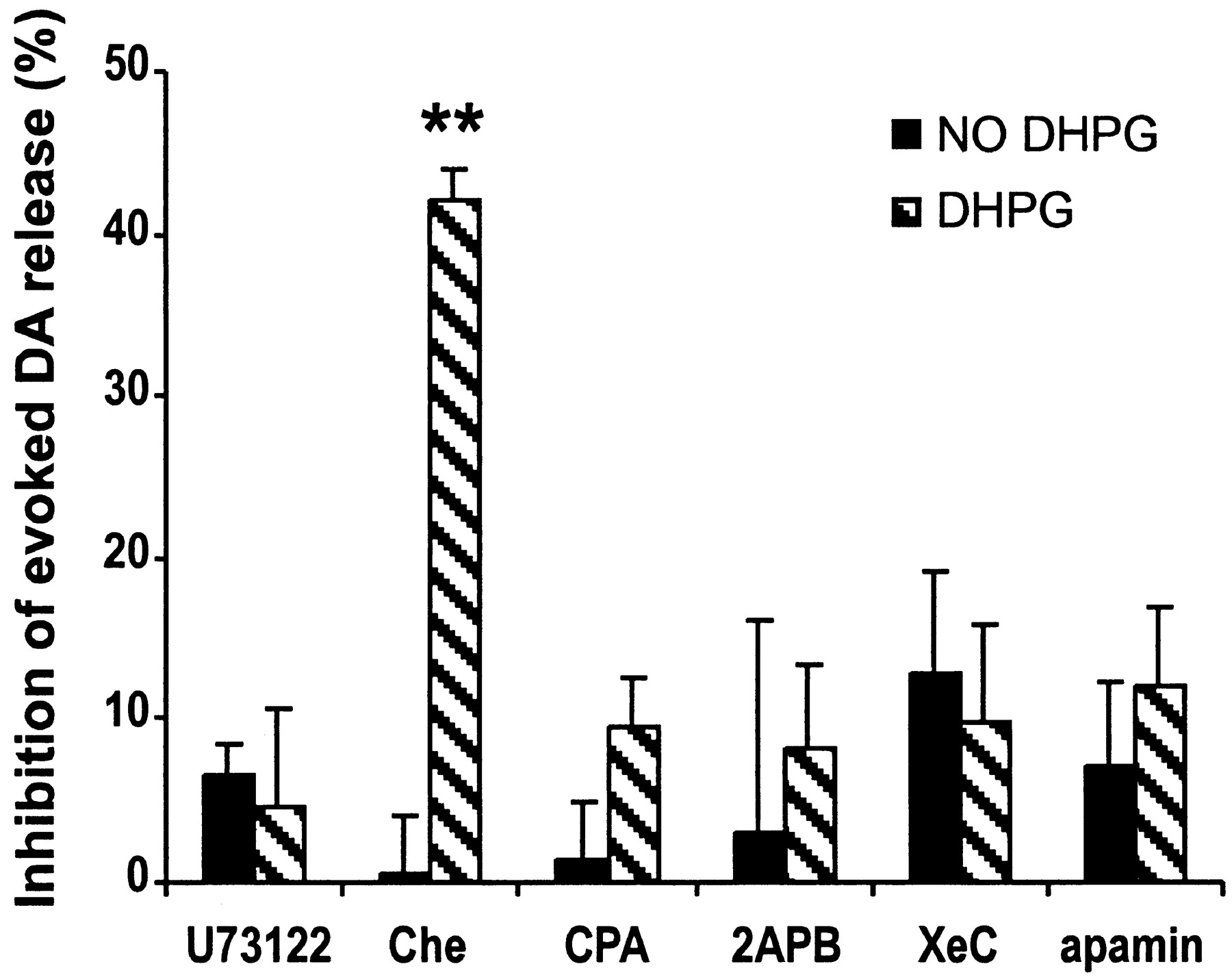
Inhibition of evoked DA release by DHPG is mediated by mobilizing internal Ca2+ stores and activation of an apamin-sensitive potassium channel. Black bars, (1 - Evoked DA release after/Evoked DA release before application of the agent) × 100%; hatched bars, (1 - Evoked DA release in the presence of 200 μm DHPG and the agent/Evoked DA release in the presence of the agent) × 100%. Inhibition of PLC by 10 μm U73122 completely blocked the inhibition by DHPG on evoked DA release. Inhibiting PKC by Che (20 μm) did not block the effect of DHPG, whereas depletion of intracellular Ca2+ store by CPA (20 μm, 20 min) or antagonizing IP3 receptor by 3 μm XeC or 100 μm 2APB blocked the effect of DHPG completely. The effect of DHPG was blocked by apamin (0.3 μm) (n = 3-10; **p < 0.01 compared with control by one-way ANOVA followed by Dunnett's test).
Because calcium release from intracellular stores can regulate neurotransmitter release and recent studies report the involvement of inositol-1,4,5-trisphosphate (IP3) in mGluR-induced Ca2+ mobilization (Finch and Augustine, 1998; Morikawa et al., 2000), we were interested in determining whether internal Ca2+ mobilization was involved. For example, it may be that the prolonged treatment with DHPG depletes internal Ca2+ stores on the DA terminals and therefore decreases evoked DA release. Pretreatment of slices with CPA (20 μm, 20 min), which depletes intracellular Ca2+ stores by blocking the ATPase that mediates Ca2+ uptake (Seidler et al., 1989; Fiorillo and Williams, 1998), completely blocked DHPG-induced depression (-9.6 ± 3.0%; n = 10). Similar results were obtained by inhibiting IP3 receptors. Application of the membrane-permeable IP3 receptor inhibitor 2APB (100 μm) completely prevented the inhibition of evoked DA release by DHPG (-8.3 ± 5.2%; n = 3). These results were confirmed by a more specific and potent membrane-permeable IP3 receptor inhibitor, XeC (3 μm; -9.8 ± 6%; n = 5) (Fig. 4) (Gafni et al., 1997). We further investigated the downstream mechanisms by applying apamin (0.3 μm), a blocker of calcium-activated potassium channels. Although apamin itself had no effect on evoked DA release (-7.1 ± 5.2%; n = 7), it blocked the DHPG-induced inhibition of evoked DA release (-12.0 ± 5.0%; n = 7). Together, these results suggest that intracellular Ca2+ mobilization and activation of apamin-sensitive potassium channels (Fiorillo and Williams, 1998) underlie the depression of DA release induced by DHPG.
DHPG may act directly on presynaptic mGluR1 on DA terminals
Our results clearly demonstrated that activating mGluR1 depressed evoked DA release. At many central synapses, release activity is thought to be inhibited by various neuromodulators acting at presynaptic receptors located on terminals (Wu and Saggau, 1997). To determine whether the DHPG-induced inhibition of evoked DA release was mediated by a direct activation of the mGluR1 on the DA terminals or by an indirect mechanism involving other transmitters, e.g., acetycholine (ACh), we performed a series of experiments using various antagonists. Neither the GABAa receptor antagonist GABAzine (10 μm) nor the GABAb receptor antagonist CGP 52432 (10 μm), blocked the effect of DHPG (-35 ± 1.6 and -39 ± 4.3%, respectively) (Fig. 5a). Similarly, neither the adenosine A1 receptor antagonist DPCPX (1 μm) nor the A2a receptor antagonist ZM241385 (3 μm) blocked the effect of DHPG (-32 ± 1.8 and -35 ± 4.0%, respectively) (Fig. 5b). The antagonists themselves had no effect on evoked DA release, indicating an absence of GABA or adenosine tone in the slice preparation.

Inhibition of evoked DA release by DHPG may be mediated by acting directly on mGluR1 on DA terminals. a, Neither the GABAa receptor antagonist GABAzine (10 μm) nor the GABAb receptor antagonist CGP 52432 (10 μm) could block the effect of DHPG. b, Neither the adenosine A1 receptor antagonist DPCPX (1 μm) nor the A2a receptor antagonist ZM241385 (3 μm) could block the effect of DHPG. c, Nether the mAChR antagonist atropine (2 μm) nor the nAChRs antagonist mecamylamine (2 μm) or the combination of both blocked the effect of DHPG. d, A mixture containing all of the antagonists (50 μmd-AP-5, 10 μm CNQX, 10 μm GABAzine, 10 μmCGP52432, 1 μm DPCPX, 3 μmZM241385, 1 μm atropine, and 2 μm mecamylamine) failed to prevent the DA depression induced by DHPG. Black bars, (1 - Evoked DA release after/Evoked DA release before application of the agent) × 100%; hatched bars, (1 - Evoked DA release in the presence of 200 μm DHPG and the agent/Evoked DA release in the presence of the agent) × 100% (n = 3-7; **p < 0.01 by one-way ANOVA followed by Dunnett's test for the agents indicated vs control for the respective group; black bars, in the absence of DHPG; hatched bars, in the presence of DHPG).
Consistent with the literature (Kudernatsch and Sutor, 1994; Zhou et al., 2001), the mAChR antagonist atropine (2 μm) had no effect on evoked DA release, whereas the nitcotinic ACh receptor (nAChR) antagonist mecamylamine (2 μm) severely inhibited evoked DA release (-55 ± 3.6%; n = 4). Nevertheless, 200 μm DHPG caused an additional inhibition (-32.0 ± 3.1%; n = 5) in mecamylamine-treated slices. Atropine and mecamylamine together also failed to block the effect of DHPG (n = 5) (Fig. 5c). To exclude an indirect effect attributable to some complicated interactive actions between the receptors, we finally applied a mixture containing all of the antagonists otherwise applied separately (50 μmd-AP-5, 10 μm CNQX, 10 μm GABAzine, 10 μmCGP52432, 1 μm DPCPX, 3 μm ZM241385, 1 μm atropine, and 2 μm mecamylamine). The mixture itself inhibited evoked DA release by 62 ± 1.8% attributable to the action of mecamylamine. However, it did not abolish the inhibitory effect of DHPG on evoked DA release (40 ± 6.0%; n = 4) (Fig. 5d). The mixture also did not block the effect of THA (data not shown).
Activation of postsynaptic mGluRs, for example, those expressed in MSNs (Testa et al., 1998) and large apical (LA) cholinergic interneurons (Takeshita et al., 1996), could retrogradely influence presynaptic functions mediated by the cannabinoid CB1 receptors (CB1R) (Kreitzer and Regehr, 2001; Robbe et al., 2002). The CB1R antagonist AM281 (3 μm) did not abolish the effect of DHPG. AM281 itself had no effect on evoked DA release. Consistently, the CB1R agonist WIN55,212-2 (15 μm) had no effect on evoked DA release (Fig. 6).

Inhibition of evoked DA release by DHPG was not CB1R mediated. The CB1R antagonist AM281 (3 μm) had no effect on evoked DA release and did not abolish the effect of DHPG. The CB1R agonist WIN55,212-2 (15 μm) had no effect on evoked DA release (n = 4-8).
Although we cannot completely exclude the possibility of an indirect pathway, our results suggest that glutamate may act directly on the mGluR1 on DA terminals to depress evoked DA release (see Discussion)
Inhibition of evoked DA release by HFS
The efficacy of 100 μm THA (Fig. 1a) suggests that even a relatively modest blockade of glutamate transporters leads to a significant activation of mGluR1. We were interested in investigating whether trains of HFS such as those used in inducing striatal long-term potentiation or long-term depression could cause glutamate spillover and in turn depress evoked DA release. Because local HFS depleted DA completely and interfered with monitoring evoked DA release (data not shown), we performed these experiments in sagittal slice preparations encompassing a more intact corticostriatal glutamatergic projection. Fiber tracts in the corpus callosum (CC) were stimulated by HFS [six trains of 100 pulses (100 Hz) at 10 sec intervals] to induce glutamate release in the striatum, whereas evoked DA release was recorded at least 500 μm away from the CC (Fig. 7a). The HFS did not elicit detectable release of DA at the recording site. In 9 of 16 slices, we observed a depression of evoked DA release within 1 min after HFS, reaching a maximum inhibition of 30 ± 4% by 5 min with a nearly complete recovery within 11 min (Fig. 7b). The development of this depression within 5 min after HFS also strongly argued against a role for undetectable depletion of DA by HFS (if HFS at the CC had depleted DA at the recording site, evoked DA release should only show recovery after HFS). In addition, in MCPG perfused slices (n = 4) (Fig. 7b) or coronal slices (n = 12), we never observed a depression of evoked DA release by HFS. The combination of the iGluRs antagonists (10 μmd-AP-5 plus 10 μm CNQX) had no effect on DA depression by HFS (data not shown).

Inhibition of evoked DA release by HFS. a, Diagram of HFS and electrochemical recording electrode placement. STR, Striatum; Hip, hippocampus; CC, corpus callosum. HFS, Six trains of 100 pulses (100 Hz) at 10 sec intervals. b, Time course of normalized evoked DA release after HFS. HFS depressed evoked DA release (filled circles; n = 9), whereas 300 μm MCPG blocked this effect (open circles; n = 4). Inset, CV recordings at different time points as indicated by a-c.
Discussion
It has been suggested that, after the accumulation of glutamate in the synaptic cleft, glutamate spillover can reach perisynaptic receptors and trigger the mGluR-mediated responses. The present results are the first to demonstrate that spillover of glutamate inhibits evoked DA release in the striatum and that this effect is mediated via activation of group I mGluRs.
Inhibition of evoked DA release by glial glutamate transporter blockers via activation of mGluR1
The level of extrasynaptic glutamate appears to be regulated via clearance by EAATs (Diamond and Jahr, 1997), particularly by glial transporters (Rothstein et al., 1996). The glial transporters GLT1 and GLAST and the neuronal transporter EAAC1 are expressed in the rat striatum (Rothstein et al., 1994; Lehre et al., 1995), with GLT1 being the dominant form (Lievens et al., 2001).
Using EAAT blockers to enhance extrasynaptic glutamate concentration, we found that glutamate could spill over from the synaptic cleft to depress evoked DA release. Selective blockade of EAATs indicated that inhibition of evoked DA release was mainly mediated through GLT1 (EAAT2) blockade. Thus, the regulation of extrasynaptic glutamate in the striatum is mostly attributable to uptake by glia.
Consistent with these findings, the depression of evoked DA release by glutamate spillover was mimicked by the direct activation of mGluR1 by its specific agonist DHPG. We were concerned, however, that because group I mGluRs are easily desensitized (Gereau and Heinemann, 1998; Rodriguez-Moreno et al., 1998; Alagarsamy et al., 2001), this effect by DHPG may not reflect the normal function of mGluR1. For instance, both nAChR agonists and antagonists depress evoked DA release in striatal slices (Zhou et al., 2001). In this case, the nAChR agonists are thought to desensitize the receptors, whereas the antagonists inhibit ongoing low-level nAChRs activation. Thus, the normal function of tonic nAChR activation appears to stimulate DA release. Similarly, if tonic mGluR1 activation is excitatory for DA release, bath application of DHPG might depress DA release via desensitization. We found, however, that AIDA, a group I mGluR antagonist, had no effect on evoked DA release, indicating that there was no tonic activation of mGluR1. In addition, glutamate spillover caused by HFS, which should imitate the physiological effects of strong high-frequency cortical inputs, depressed evoked DA release. Moreover, group I mGluRs have been shown to exhibit a dual action on glutamate release in hippocampal slices, switching from facilitation to inhibition during receptor desensitization (Rodriguez-Moreno et al., 1998). To rule out this possibility, we pretreated slices with the mGluR antagonist MCPG (300 μm, 2 hr) to remove any potential desensitization during slice preparation. We did not observe any facilitation by application of DHPG, or EAAT inhibitors, or HFS (data not shown). We therefore conclude that mGluR1 activation acts to inhibit evoked DA release.
DHPG-induced inhibition of evoked DA release is mediated by the mobilization of internal Ca2+ stores
The activation of PI-linked group I mGluRs leads, via PLC, to the hydrolysis of phosphatidylinositol-4,5-bisphosphate into two second messengers, DAG and IP3. DAG acts by stimulating PKC, whereas IP3 induces Ca2+ release from intracellular Ca2+ stores (Pin and Duvoisin, 1995). We found that, whereas inhibition of PKC failed to block the effect of DHPG on evoked DA release, depletion of intracellular Ca2+ stores by CPA blocked the effect. Thus, the effect of DHPG was mediated through Ca2+ mobilization.
This mGluR1-mediated inhibition through Ca2+ mobilization could be attributable to either depletion of internal calcium stores in the terminals by the prolonged treatment with DHPG (and therefore decreased evoked transmitter release) or activation of Ca2+-dependent pathways that inhibit DA release, for example, by activating Ca2+-dependent potassium currents. Because CPA itself had no effect on evoked DA release, it is likely that activation of mGluR1 depresses evoked DA release by activating Ca2+-dependent pathways. The involvement of Ca2+-dependent potassium currents was confirmed by the effects of apamin. Thus, the inhibition of evoked DA release by mGluR1 activation involves Ca2+-activated potassium channels. Nevertheless, given the long-lasting (several minutes) depression of evoked DA release by HFS (see below), and noting that mGluR1 can modulate Ca2+ channels (McCool et al., 1998; Stefani et al., 1998; Anwyl, 1999; Faas et al., 2002), our results do not rule out the possibility that mGluR1 may exert additional effects on evoked DA release by inhibiting presynaptic Ca2+channels.
Is DA depression mediated by a direct presynaptic mechanism or by an indirect circuit mechanism?
Although the voltammetric technique records presynaptic released DA directly, our data do not rule out a presynaptic effect mediated by other transmitters or a retrograde signal that might mediate the effect of mGluR1 activation. In addition to the D2 autoreceptor, there are various heteroreceptors expressed on DA terminals, including nAChRs (Wonnacoott et al., 2000) and GABAbR (Charara et al., 2000; Schmitz et al., 2002), and mGluRs are expressed in MSNs (Testa et al., 1998; Pisani et al., 2000) and LA cholinergic interneurons (Takeshita et al., 1996), in addition to DA terminals (Paguet and Smith, 2003). Therefore, DHPG could exhibit its effect indirectly through GABA or ACh by acting on MSN or LA neurons. For example, activation of mGluR1 may depolarize the membrane, contributing to an increased firing rate of these neurons and a subsequent augmented release of ACh and/or GABA. To rule out the possibility of an indirect effect, we performed a series of experiments using various receptor antagonists. The effect of DHPG was not mediated by iGluRs, GABAaR, GABAbR, A1R, A2AR, nAChRs, or mAChRs. A mixture containing all of these antagonists also failed to prevent the effect of DHPG. Although we could not completely exclude all indirect effects, the results are consistent with the possibility that DHPG depresses evoked DA release by acting directly on the mGluR1 on the DA terminals. There has been no previous report of a presynaptic action by this receptor in ventral midbrain DA neurons, but it is highly expressed in ventral midbrain DA cell bodies and dendrites (Hubert et al., 2001), in which it acts to depress neuronal excitability (Fiorillo and Williams, 2000). Although mGluR5 has been implicated in the modulation of DA release (Page et al., 2001), we found that the mGluR5 antagonist MPEP failed to abolish the DA depression induced by DHPG and that the effect was maintained in mGluR5-deficient mice. We conclude that the depression was mediated through mGluR1 or unidentified mGluRs with similar properties. A recent study reports that, whereas mGluR5 is almost exclusively found postsynaptically in striatal neurons, mGluR1 is the only glutamate receptor observed by ultrastructural immunolabeling on DA terminals (Paguet and Smith, 2003).
Depression of evoked DA release by HFS
Corticostriatal firing patterns are complex in anesthetized rats with firing frequencies ranging from ∼5 to >100 Hz during the up states (Stern et al., 1997), but whether glutamate spillover can occur and stimulate mGluR1 during normal corticostriatal firing patterns remains unknown. Nevertheless, extrasynaptic diffusion of glutamate to activate iGluRs has been demonstrated in the hippocampus and cerebellum (Dzubay and Jahr, 1999). Because mGluRs display higher affinity for glutamate than ionotropic receptors (Conn and Pin, 1997), it is likely that a relatively small amount of extrasynaptic spilled-over glutamate might activate mGluRs. Our results suggest that a small amount of glutamate spillover occurs during HFS, leading to mGluR1 activation. This also explains the efficacy of 100 μm THA in this study. Whereas bath application of EAAT blockers or DHPG usually requires 6-10 min to reach the maximal inhibition (mainly attributable to substation of the perfusion fluid), the time course of HFS-mediated depression was surprising, because although it was rapidly initiated, it reached a maximum value within 5 min. It may be that mGluR activation stimulates more than one downstream pathway that modulates evoked DA release. For instance, changes in DA synthesis mediated by second-messenger systems may have slowly developing effects via changes in quantal size (Pothos et al., 1998) or via Ca2+-dependent forms of modulation of Ca2+ channels (McCool et al., 1998).
Implications for physiological relevance
Our results suggest that, when glutamate spills over from corticostriatal synapses, there is a consequent depression of DA neurotransmission. The DA released from nigrostriatal synapses, under some conditions, acts on presynaptic corticostriatal D2 DA receptors to depress glutamatergic afferentation (Cepeda et al., 2001). Our results thus suggest a reciprocal depression of glutamate and DA neurotransmission on each other under conditions in which synaptic spillover is adequate to stimulate distal presynaptic group I mGluRs or D2 receptors, respectively.
Recent studies suggest that interactions between nigrostriatal DA activity and striatal group I mGluRs activation determine whether a given tetanus results in corticostriatal long-term depression or long-term potentiation (Centonze et al., 2001; Gubellini et al., 2001; Sung et al., 2001). Thus, the reciprocal interplay between these two afferent pathways in the striatum may underlie long-term plastic modulation of striatal synapses. It should be noted that, although the effects of D2 autoreceptors last only for ∼ 1 sec (Benoit-Marand et al., 2001; Phillips et al., 2002; Schimtz et al., 2002), the effect of mGluR1 activation appear to last for several minutes, perhaps underlying the factors in the timing of motor learning and the development of forms of behavioral reinforcement.
Footnotes
This work was supported by the National Association for Research on Schizophrenia and Depression, National Institute on Drug Abuse, National Institute of Neurological Disorders and Stroke, National Science Foundation, the Lowenstein Foundation, and the Parkinson's Disease Foundation. We thank Drs. Yvonne Schmitz for advice and critique, Eugene Mosharov for writing software, and Kristin Larsen for critical reading of this manuscript.
Correspondence should be addressed to Dr. David Sulzer, Columbia University, Department of Neurology, 650 West 168th Street, New York, NY 10032. E-mail ds43{at}columbia.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310585-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}