

Abstract
The cognitive hallmark of early Alzheimer's disease (AD) is an extraordinary inability to form new memories. For many years, this dementia was attributed to nerve-cell death induced by deposits of fibrillar amyloid β (Aβ). A newer hypothesis has emerged, however, in which early memory loss is considered a synapse failure caused by soluble Aβ oligomers. Such oligomers rapidly block long-term potentiation, a classic experimental paradigm for synaptic plasticity, and they are strikingly elevated in AD brain tissue and transgenic-mouse AD models. The current work characterizes the manner in which Aβ oligomers attack neurons. Antibodies raised against synthetic oligomers applied to AD brain sections were found to give diffuse stain around neuronal cell bodies, suggestive of a dendritic pattern, whereas soluble brain extracts showed robust AD-dependent reactivity in dot immunoblots. Antigens in unfractionated AD extracts attached with specificity to cultured rat hippocampal neurons, binding within dendritic arbors at discrete puncta. Crude fractionation showed ligand size to be between 10 and 100 kDa. Synthetic Aβ oligomers of the same size gave identical punctate binding, which was highly selective for particular neurons. Image analysis by confocal double-label immunofluorescence established that >90% of the punctate oligomer binding sites colocalized with the synaptic marker PSD-95 (postsynaptic density protein 95). Synaptic binding was accompanied by ectopic induction of Arc, a synaptic immediate-early gene, the overexpression of which has been linked to dysfunctional learning. Results suggest the hypothesis that targeting and functional disruption of particular synapses by Aβ oligomers may provide a molecular basis for the specific loss of memory function in early AD.
Introduction
Alzheimer's disease (AD) is a progressive dementia that manifests in early stages primarily as a profound inability to form new memories (Selkoe, 2002). The basis for this specificity is unknown, but evidence favors involvement of neurotoxins derived from the self-associating amyloid β peptide (Aβ). Aβ generates the fibrils of the hallmark amyloid plaques of AD, its production is elevated by disease-associated mutations and risk factors, and fibrils made in vitro kill cultured brain neurons (Klein, 2004). The original amyloid cascade hypothesis, developed from these core findings, thus attributed AD memory loss to neuron death caused by fibrillar Aβ (Hardy and Higgins, 1992).
Despite its intuitive appeal and strong experimental support, this hypothesis has proven inconsistent with key observations, including the poor correlation between dementia and amyloid plaque burden (Katzman et al., 1988). Studies of experimental vaccines with transgenic (tg) human amyloid protein precursor (hAPP) mice, which provide good models of early AD by developing age-dependent plaques and memory dysfunction (Dodart et al., 2002; Kotilinek et al., 2002), have been particularly illustrative. When injected with monoclonal antibodies against Aβ, these mice respond in ways unanticipated by the amyloid cascade: (1) vaccinated mice show reversal of memory loss, with recovery evident in 24 hr, and (2) cognitive benefits of vaccination accrue despite no decrease in plaque levels. Such findings are inconsistent with a mechanism for memory loss dependent on neuron death caused by amyloid fibrils.
A new version of the amyloid cascade hypothesis proposes an alternative mechanism for memory loss based on the impact of small, soluble Aβ oligomers (Klein et al., 2001; Hardy and Selkoe, 2002). Its two salient new features are that (1) early memory loss stems from synapse failure before neuron death and (2) synapse failure derives from actions of Aβ oligomers rather than fibrils. This hypothesis emerged from experiments showing that Aβ oligomers rapidly inhibit long-term potentiation (LTP), a classic experimental paradigm for memory and synaptic plasticity (Lambert et al., 1998; Walsh et al., 2002; Wang et al., 2002). Recent findings that oligomers are strikingly elevated in hAPP transgenic mice AD models (Chang et al., 2003) and, most significantly, in AD brain (Gong et al., 2003) give the hypothesis substantive clinical support.
Considerable interest is focused now on the mechanism by which oligomers interact with neurons (Caughey and Lansbury, 2003). Two prominent possibilities are that oligomers insert into membrane bilayers to create cytotoxic pores, a relatively nonspecific mechanism, or alternatively, that oligomers bind to particular membrane targets as highly specific ligands. Here, oligomer-neuron association has been examined using oligomers extracted from AD brain or made in vitro. Results show that oligomers bind to neuron surfaces in small punctate clusters that colocalize almost exclusively with a subpopulation of synaptic terminals. Binding is accompanied by ectopic induction of Arc, a synaptic immediate-early gene the overexpression of which has been linked to dysfunctional learning. We hypothesize that targeting and functional disruption of particular synapses by Aβ oligomers could underlie the specific loss of memory function in early AD.
Materials and Methods
Aβ-derived diffusible ligands and fractionation. Aβ1-42 peptide (California Peptide Research, Napa, CA) or biotin-Aβ1-42 peptide (Recombinant Peptide, Athens, GA) was used to prepare synthetic Aβ-derived diffusible ligands (ADDLs) or biotinylated ADDLs according to published protocols (Lambert et al., 2001; Klein, 2002). Molecular weight (MW) fractionation of oligomeric species was obtained using Centricon YM-100 and YM-10 concentrators (Millipore, Bedford, MA), used according to manufacturer's instructions. Size-exclusion chromatography (SEC) using the Akta Explorer HPLC apparatus with a Superdex 75 HR 10/30 column was conducted according to published protocols (Chromy et al., 2003).
Tissue extracts and CSF. Frontal cortex, cerebellum, and CSF from Alzheimer's disease and nondemented control subjects were obtained from the Northwestern Alzheimer's Disease Center Neuropathology Core (Chicago, IL). Soluble extracts from brain tissues were prepared as described previously (Gong et al., 2003).
Cell culture. Hippocampal neurons were maintained in Neurobasal medium supplemented with B27 (Invitrogen, Carlsbad, CA) for at least 3 weeks as described previously (Gong et al., 2003). Cells were incubated with vehicle, synthetic or biotinylated ADDLs (500 nm), crude human CSF (100 μl), or F12-extracted human cortex (1.0 mg protein/ml) for the indicated times.
Immunocytochemistry. Immunocytochemistry was performed as described (Gong et al., 2003). Some cells were permeabilized with 0.1% Triton X-100 in 10% normal goat serum and PBS (NGS/PBS) for 1 hr at room temperature (RT). Cells were double immunolabeled with M94 [Aβ oligomer-generated polyclonal antibody characterized previously (Lambert et al., 2001)] (1:500) and anti-αCaM kinase II (αCaMKII) (1:250) or anti-postsynaptic density (PSD)-95 (1:500) monoclonal antibodies (Affinity BioReagents, Golden, CO) or goat polyclonal anti-Arc antibody (1:200) (Santa Cruz Biotechnology, Santa Cruz, CA), NMDA-R1 (C-term, 1:200) (Upstate, Lake Placid, NY), or synaptophysin (SVP-38, 1:500) (Sigma, St. Louis, MO) overnight at 4°C, followed by an incubation with appropriate Alexa Fluor 488- or 594-conjugated IgG (Molecular Probes, Eugene, OR) (2 μg/ml) for 2 hr at RT. Double labeling for ADDLs and either NMDA-glutamate receptor subunit NR1 (1: 200) or AMPA-glutamate receptor subunit GluR1 (1:200) (Upstate Biotechnology) used synthetic biotinylated ADDLs and streptavidin-Alexa Fluor 488 conjugate. Cells were visualized using a Leica TCS SP2 Confocal Scanner DMRXE7 Microscope (Bannockburn, IL) with constant settings of laser power, detector gain, amplification gain, and offset. Images were acquired in z-series scans at 0.5 μm intervals from individual fields to determine whether ADDLs colocalized with αCaMKII, PSD-95, SVP-38, NR1, GluR1, or Arc. Morphometric quantifications were performed with Meta-Morph imaging software (Universal Imaging, West Chester, PA).
Immunohistochemistry. Autopsied brain from seven AD cases (59-87 years old) and seven nondemented elderly controls (68-78 years old) were immersion fixed in 10% buffered formalin for 30-48 hr and then transferred to a 10-40% sucrose gradient. Free-floating 40-μm-thick serial sections were obtained from frontal cortex and kept in cryoprotectant at 4°C until immunolabeling. Sections were rinsed with TBS, pretreated with 2% sodium m-periodate in TBS for 20 min, and permeabilized with 0.25% Triton X-100 in TBS (TBST). Aspecific immunoreactivity (IR) was blocked with 5% horse serum in TBST for 40 min and 1% nonfat dry milk in TBST for 30 min. Sections were subsequently incubated with M94 (1:1000) overnight at 4°C and Alexa Fluor 488 anti-rabbit IgG (1:500) for 90 min at RT. Serial sections were stained with 0.5% thioflavine-S in 50% ethanol. Confocal images were collected on the Leica confocal microscope as described above with z-series scans of 1 μm intervals. Similar sections were labeled with M94 antibody and secondary antibody-ABC complex and diaminobenzidine. Sections were counterstained with hematoxylin. Control sections with the primary or secondary antibodies omitted were negative.
Immunoblot. Cells were lysed in 1 vol PBS-protease inhibitor mixture solution and 1 vol of 2× loading buffer, pH 6.8 (80 mm Tris-HCl, 16.7% glycerol, 1.67% SDS, 1.67% β-mercaptoethanol) and sonicated briefly. Proteins were separated on 4-20% Tris-glycine gels (Bio-Rad, Hercules, CA) at 100 V and transferred to nitrocellulose membrane at 100 V for 1 hr at 4°C in transfer buffer (25 mm Tris-HCl, pH 8.3, 192 mm glycine, 20% v/v methanol). Blots were blocked with 5% nonfat dry milk in 10 mm Tris-buffered saline containing 0.1% Tween 20, pH 7.5, for 2 hr. Blots were incubated overnight at 4°C with anti-Arc antibody (1:250) and 2 hr with HRP-conjugated IgG (1:100,000). Membranes were developed with SuperSignal West Femto chemiluminescence kit (Pierce Biotechnology, Rockford, IL) and then washed, blocked, and reblotted with anti-cyclophilin B antibody (1:40,000) used as a control for protein loading. Proteins were visualized and quantified using the Kodak IS440CF Image Station (New Haven, CT).
Dot blot assay. A previously described dot blot assay was used to measure assembled forms of Aβ in soluble extracts of human frontal cortex and cerebellum (Gong et al., 2003). Nine AD samples (pathology diagnosis based on Braak and Braak, Consortium to Establish a Registry for Alzheimer's Disease (CERAD), and National Institute on Aging (NIA)/Reagan Institute Criteria) were compared with 15 non-AD control samples. Tissue (100 mg) was homogenized in 1 ml Ham's F12 phenol-free medium (BioSource, Camarillo, CA) containing protease inhibitors (Complete mini EDTA free tablet; Roche, Indianapolis, IN) on ice using a Tissue Tearor (Biospec Products, Bartlesville, OK). After centrifugation at 20,000 × g for 10 min, the supernatant was centrifuged at 100,000 × g for 60 min. Protein concentration of 100,000 × g supernatant was determined by standard BCA assay. For dot blot assay, nitrocellulose was prewetted with TBS (20 mm Tris-HCl, pH 7.6, 137 mm NaCl) and partially dried. Extracts (2 μl, 1 μg of total protein) were applied to nitrocellulose and air dried completely. The nitrocellulose membranes were then blocked in 0.1% Tween 20 in TBS (TBS-T) with 5% nonfat dry milk powder for 1 hr at RT. The membranes were incubated for 1 hr with primary antibody M93/3 in the blocking buffer (1:1000) and washed three times for 15 min with TBS-T. Incubation with HRP-conjugated secondary antibody (1:50,000, Amersham Biosciences, Piscataway, NJ) in TBS-T for 1 hr at RT was followed by washes. Proteins were visualized with chemiluminescence and analyzed on a Kodak IS440CF Imaging Station with Kodak 1D image software.
Results
Soluble Aβ species, detected by Aβ oligomer-raised antibody, are deposited around neuronal cell bodies and increased in AD cortex
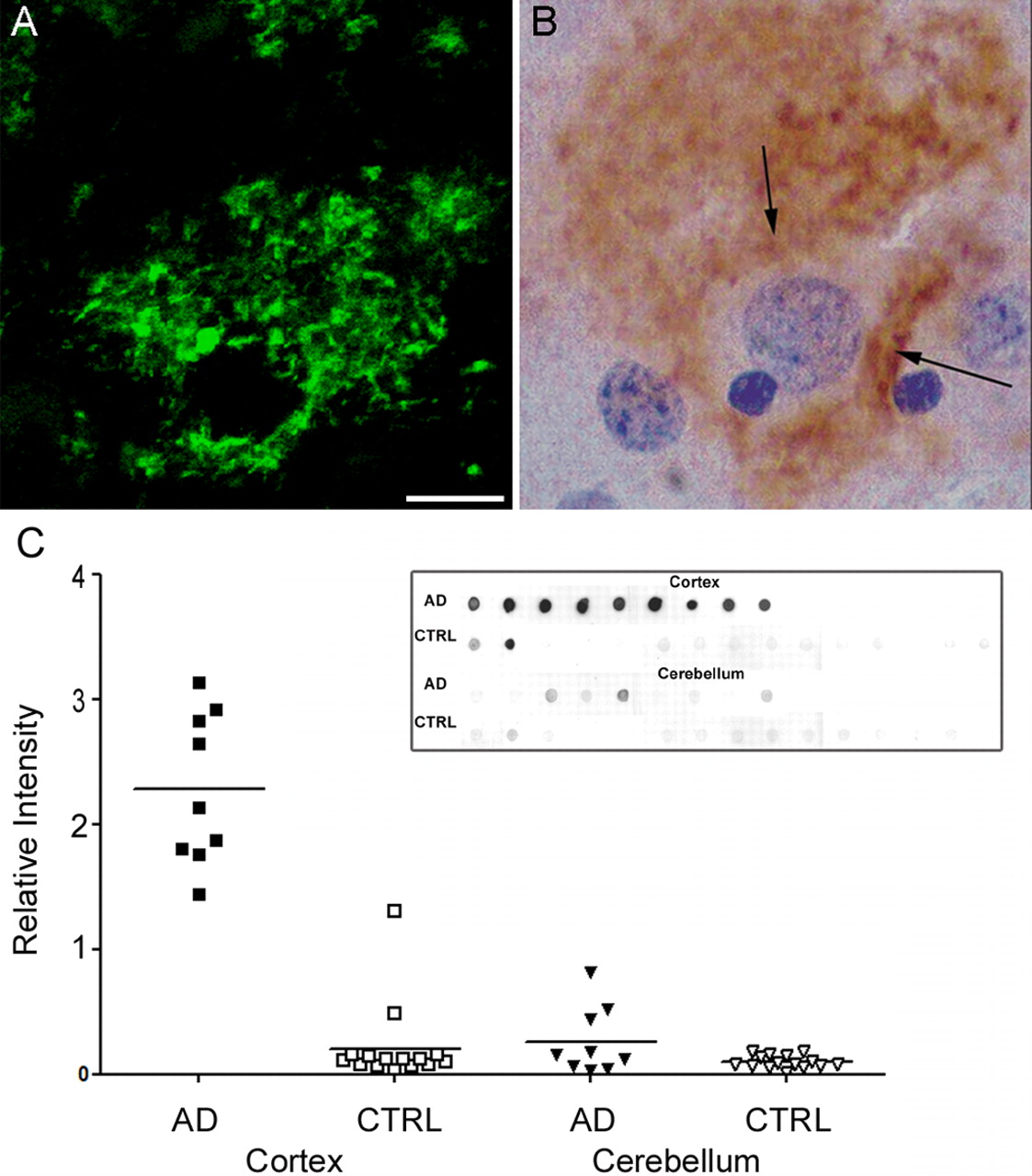
The first goal was to verify the presence of ADDLs in AD brain and to establish that the antibodies used in subsequent cell biology experiments were specific for Alzheimer's pathology. Accordingly, sections from human frontal cortex (seven AD patients and nondemented age-matched controls) were immunolabeled with M94 (an oligomer-selective antibody) (Gong et al., 2003) and assessed for fibrillar amyloid deposits with thioflavin-S. Immunolabeled AD brain sections exhibited localized immunoreactive deposits that selectively surrounded cell bodies in regions that also showed characteristic Aβ deposition in the form of senile neuritic and diffuse amyloid plaques; however, the pericellular diffuse immunoreactivity, which was found in all AD cases, was clearly distinct from fibrillar amyloid deposits (detected by thioflavin-S staining; data not shown). Representative images of individual pyramidal neurons located in cortical layer III are shown labeled by immunofluorescence (Fig. 1A) and HRP staining (Fig. 1B). One control (of seven) showed similar structures; this particular control brain was Braak stage 0 with low levels of plaques from an individual with mild cognitive impairment. No immunoreactivity was observed in non-demented age-matched control frontal cortex (data not shown). Overall, the diffuse oligomer staining in AD sections was pericellular rather than intracellular, reminiscent of the description of the diffuse synaptic-type deposit observed with prion-associated diseases (Hainfellner et al., 1997; Kovacs et al., 2002).

Aβ oligomers are deposited extracellularly around neurons and are highly elevated in Alzheimer's disease cortex. Sections from frontal cortex of AD brain were immunolabeled for ADDLs using M94 antibody and visualized with either fluorescent- or peroxidase-conjugated secondary antibodies (A and B, respectively). Note the diffuse synaptic-type labeling surrounding the cell body of a single pyramidal neuron in both labeling conditions (arrows). No immunoreactive deposits are observed in nondemented controls (data not shown). Scale bar: A, B, 10 μm. C, Scatter plot of soluble Aβ levels measured by dot blot from two brain regions [cortex (square) and cerebellum (triangle)] form 9 subjects with AD (filled symbols) or 15 subjects without AD diagnosis (CTRL; open symbols). Brain samples were assayed by dot blot (inset) and analyzed by densitometry. Each point is the relative intensity average of duplicate measurements. The bar indicates the mean of each group (AD cortex, 2.281 ± 0.202; CTRL cortex, 0.206 ± 0.083; AD cerebellum, 0.263 ± 0.090; CTRL cerebellum, 0.097 ± 0.013; values represent mean ± SEM). This plot shows that the scatter of Aβ levels in AD cortex is larger than in control cortex, as well as being significantly elevated over control (p < 0.0001; AD vs CTRL). For the cerebellum, however, the difference between AD and CTRL Aβ levels was not as pronounced and not quite significant (p = 0.1316; AD vs CTRL). Mann-Whitney test was used and a one-tailed p value was established to test for significance using GraphPad InStat 3 software.
Dot blot immunoassays were used to verify the presence of oligomeric Aβ in soluble extracts of human frontal cortex and cerebellum (Fig. 1C, dot blot), with and without diagnosis of AD. Immunoreactivity was robust in all AD frontal cortex extracts. In controls, immunoreactivity was close to assay background for all cortex and cerebellum samples, with the exception of two cortical samples, from a subject with low levels of plaques and tangles (plaque severity 1; CERAD A; Braak II; NIA/Reagan low). The mean signal in AD cortical samples is elevated ∼11-fold (p < 0.0001), although the closeness of many control samples to assay background makes the magnitude of this ratio imprecise. In contrast to the cortical samples, levels of soluble oligomers in AD cerebellum were minimally greater, but not significantly, than in nondemented controls (p = 0.1316) (Fig. 1C, scatter plot).
These results confirm and extend the report that soluble oligomers are bona fide constituents of AD pathology (Gong et al., 2003). They verify, furthermore, that the antibodies used previously to characterize soluble oligomers from AD brain specifically recognize AD brain pathology. The data thus validate the use of these antibodies and soluble AD brain extracts in cell biological experiments, described below, that are designed to characterize the nature of interactions between ADDLs and neurons.
Aβ oligomers extracted from AD brain bind specifically to clustered sites
ADDLs are small, diffusible oligomers of Aβ, an amphipathic peptide. Given their solubility, it is likely that oligomers sequester their hydrophobic domains while presenting their hydrophilic domains to the aqueous environment. Such orientation is consistent with the immunoneutralization of ADDLs in solution by conformation-sensitive antibodies (Lambert et al., 2001); however, although ADDL structure theoretically seems competent to accommodate a ligand-like specificity for memory-related neurons, the reported insertion of Aβ into artificial lipid bilayers suggests that binding to cells could be highly nonspecific (McLaurin and Chakrabartty, 1996).
To resolve the issue of specificity, we investigated ADDL interactions with neurons under physiologically relevant conditions, using as our experimental model rat hippocampal neurons maintained in culture for at least 3 weeks. These cultures are synapse generating (Fong et al., 2002) and produce mature, highly differentiated neurons with complex arbors.
Our first binding experiments were done with extracts of human brain and with human CSF. Soluble extracts of AD cortex, previously shown to contain oligomers that are structurally equivalent to oligomers prepared in vitro (Gong et al., 2003), were incubated with cultured neurons. Unbound material was removed by washes, and cells were examined by immunofluorescence microscopy. ADDL distribution was determined using polyclonal antibodies (M94) generated by vaccination with synthetic Aβ oligomers. These antibodies bind to low doses of pathogenic Aβ oligomers but not physiological monomers (Lambert et al., 2001; Chang et al., 2003; Gong et al., 2003) and, as described above, are specific for AD brain tissue.
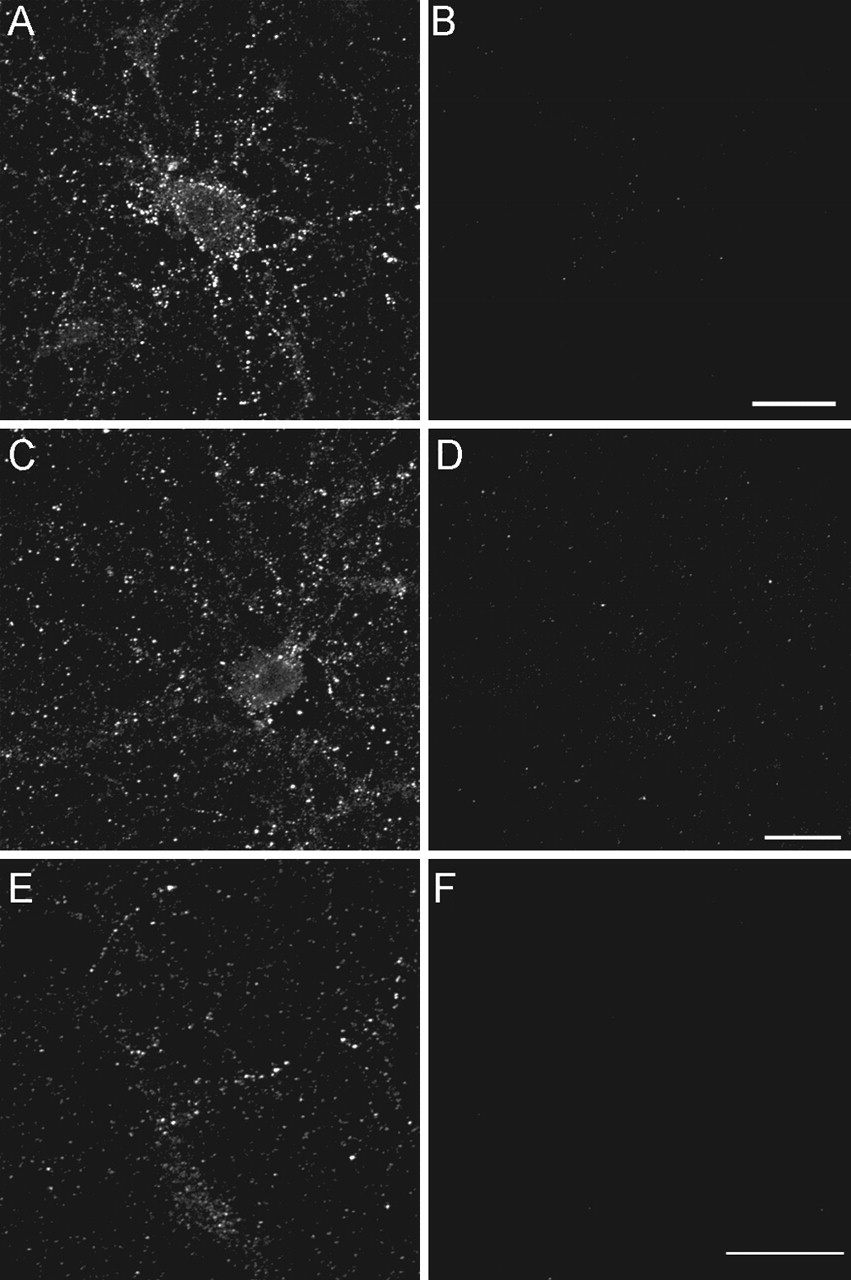
Incubation of extracts with well differentiated cultures of rat hippocampal cells, even for times as short as 5 min, resulted in membrane-type labeling along cell bodies and neurites (Fig. 2A). Under identical conditions, no signal was generated by extracts from age-matched nondemented controls (Fig. 2B). There was no indication that antibodies recognized physiological molecules such as Aβ monomer or amyloid precursor protein. Although Aβ1-42 has been reported to accumulate intracellularly in AD and in transgenic mouse models of AD (Gouras et al., 2000; Oddo et al., 2003), ADDL immunoreactivity on neurons was exclusively at cell surfaces, even after permeabilization. Distribution was distinctly punctate in nature. Centricon filter fractionation of AD extracts showed that binding activity resided with oligomers of MW between 10 and 100 kDa, consistent with previous characterization (Gong et al., 2003) (Fig. 2C,D). Unfractionated extracts of human CSF also exhibited binding activity that was AD dependent (Fig. 2E,F).

Aβ oligomers from AD brain bind neurons with punctate specificity. Primary hippocampal neurons were incubated for 30 min with soluble extracts from AD frontal cortex (A) or AD CSF (E). Some cultures were incubated with age-matched control cortical extract (B) or CSF (F). In some experiments, hippocampal neurons were incubated similarly with Centricon-fractionated soluble AD extract (see Materials and Methods) containing species with MW between 10 and 100 kDa (C) or species with MW ≤ 10 kDa (D). Unbound species were washed out, and ADDL attachment was assessed under nonpermeabilized immunolabeling conditions using the rabbit polyclonal oligomer-specific M94 antibody [as described by Lambert et al. (2001)]. Soluble extracts from AD brain (A) and CSF (E) contain ADDLs that bind selectively to neuronal surfaces with a punctate distribution. No labeling was detected with cortical extract (B) and CSF (F) from age-matched controls. Centricon filter fractionation of AD extracts containing species with an MW ranging from 10 to 100 kDa showed punctuate staining indistinguishable from unfractionated soluble extract, whereas binding species were not present in the fraction containing species with MW ≤ 10 kDa. Similar observations were obtained from three independent experiments.
Results show that human-derived ADDLs, from Alzheimer's brain and CSF, are capable of highly selective binding to neuronal cell surfaces characterized by punctate clusters of binding sites found in abundance within neuritic arbors.
Synthetic oligomeric species bind specifically to clustered sites
We next investigated the binding characteristics of ADDLs generated in vitro. Such preparations constitute the standard for investigating the neurological impact of oligomers. Use of these defined preparations eliminates unknown factors in extracts and CSF that could contribute to binding and its consequences. In addition, as tools for widespread use and convenient comparisons between laboratories, synthetic ADDLs provide a much more accessible preparation than human brain extracts or CSF.
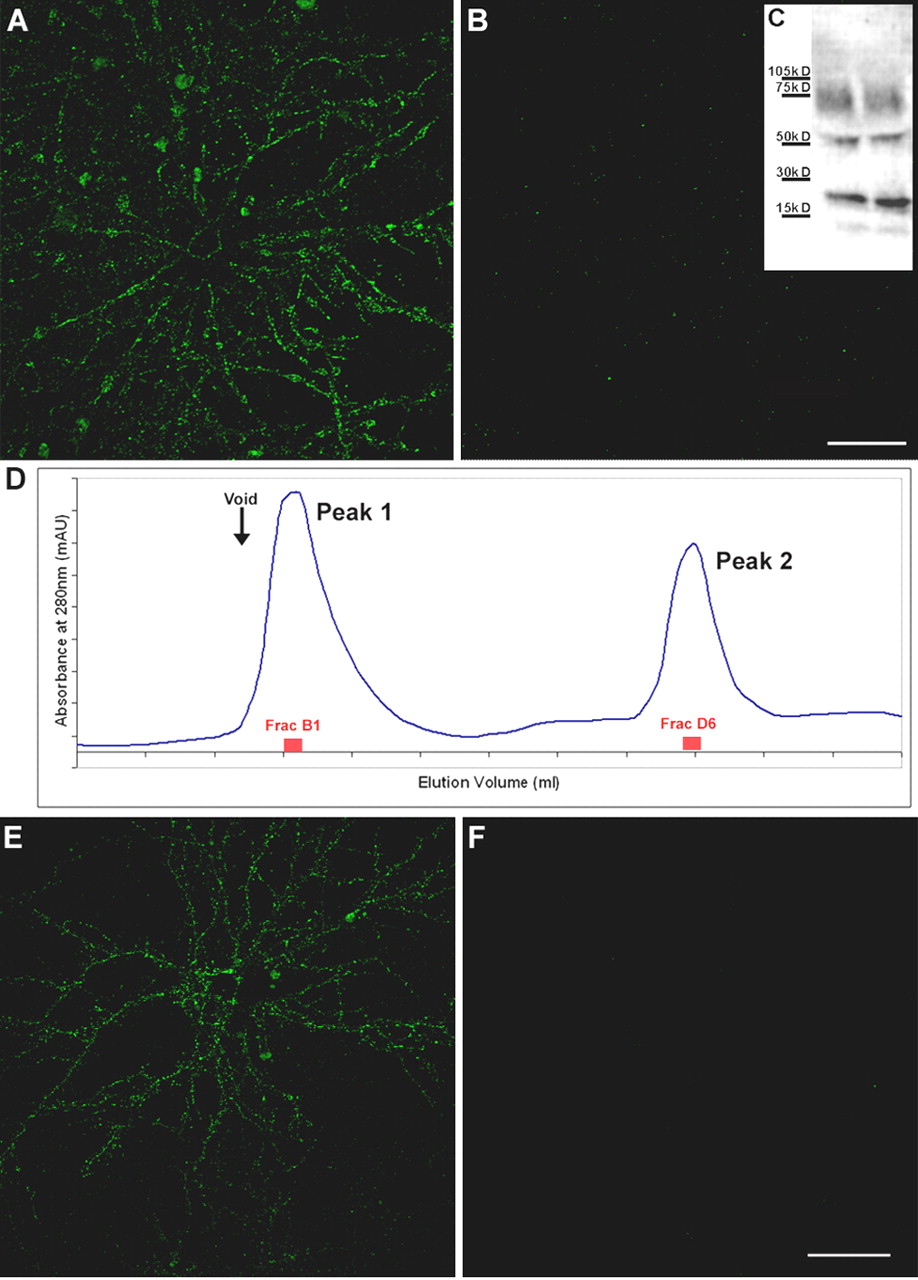
As observed with human preparations, ADDLs prepared in vitro and incubated with mature hippocampal neuronal cultures generated a specific binding pattern that exhibited abundant punctate sites within neuronal arbors. Preabsorption of antibodies with synthetic oligomers produced no detectable signal (data not shown), ruling out nonspecific antibody association. Immunolabeling using an oligomer-specific monoclonal antibody (Chromy et al., 2003) indicated that the ligands were not monomers or fibrils (data not shown), a conclusion substantiated by Centricon filter fractionation experiments. The 10-100 kDa Centricon fraction (Fig. 3A) but not the <10 kDa fraction (Fig. 3B) contained oligomers capable of binding to neuronal surfaces. As illustrated in Figure 3C, there is no change in the Aβ species present in the cultured media during incubation (up to 6 hr) with hippocampal cells nor appearance of species with molecular weights >24-mers.

Synthetic ADDLs, but not low molecular weight species, bind neurons analogously to AD-derived species. Primary hippocampal neurons were incubated with synthetic ADDLs fractionated by a Centricon filter (A, B) or biotinylated ADDLs separated by size exclusion chromatography (E, F) for 30 min [as described in Chromy et al. (2003)]. After removing unbound species by washing with fresh media, cell-bound ADDLs were assessed under nonpermeabilized immunolabeling conditions using M94 and Alexa 488-conjugated anti-rabbit secondary antibody (A, B) or Alexa 488-conjugated streptavidin (E, F). Confocal images demonstrate that oligomer immunoreactivity is at the plasma membrane of neurons and predominantly within dendritic arbors, although some binding to cell bodies is also evident. Punctate binding is reminiscent of receptor clusters and analogous to that of Alzheimer's Aβ oligomers (Fig. 2). As with fractionated soluble oligomers from AD brain, incubation of hippocampal neurons with synthetic ADDL species ranging from 100 to 10 kDa shows immunoreactive puncta (A), where as species ≤ 10 kDa, which would contain monomer and dimer, do not (B). Inset, Western blot of ADDL species present in the culture media after a 6 hr incubation with hippocampal neurons demonstrated that ADDLs are stable and contain no species with molecular weight >100 kDa (C). Lanes represent two separate culture media. Separation of biotinylated ADDLs (∼14 nmol of a 70 μm ADDL preparation) by SEC on Superdex 75 yielded two peaks (D). Histogram of elution volume versus absorbance at 280 nm depicted peak 1 at 8.1 ml with an absorbance at 16.9 mAU and peak 2 at 13.9 ml with an absorbance at 11.7 mAU. Fractions B1 and D6 with respective protein concentrations of 6.5 and 4 μm were incubated for 1 hr with mature hippocampal cells at a final concentration of 500 nm in parallel to an SEC-control fraction (taken between peaks 1 and 2). Binding of biotinylated species was detected with Alexa Fluor 488-conjugated streptavidin. Hot spots of fluorescence were observed exclusively with peak 1 fraction B1 (E), consistent with species of molecular weight >50 kDa such as 12-mers. No fluorescence was seen with the peak 2 fraction D6 (F) or the control fraction (data not shown). Confocal images were acquired with constant confocal microscope settings (laser power, filters, detector gain, amplification gain, and amplification offset). Images are representative of three different experiments. Scale bar, 40 μm.
Additionally, neuronal binding of Aβ species was examined after separation by HPLC size-exclusion chromatography on Superdex 75 [as described in Chromy et al. (2003)]. Biotinylated ADDLs, prepared from biotin-Aβ1-42 peptide, eluted in two peaks (Fig. 3D). Calibrated against known molecular weight standards, peak 1 contained species of an apparent MW >50 kDa, consistent with 12-mers and larger species, whereas peak 2 contained monomers and small oligomers. Dot blot, a method that detects all forms of assembled Aβ, was performed as in Figure 1 on the eluted fractions to identify fractions with the highest levels of immunoreactive material (data not shown). These fractions were then tested for binding capacity on mature hippocampal cell cultures. The high molecular weight Aβ species contained in peak 1 showed binding to hippocampal dendritic trees (Fig. 3E), whereas low molecular weight species from peak 2 did not bind (Fig. 3F).
Thus, results from fractionation by Centricon filters or chromatography show that the immunoreactivity imaged on neurons was not attributable to large molecules such as protofibrils or to small molecules such as monomers or dimers.
Selective binding by experimentally generated oligomers: clusters of binding sites and cell-to-cell specificity
Contrary to what would be expected if ADDLs bound by nonselective membrane adsorption, all cells did not exhibit punctate clusters of binding sites. Cell-to-cell specificity in a double-label experiment is illustrated for a pair of αCaMKII-positive neurons (Fig. 4A,B), only one of which exhibits ADDL binding. Over many experiments, the subpopulation of neurons that bound ADDLs typically comprised 30-50% of the total in a given culture. These results establish the specificity of ADDL binding at the cellular level.

ADDL binding is cell specific. Double-labeling immunofluorescence studies were performed on mature hippocampal neurons (21 DIV) with mouse monoclonal anti-αCaM kinase II and rabbit polyclonal anti-ADDL (M94) and visualized with Alexa Fluor 594 (red) and Alexa Fluor 488 (green) secondary antibodies, respectively (A, B). Similar double-labeling experiments were conducted with mouse monoclonal anti-PSD-95 (C, red) and anti-ADDL (green). Overlays (B, C) of three-dimensional reconstructed images of confocal z-series scans (taken at 0.5 μm steps) show that ADDLs bind selectively to some αCaMKII-positive neurons (overlap appears yellow), depicted here after 6 hr incubation with ADDLs. Similar cell selectivity was observed with the PSD-95 labeling. Note that only one of the two neurons is targeted by ADDLs. Similar cell-specific binding and colocalization between ADDLs and αCaMKII- or PSD-95-positive neurons was observed after 30 min incubation of ADDLs (data not shown). DIV, Days in vitro. Scale bar, 20 μm.
Clusters of ADDL binding sites are coincident with synapses
At the subcellular level, whether ADDLs bind specifically to synapses is of great significance to the hypothesis that memory loss in AD is an oligomer-induced synaptic failure (Lambert et al., 1998; Selkoe, 2002). The rapidity with which ADDLs inhibit synaptic plasticity (Lambert et al., 1998; Walsh et al., 2002) suggests that the neurologically relevant binding might occur near synapses, whereas binding that specifically targets particular synapses would not only account for memory specificity in AD but would also confer considerable constraints on the mechanism. Localization of ADDLs to punctate binding sites within dendritic arbors described above clearly is consistent with the hypothesis that ADDLs are synapse-specific ligands; however, the morphology of punctate binding sites also is consistent with other subcellular specializations such as membrane rafts or focal contacts.
To test further the nature of oligomer binding sites, we examined colocalization with PSD-95. PSD-95 is a critical scaffolding component of postsynaptic densities found in excitatory CNS signaling pathways (Sheng and Pak, 1999), and clusters of PSD-95 have been established previously as definitive markers for postsynaptic terminals (Rao et al., 1998). In mature hippocampal cell cultures, such as used here, essentially all clusters of PSD-95 occur at synapses (Allison et al., 2000). As predicted, ADDL binding sites show striking coincidence with PSD-95 puncta, shown in an overlay at low magnification (Fig. 4C). Overlay analysis at higher magnification indicated that the ADDL binding sites colocalized almost exclusively with puncta of PSD-95 (Fig. 5A-C). Identical patterns were obtained with extracts of AD brain (data not shown). ADDL binding sites also were juxtaposed to synaptophysin-positive presynaptic terminals (Fig. 5D), although complete coincidence of ADDL and synaptophysin immunoreactivities was uncommon. To verify the apparent synaptic targeting by ADDLs, the extent of colocalization between ADDLs and PSD-95 was quantified by image analysis. Quantification of 14 fields showed that synthetic ADDLs colocalized with PSD-95 in 93 ± 2% of the sites (Fig. 5F,H). ADDL binding sites thus were almost completely localized to synapses. These sites were selective, furthermore, for a synaptic subpopulation: only half of the PSD-95 puncta colocalized with ADDL binding sites (Fig. 5E,G). The synapses targeted by ADDLs thus comprised approximately half of those that were present.

ADDLs specifically target a subset of PSD-95-positive terminals. Hippocampal neurons treated with ADDLs were double immunolabeled for PSD-95 (A, red) and ADDLs (B, green). Overlay of the confocal reconstructed z-series scans shows that dendritic clusters of ADDL-immunoreactive puncta colocalized almost completely with PSD-95, as seen by the amount of yellow puncta in the merged image (C). The degree of colocalization between ADDLs and PSD-95 was quantified using MetaMorph software. Bar graphs show the number of PSD-95 sites targeted by ADDLs (E) and the number of ADDL binding sites colocalized with PSD-95 (F) for 14 different fields (40× objective). Graph (E) shows that many PSD-95 sites are unoccupied by ADDLs (yellow bar, PSD-95 colocalized with ADDLs; red bar, PSD-95 without ADDLs) (mean total oligomer binding sites per field was 1062 ± 125; mean oligomer sites that colocalized with PSD-95 was 971 ± 105). Graph (F) shows for each field the proportion of ADDL puncta localized to PSD-95 synaptic sites. The number of ADDLs at PSD-95 sites (yellow bar) greatly exceeds ADDLs at nonsynaptic sites (green bar). Pie charts illustrate the distribution summed for all fields, showing the fraction of PSD-95 sites occupied by ADDLs (G) and the fraction of ADDL binding sites colocalized with PSD-95 (H). Results are presented as an average percentage ±SEM for all 14 different fields (mean total sites per field was 1960 ± 174, with 50 ± 2% not associated with oligomer binding sites). In summary, one-half of PSD-95 puncta are targeted by ADDLs (G), whereas >90% ADDL puncta colocalize with PSD-95 (H). Analyses verify that ADDLs specifically localize to a subset of synaptic sites. Scale bar, 10 μm. D, Inset, Image of hippocampal neurons showing ADDLs (green) juxtaposed to presynaptic marker, synaptophysin (red), rather than superimposed, as above.
ADDL binding sites, moreover, were found to overlap with NMDA receptor (NR1) immunoreactivity (data not shown), consistent with the association of PSD-95 and NMDA glutamate receptors in excitatory hippocampal signaling pathways (Sheng and Pak, 1999). ADDLs were also highly colocalized with PSD-family proteins and spinophilin (data not shown). No overlap was evident between ADDLs and GluR1 (when using GluR1 C-terminal antibody, which recognizes receptors expressed in dendritic shafts), phosphorylated tau (using Tau-1, an axonal marker), and SorLa (sorting protein-related receptor containing LDLR class A repeats; also called apolipoprotein E receptor LR11; a gift from Dr. H. C. Schaller, University of Hamburg, Hamburg, Germany). Colocalization thus was selective for synaptic markers.
The molecular basis for specific synaptic targeting by ADDLs is not known, although previous studies with the B103 CNS neuronal cell line indicated specific binding to trypsin-sensitive cell-surface proteins in flow cytometry experiments (Lambert et al., 1998). A wide range of candidate proteins accumulate at PSD-95 synapses, including receptors for neuro-transmitters, trophic factors, adhesion molecules, and extracellular matrix proteins. In ligand overlay assays, which have the potential to detect binding to particular proteins that have been separated by SDS-PAGE, ADDLs bind with high affinity to two membrane-associated proteins from hippocampus and cortex (molecular weight 140 and 260 kDa) (Gong et al., 2003). These proteins are also significantly enriched in isolated synaptosomes (800-900%) (D. Khuon, personal communication).
Oligomers ectopically upregulate the synaptic memory-linked immediate-early gene protein “Arc”
Synaptic relevance of the puncta is consistent with the striking specificity of ADDL binding to the highly arborized αCaMKII-positive neuron shown in Figure 6A. This neuron shows discretely clustered sites found predominantly on dendrites; punctate binding is detectable on the cell body but at a much lower density. At higher magnification (Fig. 6B), the composite overlays show numerous oligomer puncta capping the αCaMKII-positive dendritic spines. αCaMKII is known to accumulate in postsynaptic terminals of neurons linked to memory function, where it comprises >30% of the protein in spiny postsynaptic terminals (Inagaki et al., 2000). The frequent localization of ADDL binding sites to dendritic spines suggests a potential for rapid impact on spine molecules.

Localization of ADDL binding sites to dendritic spines. Highly differentiated hippocampal cells (21 DIV) treated with synthetic ADDLs for 1 hr were double immunolabeled for ADDLs (green) and αCaMKII (red). An ADDL-bound αCaMKII-positive neuron is pictured (A). Higher magnification illustrates that many of the ADDL-IR puncta codistributed with αCaMKII-enriched dendritic spines (B). As indicated by arrows, ADDLs mainly targeted dendritic spines. Image is representative of three separate trials. Scale bars: A, 40 μm; B, 8 μm.
To investigate this possibility further, we tested the impact of oligomer binding on a synaptic immediate-early gene mechanism critically linked to long-term memory formation. The gene of interest codes for Arc (activity-regulated cytoskeletal-associated) protein (Guzowski et al., 2000). Arc mRNA is targeted to synapses where, physiologically, the protein is induced transiently by synaptic activity (Link et al., 1995; Lyford et al., 1995; Steward and Worley, 2001). Animal model studies have shown that appropriate Arc expression is essential for LTP and long-term memory formation (Guzowski et al., 2000). Besides being linked to drug abuse and sleep disruption (Cirelli and Tononi, 2000; Freeman et al., 2002), the ectopic and aberrant expression of Arc has been predicted to cause failure of long-term memory formation (Guzowski, 2002).
Hippocampal neurons were treated with synthetic oligomers or vehicle for 5 min and 1 and 6 hr, and the impact on Arc protein was determined by immunofluorescence and immunoblot. Synthetic ADDLs were used because, as discussed previously, they are obtained more readily than AD-derived species and uncontaminated by the myriad unknowns present in soluble brain extracts. At the earliest time point (5 min), double-label immunofluorescence revealed that oligomer binding colocalized with dendritic punctate Arc expression (Fig. 7). This location appears to be an ectopic induction, because low levels of Arc protein that are expressed constitutively are known to localize in cell bodies and not at synapses (Steward and Worley, 2001).

Rapid ADDL-induced synaptic expression of the immediate-early gene Arc. Hippocampal neurons were exposed to synthetic ADDLs for 5 min and then labeled for Arc (red) and ADDLs(green). Merging of the images shows points of colocalization between ADDL puncta and Arc-positive synapses (yellow). Scale bar, 8 μm.
After longer exposure to ADDLs, the expression of Arc exhibited a robust upregulation. Expression of Arc throughout spines and dendrites was striking after 1 hr (Fig. 8B) and remained so after 6 hr (Fig. 8D) compared with vehicle-treated controls (Figs. 8A,C). Elevated Arc expression also was evident in immunoblots (Fig. 8A,B, inset), with low levels of Arc-IR in controls consistent with minimal basal Arc expression in neuronal cell bodies. The ADDL-induced increase in Arc was fivefold over vehicle-treated cultures.

ADDLs promote sustained upregulation of Arc. Hippocampal cells treated with vehicle (A, C) or ADDLs (B, D) for 1 hr (A, B) or 6 hr (C, D) were fixed, permeabilized, and labeled for Arc protein. Immunofluorescence demonstrated a large ADDL-induced increase in Arc expression throughout the dendrites and dendritic spines of a subset of neurons; in vehicle-treated cells, Arc expression is restricted to the eneuronal cell body. Inset represents extracts from hippocampal cells treated with vehicle (-) or ADDLs (+) for 1 hr that were immunoblotted after SDS-PAGE using an Arc polyclonal antibody. Immunoblots show increased concentration of Arc in ADDL-treated (+) compared with vehicle-treated (-) cell extracts. Cylophilin B (cyclo) was used to normalize with respect to protein loading; ratio of Arc to cyclo IR was 0.70 ± 0.11 in controls and 3.51 ± 0.76 in oligomer-treated samples (n = 4; p = 0.01 using t test; paired 2 sample for means). Blot is representative of four independent experiments. Hippocampal cells also were treated for 6 hr with vehicle (C) or ADDLs (D) and permeabilized and labeled for Arc. High-magnification confocal images of vehicle-treated neurons show that Arc-immunoreactive puncta were localized to the dendritic shaft with only a few spine heads reaching an intensity level higher than that found in the dendritic shaft. The dendrites of ADDL-treated neurons show a punctate pattern of intense Arc-IR concentrated in dendritic spine heads as well as upregulated Arc-IR throughout the dendritic shafts and spines. Controls for the primary antibodies showed no labeling. Identical results were obtained in three independent trials. Scale bars: A, B, 20 μm; C, D, 6 μm.
Arc protein is coupled to F-actin and linked functionally to spine morphology, and its chronic overexpression has been suggested to generate abnormal spine structure (Kelly and Deadwyler, 2003). Examination of spine morphology in the current experiments indicated that Arc-positive spines in oligomer-treated groups differed from the few Arc-positive spines incontrol groups. Control spines expressing a low level of Arc were stubby and lay close along dendritic shafts (Fig. 8C), whereas ADDL-treated spines were longer andappeared to extend from the dendritic shaft (Fig. 8D). Similar protruded spine structures were evident in treated cultures immunolabeled with anti-spinophilin (data not shown).
Discussion
Soluble Aβ oligomers are neurologically harmful molecules that accumulate in AD brain (Gong et al., 2003). The current data show that ADDLs act as specific ligands for synaptic terminals, where they disrupt normal expression of a synaptic immediate-early gene essential for long-term memory formation. The data provide a new molecular mechanism to support the emerging hypothesis that early AD memory loss is an oligomer-induced synapse failure, independent of neuron death and amyloid fibrils (Lambert et al., 1998; Klein et al., 2001; Hardy and Selkoe, 2002; Kirkitadze et al., 2002; Walsh and Selkoe, 2004).
Aβ oligomers have been implicated previously in AD memory loss. Roher and colleagues (Kuo et al., 1996) first showed that soluble oligomers are elevated in AD, although they initially were thought to be neurologically irrelevant, existing only as transient species en route to formation of toxic amyloid fibrils. Oligomers now are established as metastable molecules that exist for prolonged periods without conversion to fibrillar structures (Chromy et al., 2003), and they are known to be potent CNS neurotoxins. Most relevant to early AD, oligomers inhibit LTP, an impact that is rapid, selective, and nondegenerative, (Lambert et al., 1998; Walsh et al., 2002; Wang et al., 2002). Such impairment of synaptic plasticity may account for plaque-independent cognitive failures seen in hAPP transgenic mice (Hsia et al., 1999; Mucke et al., 2000; Buttini et al., 2002; Van Dam et al., 2003), which accumulate oligomers in an age-, region-, and transgene-dependent manner (Chang et al., 2003). Moreover, oligomers likely are the targets of therapeutic antibodies that reverse memory loss in hAPP mice, a recovery that is both rapid and unrelated to plaque burden (Dodart et al., 2002; Kotilinek et al., 2002).
In AD brain sections, oligomer-specific antibodies have identified antigens at locations distinct from neuritic plaques (Kayed et al., 2003), establishing the in situ presence of oligomers independent of fibrils. Moreover, current data show a perineuronal distribution, indicating that oligomers likely accumulate within dendritic arbors. Oligomers extracted from AD brain were found previously to attach selectively to dendrites in culture (Gong et al., 2003). Fractionation of extracts shows the size of these dendritic ligands to be between 10 and 100 kDa, consistent with prominent 12-mers (56 kDa) previously observed in two-dimensional gel immunoblots. Overall, AD brain 12-mers are indistinguishable from 12-mers in synthetic oligomer preparations with respect to isoelectric point, recognition by conformation-sensitive antibody, and selective dendritic binding.
Colocalization data presented here establish that the dendritic targeting of oligomers occurs at synaptic terminals. Oligomers, whether synthetic or from AD brain, colocalized with PSD-95, found exclusively at synapses in mature hippocampal cultures (Rao et al., 1998). Whether diffuse amyloid immunoreactivity in AD brain has a synaptic origin is under investigation. In culture, oligomer binding is highly selective, targeting only certain neurons and synapses, but the particular phenotypes that are targeted remain to be established. The targeted synapses, however, contain glutamate receptors (P. Lacor and M. Buniel, unpublished observations), and preliminary competition experiments (K. Viola, D. Khuon, and Y. Gong, unpublished observations) suggest the hypothesis that glutamate receptors themselves might mediate synapse-specific ADDL targeting. Other candidates also have been proposed, however, and the identity of oligomer binding sites is still unknown (Verdier et al., 2004).
If synapses were targeted in situ, the impact on memory ultimately would depend on the number of synapses targeted, the extent to which individual synapses are compromised, and the relevance of affected synapses to the overall process of memory formation. Synaptic impact could even be reversed spontaneously if ligands dissociated or their binding sites were turned over. Such complexities, although in harmony with the concept of synaptic reserve and day-to-day fluctuations in cognitive function, make it difficult to predict a simple relationship between oligomer levels and memory loss. It seems likely, however, that a threshold of occupancy must be surpassed before memory loss would manifest.
The response of Arc to oligomers is particularly intriguing because of the putative involvement of Arc in long-term memory formation (Guzowski, 2002). Physiologically, Arc expression is controlled by patterned synaptic activity (Link et al., 1995; Lyford et al., 1995) and occurs in recently activated dendritic spines (Moga et al., 2004). In dendrites, Arc mRNA localizes to synaptic spines, consistent with the initial colocalization of Arc and oligomers. Although Arc mRNA decreases with age in a tg-mouse AD model, the relevance to synaptic Arc protein expression was not determined (Dickey yet al., 2003). In the current study, oligomers caused sustained Arc induction, leading to ectopic protein diffusion throughout the dendritic arbor. Normally, Arc protein functions in a transient manner, and it has been proposed that sustained Arc expression would generate synaptic noise and thereby inhibit long-term memory formation (Guzowski et al., 2000). Supporting this prediction, findings with Arc tg-mice indicate that chronically elevated Arc is linked to slow learning (Kelly and Deadwyler, 2003).
How ectopic Arc could lead to synapse failure is unknown but may involve spine shape or receptor trafficking. It has been proposed, e.g., that elevated Arc in tg-mice may interfere with learning by making spines more rigid (Kelly and Deadwyler, 2003), consistent with the association of Arc with cytoskeletal and postsynaptic proteins (Lyford et al., 1995; Fujimoto et al., 2004). Changes in spine structure may rapidly alter synaptic signal processing and related information storage (Crick, 1982; Rao and Craig, 2000; Yuste and Bonhoeffer, 2001), and spine abnormalities are common to various brain dysfunctions (Fiala et al., 2002), including mental retardation, in which spines are atypically bent and protruding (Kaufmann and Moser, 2000). Elevated Arc also could disrupt cycling of receptors required for synaptic plasticity, e.g., blocking upregulation of AMPA receptors. Consistent with Arc cell biology, this disruption could derive from effects on cytoskeletal organization (e.g., f-actin or PSD proteins) or signaling pathways (e.g., via CaMKII) through a mechanism that may concomitantly alter spine structure.
Other synaptic signal transduction pathways also are affected by oligomers in culture models. In cortical cultures, low doses of oligomers inhibit glutamate-stimulated cAMP response element-binding protein phosphorylation (Tong et al., 2001), a signaling pathway associated with synaptic plasticity (Sweatt, 2001). In hippocampal slice cultures used to study the impact of oligomers on LTP, pharmacological studies suggest roles for several protein kinases. Potentially of great interest, inhibitors of p38MAPK (p38 mitogen-activated protein kinase), JNK (c-Jun N-terminal protein kinase), and cdk5 (cyclin-dependent kinase 5) block the ability of oligomer to inhibit LTP, as do antagonists of type 5 metabotropic glutamate receptors (Wang et al., 2004). The authors also suggest putative involvement of unknown oligomer receptor(s). Whether the signaling events described above are parallel or sequential with respect to the impact of oligomers on Arc remains to be determined.
The action of ADDLs as pathogenic synaptic ligands provides an intuitively appealing hypothesis for AD synapse failure. A key facet is that synapses themselves are targeted, giving a parsimonious mechanism in which memory-initiating events could be specifically and locally disrupted. There is no need to explain how memory-specific dysfunction might derive from nonspecific cellular associations (e.g., random insertion into cell membranes) (Gibson et al., 2003). Ultimately, with prolonged exposure, the synapses targeted by oligomers may undergo physical degeneration. It has been proposed that oligomers account for plaque-independent loss of terminals in some transgenic models of early AD (Mucke et al., 2000), potentially involving the synaptic signaling molecule Fyn (Chin et al., 2004). Extensive precedents from studies of fibrillar preparations suggest as well the possible involvement of oxidative stress and calcium dysfunction (Butterfield, 2003; Mattson, 2004), whereas recent experiments with Caenorhabditis elegans containing inducible Aβ1-42 indicate that oxidative stress can precede fibrillogenesis (Drake et al., 2003). Possible early-stage synaptic deterioration may explain age-dependent decreases in Arc mRNA in some tg-mice, although frank loss of terminals does not appear to occur in these strains (Dickey et al., 2003). It also has been hypothesized that damage initiated at synapses could lead to neuron death (Klein, 2004; Mattson, 2004).
In end-stage AD, cognitive degeneration extends far beyond memory loss (Coyle, 1987). Ideally, neutralization of early pathogenic events might prevent this downstream catastrophic cascade. Results from human vaccine trials indicate that the therapeutic antibodies that target Aβ-derived neurotoxins might indeed halt disease progression (Hock et al., 2003), although the incidence of significant brain inflammation with active vaccines complicates this strategy (Schenk, 2002; Weiner and Selkoe, 2002). Passive vaccination, although more expensive, would have fewer side effects and also overcome the problem of compromised immune response common in the elderly. Monoclonal antibodies that immunoneutralize soluble Aβ species have been shown in two independent studies to reverse memory loss in tg-mice models of early AD (Dodart et al., 2002; Kotilinek et al., 2002). It has been possible, moreover, to use ADDLs to generate antibodies that are specific for toxic forms of Aβ with minimal affinity for physiological monomers (Lambert et al., 2001). Recent hybridoma work further indicates that it is possible to generate antibodies that bind oligomers but not amyloid fibrils, reducing concerns of inflammation caused by antibodies bound to plaques (Chromy et al., 2003). The prospects for developing human therapeutic antibodies that target memory-relevant Aβ assemblies thus seem encouraging.
Footnotes
This work was supported by grants from the Illinois Department of Public Health (P.N.L.); the Alzheimer's Disease Research Fund; National Institutes of Health-National Institute on Aging Grants RO1-AG18877, RO1-AG22547, and RO1-AG11385 (W.L.K.), RO3-AG022237 (P.N.L.), and P30AG13854 (Northwestern Alzheimer's Disease Center); the Boothroyd, Feiger and French Foundations; and the Institute for the Study of Aging. We are grateful to Pierce Cain for assistance with immunoblotting and William Russin of the Biological Imaging Facility of Northwestern University.
Correspondence should be addressed to William L. Klein, Northwestern University, Neurobiology and Physiology Department, O. T. Hogan Biological Sciences, 5-110, 2205 Tech Drive, Evanston, IL 60208. E-mail: wklein{at}northwestern.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410191-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}