

Abstract
Glutamate receptor (GluR) δ2 selectively expressed in cerebellar Purkinje cells (PCs) plays key roles in long-term depression (LTD) induction at parallel fiber (PF)–PC synapses, motor learning, the matching and connection of PF–PC synapses in developing and adult cerebella, the elimination of multiple climbing fibers (CFs) during development, and the regulation of CF territory on PCs. However, it remains unsolved how GluRδ2 regulates cerebellar synaptic plasticity, PF–PC synapse formation, and CF wiring. One possible signaling mechanism through GluRδ2 is signaling by protein–protein interactions. The C-terminal region of GluRδ2 contains at least three domains for protein–protein interactions. The PDZ (postsynaptic density-95/Discs large/zona occludens 1)-binding domain at the C terminal, named as the T site, interacts with several postsynaptic density proteins. Here, we generated GluRδ2ΔT mice carrying mutant GluRδ2 lacking the T site. There were no significant differences in the amount of receptor proteins at synapses, histological features, and the fine structures of PF–PC synapses between wild-type and GluRδ2ΔT mice. However, LTD induction at PF–PC synapses and improvement in the accelerating rotarod test were impaired in GluRδ2ΔT mice. Furthermore, CF territory expanded distally and ectopic innervation of CFs occurred at distal dendrites in GluRδ2ΔT mice, but the elimination of surplus CF innervation at proximal dendrites appeared to proceed normally. These results suggest that the C-terminal T site of GluRδ2 is essential for LTD induction and the regulation of CF territory but is dispensable for PF–PC synapse formation and the elimination of surplus CFs at proximal dendrites during development.
- cerebellum
- climbing fiber territory
- glutamate receptor δ2
- long-term depression
- parallel fiber synapse
- PDZ-binding domain
Introduction
The pattern of intrinsic neural connections in the cerebellum is known in considerable detail (Altman and Bayer, 1997). Various studies have suggested the important roles of the cerebellum in fine motor control and motor learning (Christian and Thompson, 2003; Boyden et al., 2004). These features make the cerebellum an ideal system for studying the molecular and cellular mechanisms of brain wiring and function. We found the δ subfamily of the glutamate receptor (GluR) by molecular cloning (Yamazaki et al., 1992). From the amino acid sequence identity, the GluRδ subfamily positions between the classical AMPA/kainate and NMDA subtypes of GluR. The second member of this subfamily, GluRδ2, is selectively expressed in cerebellar Purkinje cells (PCs) (Araki et al., 1993; Lomeli et al., 1993). In PCs, GluRδ2 is exclusively localized at parallel fiber (PF)–PC synapses (Takayama et al., 1996; Landsend et al., 1997). Long-term depression (LTD) at PF–PC synapses, motor learning, and motor coordination are impaired in GluRδ2 mutant mice (Funabiki et al., 1995; Kashiwabuchi et al., 1995; Kishimoto et al., 2001). Furthermore, a significant number of PC spines lack synaptic contacts with PF terminals (Kashiwabuchi et al., 1995; Kurihara et al., 1997). In addition, multiple climbing fiber (CF) innervation to PCs is sustained and CF territory expands distally in GluRδ2 mutant mice (Kashiwabuchi et al., 1995; Hashimoto et al., 2001; Ichikawa et al., 2002). Inducible ablation of GluRδ2 has shown a key role of GluRδ2 in matching and connecting PF–PC synapses in the adult brain (Takeuchi et al., 2005).
However, it remains unsolved how GluRδ2 regulates cerebellar synaptic plasticity, PF–PC synapse formation and CF wiring. There is no evidence for GluRδ2 channel activities, although lurcher mutation transformed GluRδ2 to constitutively active channels (Zuo et al., 1997). One possible signaling mechanism through GluRδ2 is signaling by protein–protein interactions. The C-terminal region of GluRδ2 contains at least three domains for protein–protein interactions (Roche et al., 1999; Uemura et al., 2004; Yawata et al., 2006). The PDZ [postsynaptic density (PSD)-95/Discs large/zona occludens 1]-binding domain at the very end of the C terminal, designated as the T site, interacts with PSD-93, PTPMEG (protein tyrosine phosphatase), Delphilin, nPIST (Golgi-resident PDZ protein), and S-SCAM (synaptic scaffolding molecule) (Roche et al., 1999; Hironaka et al., 2000; Miyagi et al., 2002; Yue et al., 2002; Yap et al., 2003). In the middle of the C-terminal region, there is the domain that interacts with Shank scaffold proteins, designated as the S segment (Uemura et al., 2004). The membrane-proximal domain of the C-terminal region of GluRδ2 interacts with protein interacting with C kinase 1 (Yawata et al., 2006). Here, we generated GluRδ2ΔT mice carrying mutant GluRδ2 lacking the T site to investigate the molecular mechanisms underlying diverse GluRδ2 functions. The C terminal truncation impaired LTD induction at PF–PC synapses and caused CF territory expansion but had little effect on PF–PC synapse formation and elimination of surplus CFs at proximal dendrites. These results suggest that signaling through the T site of GluRδ2 underlies the heterosynaptic competition between PF and CF.
Materials and Methods
Generation of mutant mice with GluRδ2 lacking the C terminus.
We identified a bacterial artificial chromosome (BAC) clone, RP23-37704 (BACPAC Resources Center, Oakland, CA) prepared from the C57BL/6 strain, carrying the C-terminal region of GluRδ2 using basic local alignment search tool searches against the mouse genome sequence database. The 11.9 kb genomic DNA fragment carrying exon 16 encoding the C-terminal region of GluRδ2 was introduced into the pMC1DTpA (Taniguchi et al., 1997) by Red/ET recombination using BAC subcloning kit (Gene Bridges, Dresden, Germany) to yield pδ2–11.9. Genomic DNA fragment carrying the exon 16 of GluRδ2 gene having a deletion of the 21 nucleotides encoding the C-terminal 7 aa residues (T site) was prepared by PCR and introduced into pδ2–11.9 by Red/ET recombination to yield pδ2–11.9ΔT. The 3.0 kb DNA fragment carrying phosphoglycerate kinase Pgk-1 promoter-driven neomycin phosphotransferase gene (neo) and Tn5 promoter-driven chloramphenicol-acetyltransferase gene (cam) flanked by two Flp recognition target (flp) sites was inserted into 1.6 kb downstream of exon 16 of pδ2–11.9ΔT by Red/ET recombination to yield pTVΔT. The targeting vector pTVΔT contained exon 16 having a deletion of the T site, neo and cam genes flanked by two frt, the 5.9 kb upstream and the 3.0 kb downstream genomic sequences, and 4.3 kb pMCDTpA. The targeting vector pTVΔT was linearized by NotI and electroporated into embryonic stem (ES) cell line RENKA derived from C57BL/6N strain (Mishina and Sakimura, 2007) as described previously (Takeuchi et al., 2005). G-418 (150 μg/ml)-resistant clones were picked, and targeted clones were identified by Southern blot hybridization analysis using PCR-amplified 709 bp fragment, PCR-amplified 793 bp fragment, and 0.6 kb PstI fragment from pLFNeo (Takeuchi et al., 2002) as 5′, 3′, and neo probes, respectively. The 5′ probe and 3′ probes were amplified by PCR using 5′-CCATGGTTGCAGGGACGC-3′ and 5′-CCATGGGAAGACTTTGGTTG-3′, 5′-AGATCTCCAAGAAAAAAGATAG-3′ and 5′-GAATTCAGATTCATCTGCTTC-3′ as primers, respectively. Recombinant ES cells were injected into eight-cell stage embryo of CD-1 mouse strain. The embryos were cultured to blastocysts and transferred to the pseudopregnant CD-1 mouse uterus. Resulting chimera mice were mated to C57BL/6N mice to yield heterozygous (GluRδ2+/ΔT) mice. Homozygous GluRδ2ΔT/ΔT and GluRδ2+/+ mice were obtained by crossing heterozygous pairs. The GluRδ2+/ΔT allele was identified by PCR using primers 5′-GGCTCAATCTGGGCAATGAC-3′ (P1) and 5′-AAATGGAGGGTGATAGACAGAG-3′ (P2). Animal care was performed in accordance with institutional guidelines. Mice were fed ad libitum with standard laboratory chow and water in standard animal cages under a 12 h light/dark cycle. All animal procedures were approved by the Animal Care and the Use Committee of Graduate School of Medicine, the University of Tokyo (Approval number 1721T062), and the Animal Care and Use Committee of Hokkaido University (Approval number 17-96). For subsequent analyses, we used GluRδ2+/+ and GluRδ2ΔT/ΔT mice of 4 weeks old unless otherwise specified.
Western blot analysis.
Homogenates and PSD fractions were prepared essentially according to the procedure as described previously (Carlin et al., 1980; Cho et al., 1992). Triton X-100-treated synaptosomes were centrifuged (1 h, 70,000 × g) to obtain the PSD fraction (Steigerwald et al., 2000). Proteins were separated by SDS-PAGE and analyzed by Western blot with anti-GluRδ2 (Araki et al., 1993), anti-GluRδ2 (C-19) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-GluRε1 (Watanabe et al., 1998), or anti-neuron-specific enolase (NSE) (Chemicon, Temecula, CA) as described previously (Uemura et al., 2004). For quantitative analysis, signals were analyzed by LAS-1000plus image analyzer (Fujifilm, Tokyo, Japan) with Multi Gauge version 3.1 software (Fujifilm).
Histology and immunohistochemistry.
Under deep pentobarbital anesthesia (100 μg/g body weight, i.p.), mice were perfused transcardially with 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB), pH 7.2, for immunohistochemistry and postembedding immunogold electron microscopy, or 2% paraformaldehyde/2% glutaraldehyde in sodium cacodylate buffer, pH 7.2, for conventional electron microscopy. Sections were prepared with microslicer (VT1000S; Leica, Wetzlar, Germany) for immunofluorescence (50 μm in thickness) and for electron microscopy (400 μm in thickness). Sections were stained with guinea pig anti-vesicular glutamate transporter 2 (VGluT2) (0.5 μg/ml) antibody (Miyazaki et al., 2003) and rabbit anti-calbindin serum (1:10,000 dilution) (Nakagawa et al., 1998), followed by incubation with Alexa Fluor 488-conjugated (Invitrogen, Carlsbad, CA) and cyanine 3 (Cy3)-conjugated (Jackson ImmunoResearch, West Grove, PA) secondary antibodies. Images of double immunofluorescence were taken with a confocal laser-scanning microscope (Radiance 2100; Bio-Rad, Hercules, CA). Paraffin sections (5 μm in thickness) were prepared with a sliding microtome (SM2000R; Leica). Paraffin sections were stained with hematoxylin or immunostained with goat anti-VGluT2 (1 μg/ml), guinea pig anti-calbindin (1 μg/ml) (Nakagawa et al., 1998), and rabbit anti-GluRδ2 (1 μg/ml) (Araki et al., 1993) antibodies or goat anti-VGluT1 (1 μg/ml) (Miura et al., 2006), guinea pig anti-calbindin (1 μg/ml), and rabbit anti-GluRδ2 (1 μg/ml) antibodies, respectively, followed by incubation with species-specific Alexa Fluor 488-conjugated (Invitrogen), Cy3- and Cy5-conjugated (Jackson ImmunoResearch) secondary antibodies. Immunoperoxidase staining with anti-calbindin antibody was performed essentially as described previously (Nakagawa et al., 1998). Images of hematoxylin staining, immunoperoxidase staining, and triple immunofluorescence were taken with a fluorescence microscope (DM 2500; Leica) equipped with a CCD camera (DFC 480; Leica) and a confocal laser-scanning microscope (TCS SP5; Leica), respectively. The CF-terminal reach and the thickness of the molecular layer were measured using NIH ImageJ 1.36b software. For a quantitative analysis, eight CFs and eight points of molecular layers in the straight portion of lobules 4/5 per section were measured, and nine sections from nine mice for each genotype were analyzed.
Anterograde labeling.
Under anesthesia with chloral hydrate (350 mg/kg body weight, i.p.), a glass pipette (G-1.2; Narishige, Tokyo, Japan) filled with 2–3 μl of 10% solution of dextran Texas red (DTR) or biotinylated dextran amine (3000 molecular weight; Invitrogen) in PBS, pH 7.4, was inserted to the inferior olive by the dorsal approach. The tracer was injected by air pressure at 5 psi with 5 s intervals for 1 min (Pneumatic Picopump; World Precision Instruments, Tokyo, Japan). After 4 d of survival, mice were anesthetized and fixed by transcardial perfusion. DTR-labeled microslicer sections were further subjected to incubation with goat anti-calbindin antibody (1 μg/ml) or with a mixture of goat anti-calbindin antibody (1 μg/ml) and guinea pig anti-VGluT2 antibody (0.5 μg/ml) and finally with fluorescent secondary antibodies for 2 h. Images of double or triple fluorescent labeling were taken with a confocal laser-scanning microscope (FV1000; Olympus Optical, Tokyo, Japan).
Conventional electron microscopy.
For qualitative and quantitative analysis by electron microscopy, microslicer sections were postfixed with 1% osmium tetroxide in 0.1 m cacodylate buffer for 1 h, stained in block with uranyl acetate, dehydrated in graded alcohols, and embedded in Epon 812. Ultrathin sections (70 nm in thickness) cut in the parasagittal plane were prepared from the straight portion of lobules 4/5 using an Ultracut ultramicrotome (Leica). Serial ultrathin sections were mounted on Formvar-supported copper grids and stained with 2% uranyl acetate for 5 min and mixed leads solution for 2 min. In each mouse, serial electron micrographs were sampled randomly from the neuropil of the molecular layer. Electron micrographs were taken at an original magnification of 4000× or 8000× using an electron microscope (H-7100; Hitachi High Technologies, Tokyo, Japan). For quantitative analysis, negative films of electron micrographs were scanned and analyzed with MetaMorph software (Molecular Devices, Sunnyvale, CA).
Postembedding immunogold electron microscopy.
For postembedding immunogold electron microscopy, microslicer sections were cryoprotected with 30% glycerol in 0.1 m PB, pH 7.4, and frozen rapidly in liquid propane in a cryofixation unit (CPC; Leica). Frozen sections were transferred to 0.5% uranyl acetate in methanol in a cryosubstitution unit (AFS; Leica), embedded in Lowicryl HM20 resin (Electron Microscopy Sciences, Hatfield, PA), and polymerized with UV light. Ultrathin sections were mounted on nickel grids and etched with a saturated solution of NaOH in absolute ethanol for a few seconds. After blocking with 2% human serum albumin (Wako Pure Chemicals, Osaka, Japan) in 10 mm Tris-buffered saline (TBS), pH 7.6, grids were immunoreacted with a mixture of rabbit anti-GluRδ2 (20 μg/ml) and guinea pig anti-VGluT2 (20 μg/ml) antibodies overnight. After washing with TBS, grids were further incubated with a mixture of colloidal gold (10 nm)-conjugated anti-rabbit IgG and colloidal gold (20 nm)-conjugated anti-guinea pig IgG (British Biocell International, Cardiff, UK) for 2 h. Finally, grids were stained with 2% uranyl acetate for 5 min and mixed lead solution for 30 s. Electron micrographs were taken randomly by an H-7100 electron microscope at magnification of 10,000×. For quantitative analysis, the number of metal particles and the length of postsynaptic density were measured on scanned electron micrographs with MetaMorph software (Molecular Devices).
Electrophysiology.
Four-week-old male mice were killed by cervical dislocation under deep anesthesia with diethyl ether. The cerebellum was excised, and parasagittal cerebellar slices (250 μm thick) were prepared from the vermis (Edwards et al., 1989; Kakizawa et al., 2000, 2005). Whole-cell recordings were obtained from visually identified PCs under an upright microscope (BX51WI; Olympus Optical) using a 40× water-immersion objective at room temperature (23–25°C). The resistances of patch pipettes were 2.0–3.5 MΩ when filled with an intracellular solution composed of the following (in mm): 60 CsCl, 10 Cs d-gluconate, 20 tetraethylammonium (TEA)-Cl, 20 BAPTA, 4 MgCl2, 4 ATP, 0.4 GTP, and 30 HEPES, adjusted to pH 7.3 with CsOH. The standard bathing solution was composed of the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose (bubbled with 95% CO2 and 5% CO2). Bicuculline (10 μm) was always added to block IPSCs. There were no significant differences in passive electrophysiological membrane properties in PCs (Llano et al., 1991) between wild-type and GluRδ2ΔT mice (data not shown). A stimulation pipette (5–10 μm in tip diameter) was filled with the standard bathing solution and used in applying square pulses (0.1 ms in duration) for the focal stimulation of PFs (0–20 V in amplitude) in the molecular layer at the deeper one-third from the pial surface or CFs in granular layer (0–90 V in amplitude). Ionic current was recorded from PCs with a patch-clamp amplifier (EPC-9; HEKA Elektronik, Lambrecht/Pfalz, Germany) at a holding potential of −90 or −80 mV (for PF–EPSC) or −20 or −10 mV (for CF–EPSC), after the compensation of liquid junction potential. The signals were filtered at 2 kHz and digitized at 20 kHz. On-line data acquisition and off-line data analysis were performed using PULSE (HEKA Elektronik) software. For the LTD experiment (see Fig. 5C), a pipette solution with the following composition was used (in mm): 60 CsCl, 40 Cs d-gluconate, 20 TEA-Cl, 1 EGTA, 4 MgCl2, 4 ATP, 0.4 GTP, and 30 HEPES, adjusted to pH 7.3 with CsOH. LTD was induced by conjunctive stimulation (PF stimuli concomitant with 50 ms depolarizations of the PC to 0 mV at 1 Hz, 300 pulses) after the acquisition of baseline responses (Fujiwara et al., 2007). Test stimulus was applied to PF every 10 s. The amplitude of PF–EPSC was averaged every 60 s and normalized to the mean value observed for 10 min before an LTD-inducing stimulus. A 100 ms, −5 mV hyperpolarizing test pulse preceded each PF stimulus to monitor the series resistance and input resistance of PCs throughout the experiment, the data of which were discarded if the resistance changed by >10% (Namiki et al., 2005; Kakizawa et al., 2007). The data were also discarded when the slope of PF–EPSC amplitude averaged every minute during the initial recording for 10 min was larger than 2% or when the amplitude did not become stable within 30 min after the onset of whole-cell configuration (Namiki et al., 2005; Kakizawa et al., 2007).
Motor behavioral test.
The accelerating rotarod apparatus consisted of a 3.2-cm-diameter rod (RRAC-3002; O'Hara & Company, Tokyo, Japan) was used to measure motor coordination. The rotarod test was performed essentially according to the procedure described previously (Nolan et al., 2003). Male mice of 5 weeks old were used for motor behavioral test. During the training period, mice were placed on the rotating rod starting at 5 rpm and gradually accelerated to 50 rpm at a rate 0.15 rpm/s. The latency to fall (retention time) was measured with cutoff time of 5 min. Mice were trained for 3 consecutive days, receiving four trials per day with a 1 h intertrial interval.
Statistical analysis.
Statistical significance was evaluated by one-way ANOVA or two-way ANOVA. When the interaction was significant, Student's t test or Scheffé's post hoc test was used. Statistical significance was assumed when p < 0.05.
Results
Generation of GluRδ2ΔT mice carrying C terminal-truncated GluRδ2 on pure C57BL/6 genetic background
To investigate the molecular basis of the multiple and diverse functions of GluRδ2, we generated mice with mutant GluRδ2 lacking the C terminal. The targeting vector carried the mutant GluRδ2ΔT gene with a deletion of the 21 nt sequence encoding the C-terminal 7 aa residues (T site) (Fig. 1A). We obtained recombinant GluRδ2+/ΔT mice (Fig. 1B,C) by homologous recombination in ES cells derived from the C57BL/6 strain (Mishina and Sakimura, 2007). Crossing of heterozygous GluRδ2+/ΔT mice with each other yielded homozygous GluRδ2ΔT/ΔT mice, designated as GluRδ2ΔT mice (Fig. 1B,C). The mutant mice grew normally with no obvious ataxic gaits. Subsequent analyses were performed with 4-week-old GluRδ2ΔT mice unless otherwise specified, and wild-type offspring of heterozygous intercrosses served as controls.

Generation of GluRδ2ΔT mutant mice by homologous recombination in ES cells derived from the C57BL/6 strain. A, Schematic representation of GluRδ2 cDNA, the GluRδ2 gene, targeting vector, and targeted gene. Relevant restriction enzyme sites are indicated. BSK, plasmid pBluescript; Cam, chloramphenicol-acetyltransferase gene; DT, diphtheria toxin gene; M1–M4, four putative transmembrane segments; Neo, neomycin phosphotransferase gene; SP, putative signal peptide; T site, C-terminal PDZ domain recognition site; EI, EcoRI; EV, EcoRV. Gray shadow boxes indicate exons. Hatched bars indicate the location of probes for Southern blot analysis (probe 5′, 3′, and Neo). Arrows indicate PCR primers (P1 and P2). B, Southern blot analysis of genomic DNA from GluRδ2+/+ (+/+), GluRδ2+/ΔT (+/ΔT), and GluRδ2ΔT/ΔT (ΔT/ΔT) mice. Left, EcoRI-digested DNA hybridized with 3′ probe; middle, EcoRI-digested DNA hybridized with Neo probe; right, EcoRV-digested DNA hybridized with 5′ probe. C, Agarose gel electrophoresis of DNA fragments amplified by PCR from GluRδ2+/+, GluRδ2+/ΔT, and GluRδ2ΔT/ΔT mice. The amplified DNA fragments derived from the wild-type and truncated GluRδ2 genes were 232 and 211 bp, respectively.
The expression level of truncated GluRδ2ΔT in cerebellar homogenates was examined by Western blot analysis using antibody against amino acid residues 837–916 of GluRδ2 (Araki et al., 1993). This antigenic sequence does not include the C-terminal 76 aa. Thus, the antibody is expected to react with both GluRδ2 and GluRδ2ΔT at similar potencies. The amount of GluRδ2ΔT in mutant mice was 76.3 ± 3.0% (mean ± SEM; n = 6) of GluRδ2 in wild-type mice as estimated by comparing that of NSE as an internal standard (Fig. 2A). This value was higher than the expression level of GluRδ2 in GluRδ2+/CrePR mice (61.9 ± 1.2%, n = 6; p = 0.0089, Scheffé's test), in which one of the GluRδ2 alleles was disrupted by inserting the CrePR gene (Takeuchi et al., 2005). Previous studies showed that PF–PC synaptic structures of GluRδ2+/CrePR mice are indistinguishable from those of wild-type mice (Takeuchi et al., 2005). As expected, antibody against the very end of the GluRδ2 C-terminal domain detected no GluRδ2ΔT (Fig. 2B). The amount of GluRδ2ΔT in the PSD fraction from mutant mice was comparable with that of GluRδ2 from wild-type mice (87.6 ± 10.3%; n = 6; p = 0.32, t test) as estimated by comparing that of NMDA receptor GluRε1 that is expressed in cerebellar granule cells but not in PCs (Yamada et al., 2001) as an internal standard (Fig. 2C).

Expression of GluRδ2ΔT in the cerebellum. A, Cerebellar homogenates from GluRδ2+/+ (+/+), GluRδ2+/CrePR (+/−), and GluRδ2ΔT/ΔT (ΔT/ΔT) mice were separated by SDS-PAGE and immunoblotted with anti-GluRδ2 and anti-NSE antibodies, respectively. B, Cerebellar homogenates from wild-type and GluRδ2ΔT mice were separated by SDS-PAGE and immunoblotted with antibody against the most C-terminal domain of GluRδ2 and anti-NSE antibody, respectively. C, PSD fractions from wild-type and GluRδ2ΔT mice were separated by SDS-PAGE and immunoblotted with anti-GluRδ2 and anti-GluRε1 antibodies, respectively. WB, Western blot.
Histological features of the cerebellum
The foliation and laminar organization of the cerebellum were indistinguishable between wild-type and GluRδ2ΔT mice (Fig. 3A,B). The arborization of PC dendrites was also comparable as revealed by immunostaining with anti-calbindin antibody (Fig. 3A,B). VGluT1 is predominantly expressed in PF terminals (Fremeau et al., 2001). Double immunostaining for GluRδ2 and VGluT1 showed numerous punctate signals in the molecular layer of both wild-type and GluRδ2ΔT mice (Fig. 3C,D). Thus, there were no detectable differences in the histological features of the cerebellum between wild-type and GluRδ2ΔT mutant mice.

Histological and immunohistochemical analysis of cerebella of wild-type and GluRδ2ΔT mice. A, B, Sections from wild-type (+/+) (A) and GluRδ2ΔT (ΔT/ΔT) (B) mice were stained with hematoxylin (left panels) or immunostained with anti-calbindin antibody (right panels). Higher-magnification image of left panels were shown in middle panels. C, D, Sections from wild-type (C) and GluRδ2ΔT (D) mice were double-immunostained with anti-GluRδ2 antibody (green, left panels) and anti-VGluT1 antibody (red, middle panels). The images were merged on the right. Scale bars: A, left, 500 μm; A, middle, right; C, 50 μm.
Fine structures of PF–PC synapses
The ablation of GluRδ2 hinders PF–PC synapse formation, resulting in the appearance of free spines and mismatched synapses in a substantial fraction of PC spines (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Lalouette et al., 2001; Takeuchi et al., 2005). Thus, we further examined the fine structures of PF–PC synapses in wild-type and GluRδ2ΔT mice by electron microscopy. In both genotypes, most synapses in the molecular layer were asymmetrical synapses between presynaptic terminals containing round clear vesicles and postsynaptic dendritic spines with thickened PSDs; they were identified as PF–PC synapses from their characteristic morphology (Fig. 4A,B). When quantified on randomly taken electron micrographs, the numbers of PF–PC synapses per 100 μm2 were 21.2 ± 0.83 for wild-type mice (mean ± SEM; n = 1641, 3 animals) and 20.1 ± 1.6 for GluRδ2ΔT mice (n = 1827, 3 animals), showing no significant difference (p = 0.58).

Structure of PF–PC synapses and localization of GluRδ2ΔT at PF–PC synapses in mutant mice. A, B, Electron micrographs showing the fine structure of PF–PC synapses in wild-type (+/+) (A) and GluRδ2ΔT (ΔT/ΔT) (B) mice. Asterisks indicate the head of PC spines forming asymmetric synapse with PF axon terminals. C–F, Postembedding double immunogold using anti-GluRδ2 antibody raised against amino acid residues 837–916 and anti-VGluT2 antibody. C, E, PF–PC synapses of wild-type (C) and GluRδ2ΔT (E) mice. Arrowheads indicate the edge of PSD. GluRδ2ΔT accumulates in PSD, in a similar manner with GluRδ2 in PSD of wild-type synapses. D, F, CF–PC synapses of wild-type (D) and GluRδ2ΔT (F) mice. CF terminals are intensively labeled for VGluT2 (large particles, 20 nm). In clear contrast to PF–PC synapses, small GluRδ2 particles (10 nm) are hardly detectable at CF–PC synapses of both genotypes. Arrowheads indicate the edge of PSD. G, The labeling density of GluRδ2 particles per 1 μm of PSD at PF and CF synapses in wild-type and GluRδ2ΔT mice. Scale bars: A, 500 nm; C, D, 100 nm. All values represent mean ± SEM. ***p < 0.001, Student's t test.
We further examined the fine structures of PF–PC synapses by serial electron microscopy on randomly sampled PC spines. Most of these spines protruded from distal dendrites, which were <2 μm in caliber, abundant in mitochondria, and enclosed by ruffled cell membrane (Ichikawa et al., 2002). Only 1 of 513 spines counted from three GluRδ2ΔT mice was a free spine, resulting in high synaptic contact ratio (99.8 ± 0.19%). This value was not significantly different from that of wild-type mice (100 ± 0%; n = 486, 3 animals; p = 0.42). For quantitative comparison, we defined the mismatched synapse when the edges of the active zone and PSD were >100 nm apart in any of its serial sections (Takeuchi et al., 2005). Under this criterion, we found that almost all the PC spines were matched synapses in both wild-type (99.4 ± 0.38%; n = 486, 3 animals) and GluRδ2ΔT (98.9 ± 0.36%; n = 513, 3 animals) mice, showing no significant difference (p = 0.47). Thus, no structural abnormalities of PF–PC synapses were found in GluRδ2ΔT mice. We also examined the spine density at proximal dendrites of PCs because hyperspiny transformation was observed in voltage-dependent Ca2+ channel (VDCC) α1A mutant mice exhibiting abnormalities in the territory of PF–PC synapses (Miyazaki et al., 2004). There was no significant difference in the number of spines per micrometer of proximal dendrites (>2 μm in caliber) between wild-type (0.95 ± 0.26 calculated from 782 μm of dendrites) and GluRδ2ΔT (0.74 ± 0.13 calculated from 882 μm) mice (three animals each).
Localization of GluRδ2ΔT at PF–PC synapses
We examined the synaptic localization of GluRδ2ΔT by postembedding double immunogold using anti-GluRδ2 and anti-VGluT2 antibodies. At PF–PC synapses of both wild-type and GluRδ2ΔT mice, small gold particles (diameter, 10 nm) accumulated exclusively in PSD (Fig. 4C,E), indicating the selective localization of GluRδ2ΔT at the postsynaptic site. The labeling density per micrometer of PSD was determined by measuring the total number of gold particles and the length of PSD at a given synapse. There was no significant difference in mean labeling density between wild-type (32.5 ± 1.1 particles/μm; n = 119, 3 animals) and GluRδ2ΔT (32.6 ± 1.2 particles/μm; n = 130, 3 animals) mice (p = 0.97). We also examined the localization of GluRδ2ΔT at CF–PC synapses by identifying the CF terminals by VGluT2 labeling (large particles; diameter, 20 nm) (Fig. 4D,F). GluRδ2 gold particles were hardly detectable at CF–PC synapses in both genotypes. There was no significant difference in mean labeling density between wild-type (2.0 ± 0.5 particles/μm; n = 53, 3 animals) and GluRδ2ΔT (2.4 ± 0.4 particles/μm; n = 52, 3 animals) mice (p = 0.64) (Fig. 4G). Little effect of the C-terminal truncation on the postsynaptic localization of GluRδ2 is consistent with previous observations that mutant GluRδ2 lacking the C-terminal 986–992 aa was efficiently localized at the membrane in cultured cells and at the synapse in cultured PCs and cerebellar slices (Matsuda and Mishina, 2000; Yawata et al., 2006; Kohda et al., 2007). In contrast, the deletion of the C-terminal region disturbed the efficient synaptic localization of NMDA receptor GluRε2/NR2B in the hippocampal CA1 region and cultured neurons (Mori et al., 1998; Mohrmann et al., 2002). The apparent discrepancy may be ascribed to the size of deletion because the C-terminal truncation was much larger in GluRε2 than in GluRδ2. Consistent with this notion, the membrane-proximal segment in the C-terminal region is essential for the efficient membrane localization of GluRδ2 (Matsuda and Mishina, 2000; Matsuda et al., 2004).
Impairments of LTD induction at PF–PC synapses and improvement in accelerating rotarod test
We examined synaptic transmission at PF–PC synapses in GluRδ2ΔT mice by stimulating PFs at different intensities and by plotting the amplitude of PF–EPSCs as a function of stimulus intensity. As shown in Figure 5A, there was no significant difference in the slope of the amplitude–intensity curve between wild-type and GluRδ2ΔT PCs (p = 0.34, t test). We also examined paired-pulse ratio (PPR), an index of change in transmitter release probability at presynaptic terminals (Zucker and Regehr, 2002). When the interpulse interval was within 10–300 ms, the PF–EPSCs of wild-type and GluRδ2ΔT mice displayed prominent facilitation in response to the second stimuli of paired pulses (Fig. 5B), as reported previously in wild-type rats (Konnerth et al., 1990). The PPRs of PF–EPSC in GluRδ2ΔT mice were slightly higher than those in wild-type mice at shorter interpulse intervals (Fig. 5B).
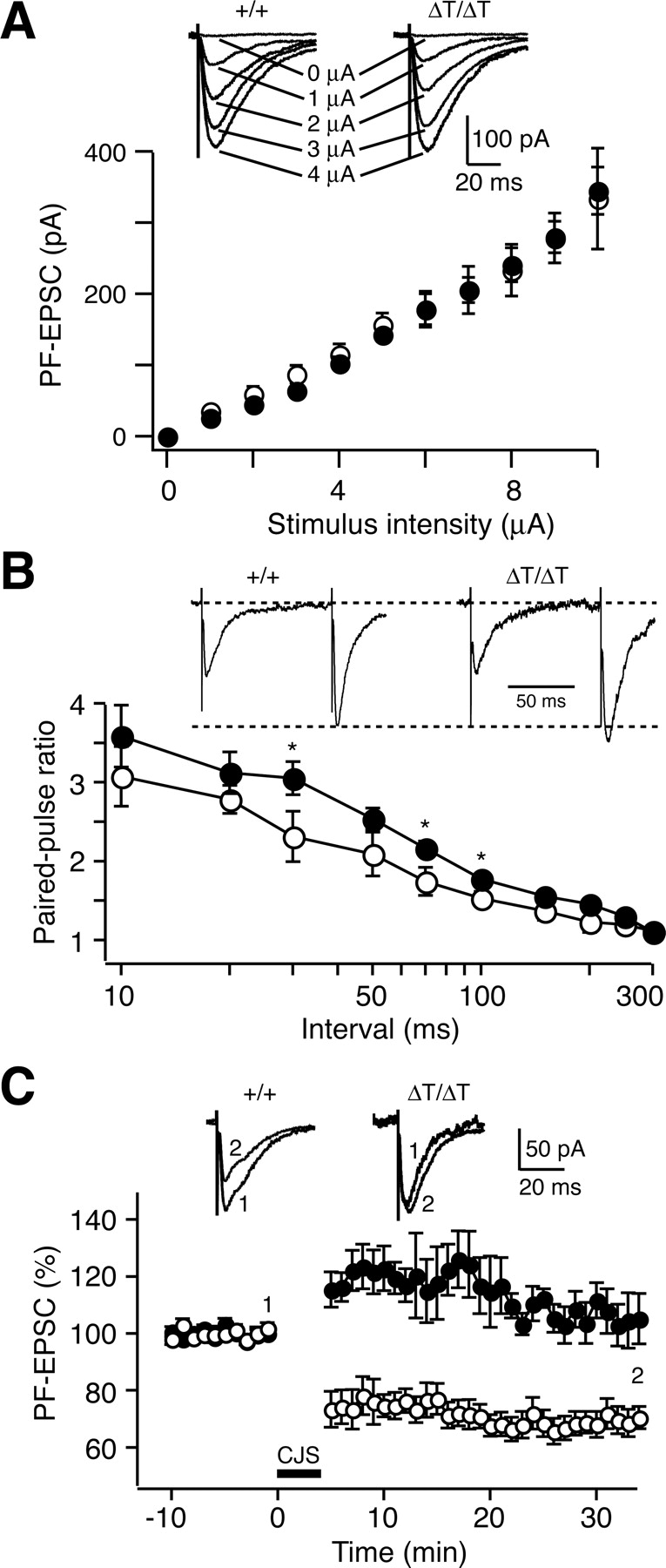
Impairment of cerebellar long-term depression in GluRδ2ΔT mice. A, Input–output relationship of the PF–EPSCs. Amplitudes of PF–EPSCs plotted as a function of stimulus intensity in the wild-type (+/+, open circles; n = 8) and GluRδ2ΔT (ΔT/ΔT, filled circles; n = 6) PCs. Representative traces of PF–EPSCs with increasing stimulus intensities (0–8 μA) recorded from wild-type and GluRδ2ΔT PCs are shown on the top. Membrane voltages were held at −80 mV. B, PPR of PF–EPSCs recorded from PCs in wild-type (open circles; n = 5) or GluRδ2ΔT (filled circles; n = 6) mice at several interpulse intervals. PPRs in GluRδ2ΔT mice are slightly higher than those in wild-type mice at relatively shorter interpulse intervals. Representative traces of PF–EPSC in response to paired stimuli (100 ms interval) recorded from wild-type and GluRδ2ΔT PCs are shown on the top. Membrane voltages were held at −80 mV. C, Time course of changes in PF–EPSCs recorded from PCs in wild-type (open circles; n = 8) or GluRδ2ΔT (filled circles; n = 5) mice. After the conjunctive stimulation (CJS; bold horizontal bar) of PF and depolarization (1 Hz, 300 s), typical LTD was induced in wild-type PCs but not in GluRδ2ΔT PCs. Representative recordings of PF–EPSC before (1) or 30 min after (2) CJS are shown on the top. All values represent mean ± SEM. *p < 0.05, Student's t test.
We then examined LTD at PF–PC synapses. After stable recordings of PF–EPSCs for 10 min, LTD was induced by a conventional conjunction protocol, which consisted of 300 single PF stimuli in conjunction with a depolarizing pulse repeated at 1 Hz (Fujiwara et al. 2007). In wild-type mice, LTD was readily induced by conjunctive stimulation (Fig. 5C). Twenty-one to 30 min after conditioning, the mean amplitude of PF–EPSCs was reduced to 68.7 ± 3.6% (mean ± SEM) of the control value measured before conjunctive stimulation. However, the same conjunctive stimulation failed to induce LTD in GluRδ2ΔT mice. The mean PF–EPSC amplitude 21–30 min after conditioning in GluRδ2ΔT mice (106.0 ± 5.4%) was significantly higher than that in wild-type mice (p = 4.4 × 10−5, Student's t test) (Fig. 5C). Thus, the T site of GluRδ2 is indispensable for LTD induction at PF–PC synapses.
GluRδ2ΔT mice showed no ataxic gait and could walk along a straight line, as wild-type mice did. The footprint pattern of GluRδ2ΔT mice was indistinguishable from that of wild-type mice (Fig. 6A). In the accelerating rotarod test (Crawley, 2000), wild-type and GluRδ2ΔT mice performed equally well in the first training session (p = 0.44) (Fig. 6B). In subsequent sessions, both wild-type and GluRδ2ΔT mice showed gradual increases in retention time on the rod during training. However, there was a significant difference in the retention time between two genotypes (ANOVA with repeated measures: genotype effect, F(1,46) = 19.5, p = 6.1 × 10−5; genotype × session interaction, F(11,506) = 2.22, p = 0.012), suggesting that the improvement in performance was impaired in GluRδ2ΔT mice.

Footprint patterns and accelerating rotarod test. A, Representative footprint patterns from wild-type (+/+) and GluRδ2ΔT (ΔT/ΔT) mice. B, Accelerating rotarod test. Time that mice remained on an accelerating rotarod before falling was measured as a function of training session. Open and filled circles represent wild-type (n = 21) and GluRδ2ΔT (n = 27) mice, respectively. All values represent mean ± SEM.
Distal extension of CF territory
GluRδ2 null mutation affects the innervation patterns of CFs to PCs (Kashiwabuchi et al., 1995; Ichikawa et al., 2002). We thus examined CF innervation patterns in GluRδ2ΔT mice by immunostaining for VGluT2, which is predominantly expressed in CF terminals (Fremeau et al., 2001). The VGluT2 immunostaining signals in the molecular layer of GluRδ2ΔT mice were more expanded toward the superficial region than those in the case of wild-type mice (Fig. 7A). CFs grow upward in the molecular layer along expanding PC dendritic arbors (Altman and Bayer, 1997). The thickness of the molecular layer in wild-type mice significantly increased from postnatal day 16 (P16) to P29–P31 (one-way ANOVA, F(2,24) = 27.0, p = 7.2 × 10−7) (Fig. 7B,C). The thicknesses of the molecular layer were comparable between GluRδ2ΔT and wild-type mice (two-way ANOVA: genotype effect, F(1,48) = 1.13, p = 0.29; age × genotype interaction, F(2,48) = 0.442, p = 0.65) (Fig. 7B,C). Then, we analyzed the developmental changes of the distal extension of CF innervation by measuring the CF-terminal reach defined as the vertical height of the most distal tip of continuous VGluT2 immunostaining signals along the dendritic tree of single PCs from the bottom of the molecular layer. From P16 to P29–P31, CF-terminal reach value gradually increased in both genotypes. However, there was a significant difference in CF-terminal reach value between wild-type and GluRδ2ΔT mice (two-way ANOVA: genotype effect, F(1,48) = 26.6, p = 4.7 × 10−6; age × genotype interaction, F(2,48) = 5.59, p = 0.0066) (Fig. 7B,C). At P29–P31, the CF-terminal reach value in GluRδ2ΔT mice was significantly larger than that in wild-type mice (t test, p = 1.6 × 10−5). We also analyzed the CF innervation territory by measuring the ratio of CF-terminal reach to molecular layer thickness during development. There was a significant difference in the CF innervation territory between wild-type and GluRδ2ΔT mice (two-way ANOVA: genotype effect, F(1,48) = 88.1, p = 1.9 × 10−12; age × genotype interaction, F(2,48) = 18.0, p = 1.5 × 10−6) (Fig. 7B,C). In GluRδ2ΔT mice, CF innervation territory was significantly larger than that in wild-type mice at P21 and P29–P31 (t test, p = 5 × 10−5 and 2.1 × 10−10, respectively). These results suggest that the territory of CF innervation along the PC dendritic trees is expanded in GluRδ2ΔT mice, despite the absence of any detectable structural abnormalities at PF–PC synapses.

Distal extension of CF innervation territory in GluRδ2ΔT mice. A, Cerebellar sections of wild-type (+/+) and GluRδ2ΔT (ΔT/ΔT) mice were triple immunostained with anti-GluRδ2 antibody (green), anti-VGluT2 antibody (red), and anti-calbindin antibody (blue). Triple-immunostained sections are shown as merged images (right panels) or separate images (left and middle panels). Calbindin, A marker for PC; VGluT2, a marker for CF terminals. B, Developmental change of CF territory in wild-type and GluRδ2ΔT mice. Cerebellar sections of wild-type and GluRδ2ΔT mice were double immunostained with anti-VGluT2 antibody (red) and anti-calbindin antibody (green) at P16, P21, and P29–P31, respectively. Representative images of double-immunostained sections are shown as merged images (right panels) or separate images (left panels). Dotted lines in B indicate the pial surface. Asterisks in B indicate the soma of PCs. Scale bars: A, 50 μm; B, 20 μm. C, Quantitative analysis of CF territory in wild-type (open circles; n = 9) and GluRδ2ΔT (filled circles; n = 9) mice. Developmental change of molecular layer thickness, CF-terminal reach, and CF innervation territory are shown in top, middle, and bottom, respectively. All values represent mean ± SEM (n = 9). ***p < 0.001, Student's t test.
Ectopic CF innervation at distal dendrites
We further examined CF innervation patterns by anterograde CF labeling with DTR (red), combined with immunofluorescence for VGluT2 (green) and calbindin (blue) (Figs. 8, 9). DTR-labeled CFs in GluRδ2ΔT mice extended their terminals more closely to the pial surface than those in wild-type mice (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material), suggesting the expansion of the CF innervation territory.

Triple fluorescent labeling showing mono-CF innervation in wild-type PCs. A, B, Triple-labeling images for DTR (red), VGluT2 (green), and calbindin (Calb; blue) are shown as merged low-power images (A) and high-power images with merged (B1) and separate images (B2–B4). CF1 and CF2 represent DTR/VGluT2 double-labeled or VGluT2 single-labeled CF, respectively. PC1 and PC2 receive mono-CF innervation by CF1 and CF2, respectively. Scale bars: A, 10 μm; B, 5 μm.

Triple fluorescent labeling showing ectopic CF innervation in GluRδ2ΔT PCs. A–C, Triple-labeling images for DTR (red), VGluT2 (green), and calbindin (blue) are shown as merged low-power images (A) and high-power images with merged (B1, C1) and separate images (B2–B4, C2). CF1 and CF2 represent DTR/VGluT2 double-labeled or VGluT2 single-labeled CF, respectively. Multiple CF innervation occurs in the boxed region in A, which is enlarged in B1–B4. Proximal dendrites mainly innervated by CF1 receive additional innervation by CF2 at their distal dendrites (green arrows). C, Another example of multiple CF innervation. A collateral of CF1 innervating PC1 invades a distal dendrite of PC2 (bottom red arrow), resulting in multiple CF innervation. Trajectory of the CF1 collateral is indicated by arrowheads. Furthermore, PC1 receives additional innervation by CF2 at distal dendrites (top green arrows). Scale bars: A, 10 μm; B, C, 5 μm.
In wild-type mice, proximal dendrites of given PCs were innervated by either DTR/VGluT2-double-labeled CFs (hereafter termed as CF1) (Fig. 8A,B, PC1) or VGluT2-single-labeled CFs (CF2) (Fig. 8A,B, PC2). Accordingly, almost all CF terminals along PC1 dendrites became white or yellow, whereas those along PC2 dendrites remained green or light blue, suggesting that the monoinnervation of PCs by CFs is established in wild-type mice from morphological view points. These results are consistent with previous observations that most PCs become monoinnervated by single CFs by the end of the third postnatal week in wild-type mice (Crépel, 1982; Hashimoto and Kano, 2005). Furthermore, the CF innervation territory was restricted up to the border of proximal and distal dendrites of the target PCs (Fig. 8B).
In GluRδ2ΔT mice, proximal dendrites of a given PC were innervated by either DTR/VGluT2-double-labeled or VGluT2-single-labeled CFs, similarly to those in wild-type mice. However, in contrast to wild-type mice, CFs in GluRδ2ΔT mice extended their terminals beyond the proximal-to-distal border to innervate distal dendrites of the target PCs and often jumped aberrantly to innervate distal dendrites of nearby PCs (Fig. 9). As shown in Figure 9, A and B, a proximal dendrite of PC1 was innervated by CF1 (red), whereas a distal dendrite stemming from this proximal dendrite was innervated by CF2 (green arrows). In another case shown in Figure 9C, a proximal dendrite of PC2 was innervated mainly by CF2 (bottom green arrows), but a distal dendrite stemming from this proximal dendrite of PC2 was innervated by a collateral emitting from CF1 that mainly innervated the adjacent PC1 (bottom red arrow). Furthermore, a distal dendrite of PC1 was also innervated by CF2 (Fig. 9C, top green arrows). These results suggest that, although single main CFs innervate the proximal dendrites of PCs, additional CFs frequently form ectopic innervation to distal dendrites of the same PCs. Consistently, by serial electron microscopy, some typical distal dendrites of PCs in GluRδ2ΔT mice were found to have both PF-innervating spines and CF-innervating ones (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus, ectopic CF innervation at the distal dendritic compartment is a distinct feature of GluRδ2ΔT mice.
CF–EPSCs with slow kinetics
We next analyzed CF–EPSCs, which were identified from their characteristics, such as a clearly discernible current elicited in an all-or-none manner and paired-pulse depression (Konnerth et al., 1990). PCs are innervated by multiple CFs in early postnatal days, but most PCs become monoinnervated by single CFs by the end of the third postnatal week by the elimination of supernumerary CFs (Crépel, 1982; Hashimoto and Kano, 2005). In wild-type mice, 78% of PCs (25 of 32) were innervated by single CFs. The rest (seven PCs) had one supernumerary CF in addition to the main CF. The main CF–EPSCs had much larger amplitudes than the surplus CF–EPSCs, although the two types of CF–EPSC were comparable in terms of kinetics (Fig. 10A).
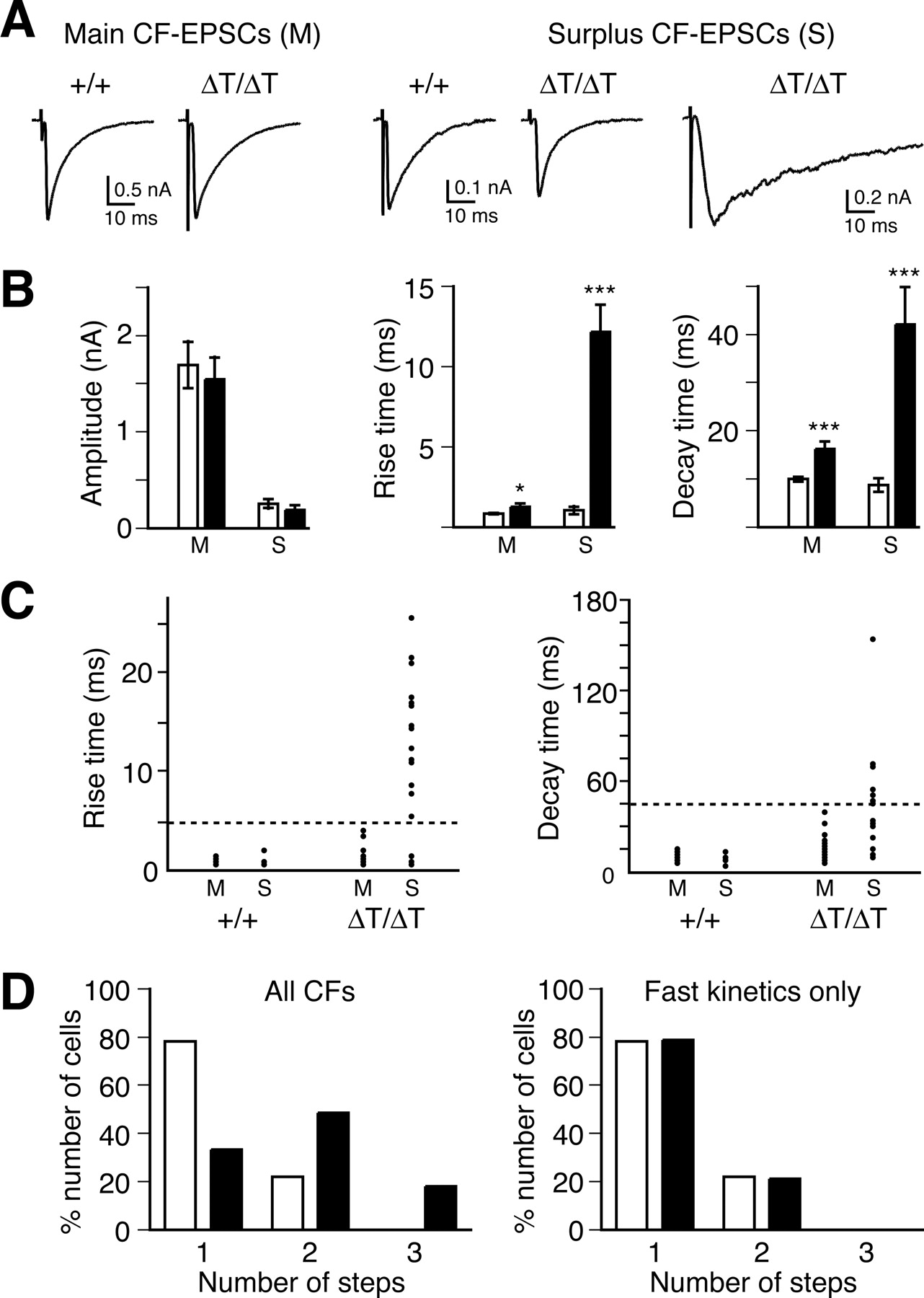
CF–EPSCs in GluRδ2ΔT mice have slower time course. A, Representative recordings of CF–EPSCs from wild-type (+/+) and GluRδ2ΔT (ΔT/ΔT) mice. EPSCs recorded from PCs in both genotypes were divided into two categories, main CF–EPSCs (M) of singly or multiply innervated PCs and surplus CF-EPSCs (S) of multiply innervated PCs. Some surplus CF–EPSCs from GluRδ2ΔT mice (right) show slower time course than other CF–EPSCs. Membrane voltages were held at −20 mV except for surplus CF–EPSCs with slower time course from GluRδ2ΔT mice (−80 mV). B, Amplitude, 10–90% rise time, and decay time constant of CF–EPSCs recorded from wild-type PCs (open column; M, n = 32; S, n = 7) and GluRδ2ΔT PCs (filled column; M, n = 33; S, n = 25). All values represent mean ± SEM. *p < 0.05; ***p < 0.001, Student's t test. C, Distribution of CF–EPSCs plotted by rise time (left) and decay time (right). CF–EPSCs with rise time longer than 5 ms and decay time longer than 45 ms are found only in surplus CF–EPSCs of GluRδ2ΔT mice. D, Frequency histograms of PCs in terms of the number of discrete CF–EPSC steps from wild-type (open column; n = 32) and GluRδ2ΔT (filled column; n = 33) mice. When all CF–EPSCs were taken into account (left), the distribution was significantly different between wild-type and GluRδ2ΔT PCs (p < 0.0001, χ2 test). However, when the slow CF–EPSCs (data distributed above bold horizontal line in C) were eliminated (right), no significant difference was seen (p = 0.86, χ2 test).
In GluRδ2ΔT mice, 33% of PCs (11 of 33) were singly innervated by a CF, and the majority had one or two supernumerary CFs in addition to the main CF (Fig. 10A). No significant differences were found in the amplitude of the main CFs between wild-type and GluRδ2ΔT mice as well as in that of surplus CF–EPSCs (Fig. 10B, left). There was no significant difference in the reversal potential of CF–EPSC between wild-type and GluRδ2ΔT PCs (data not shown). However, both the rise and decay times of CF–EPSCs were significantly longer in GluRδ2ΔT mice than in wild-type mice (Fig. 10A,B, middle and right). The slower time course of the main CF–EPSCs with large amplitudes in GluRδ2ΔT mice may be ascribed to the distal extension of CF innervation (Figs. 7, 9) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Furthermore, some surplus CF–EPSCs in GluRδ2ΔT mice (Fig. 10C, above the bold horizontal line) had rise time longer than 5 ms and decay time longer than 45 ms (Fig. 10A,C), whereas all the main CF–EPSCs in GluRδ2ΔT mice showed rise time shorter than 5 ms and decay time shorter than 45 ms. Thus, surplus CF–EPSCs in GluRδ2ΔT mice can be divided into two groups according to their kinetics (Fig. 10C). Atypical CF–EPSCs with slower kinetics represented surplus CF–EPSCs in GluRδ2ΔT mice (Fig. 10C, above the bold horizontal line) that showed much slower kinetics than the main CF–EPSCs, whereas surplus CF–EPSCs with faster kinetics had kinetics comparable with those of the main CF–EPSCs. Two populations of CF–EPSCs with different kinetics were also found in GluRδ2 null mutant mice. Atypical CF–EPSCs with slow kinetics in GluRδ2 null mutant mice were ascribed to ectopic CF innervation to neighboring PCs (Hashimoto et al., 2001; Ichikawa et al., 2002). Thus, PCs of GluRδ2ΔT mice could be classified into four categories by the pattern of CF innervation. The first group of PCs was innervated by a single main CF, which generated CF–EPSCs with faster kinetics (11 of 33 PCs). The second group of PCs was innervated by one main CF with faster kinetics and also by one or more surplus CFs with slower kinetics generating atypical CF–EPSCs (15 of 33 PCs). The third pattern was the innervation of PCs by one main CF with faster kinetics and one surplus CF with faster kinetics (5 of 33 PCs). The fourth pattern was the innervation of PCs by one main CF with faster kinetics, one surplus CF with faster kinetics, and another surplus CF with slower kinetics (2 of 33 PCs). Thus, atypical CF–EPSCs with slower kinetics significantly contributed to the high percentage of surplus CF innervation to PCs in GluRδ2ΔT mice (Fig. 10D).
In addition to the existence of ectopic CF synapses at distal dendrites, failure in the elimination of surplus CF synapses at proximal dendrites was observed in GluRδ2 null mutant mice (Kashiwabuchi et al., 1995; Hashimoto et al., 2001). Thus, we finally examined whether the deletion of the T site of GluRδ2 affects the elimination of surplus CF synapses at proximal dendrites during development. When atypical CF–EPSCs with slower kinetics that most likely represent ectopic innervation at distal dendrites (Fig. 10C, above the bold horizontal line) were omitted, most PCs had a single CF–EPSC, and the rest had one main CF–EPSC with a large amplitude and one surplus CF–EPSC with a small amplitude (Fig. 10D, right). As regards CF–EPSCs with faster kinetics, the ratios of singly to multiply innervated PCs were comparable between wild-type and GluRδ2ΔT mice (Fig. 10D, right). In contrast, the ratio was higher in GluRδ2 or mGluR1 null mutant mice than in wild-type mice (Hashimoto et al., 2001), suggesting the failure of surplus CF elimination at proximal dendrites in these mutant mice. These results suggest that the innervation pattern of CFs with faster EPSCs in GluRδ2ΔT mice is similar to that in wild-type mice but is different from those in GluRδ2 and mGluR1 null mutant mice.
Discussion
Here, we generated GluRδ2ΔT mice carrying mutant GluRδ2 lacking the T site comprising seven amino acids at the C terminal. There were no significant differences in the amount of receptor proteins in the PSD fraction and in the density of GluRδ2 immunogold particles at PF–PC synapses between wild-type and GluRδ2ΔT mice. Thus, the C-terminal truncation had little effect on the synaptic localization of receptor proteins, and GluRδ2ΔT mouse should be a valuable tool for studying the role of the T site in diverse GluRδ2 functions. PF–PC synaptic connection is critically dependent on GluRδ2 in both developing and adult cerebella (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Takeuchi et al., 2005). The direct relationship between GluRδ2 density and synaptic contact suggests that postsynaptic GluRδ2 is essential for the maintenance of the presynaptic active zone, possibly by a physical linkage between the active zone and the postsynaptic GluRδ2 complex (Takeuchi et al., 2005). Synaptic connections between PF terminals and PC spines were intact in GluRδ2ΔT mutant mice as revealed by electron microscopy. Thus, the interactions between the T site of GluRδ2 and PSD proteins such as PSD-93 appear to be dispensable for PF–PC synapse formation. Consistently, there were no detectable alterations in structures at PF–PC synapses in PSD-93 mutant mice (McGee et al., 2001).
However, LTD induction at PF–PC synapses was impaired in GluRδ2ΔT mice. Correspondingly, the improvement in the performance of mutant mice in the accelerating rotarod test was less than that of wild-type mice. The impairment of motor learning in cerebellar LTD-deficient mutant mice was reproducibly observed (Ito, 2006). Although GluRδ2 null mutant mice were strongly ataxic, GluRδ2ΔT mice showed no ataxic gait and could walk along a straight line as wild-type mice did. The essential role of the GluRδ2 C terminal in cerebellar LTD is consistent with the observation that PTPMEG mutant mice showed impairment of cerebellar LTD induction (Kina et al., 2007). In contrast, controversial results were reported on the requirement of GluRδ2 C terminal for LTD induction by cDNA transfection studies in cultured PCs and cerebellar slices (Yawata et al., 2006; Kohda et al. 2007).
It has been well established since Cajal (1911) that different classes of inputs are not randomly intertwined along target neurons; rather, they are organized along restricted and in some cases highly discrete subcellular compartments. PC dendrites are characterized by proximal and distal compartments, on which CFs and PFs impinge, respectively (Larramendi and Victor, 1967; Palay and Chan-Palay, 1974). Several studies suggested the competitive formation of the innervation territory between CF and PF (Bravin et al., 1999; Morando et al., 2001; Ichikawa et al., 2002; Cesa et al., 2003, 2007). The proper formation of PF–PC synapses seems to be necessary for restricting CF innervation to proximal dendrites of PC (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Zagrebelsky and Rossi, 1999; Ichikawa et al., 2002, Hirai et al., 2005). In GluRδ2ΔT mice, we found that the territory of CF innervation expanded distally to spiny branchlets by the VGluT2 immunostaining of CF terminals and anterograde CF labeling. Furthermore, CFs often extended their axon terminals over their target PCs to adjacent spiny branchlets of neighboring PCs, resulting in ectopic CF innervation. Thus, CF territory expansion and ectopic innervation are characteristic features of GluRδ2ΔT mice. Correspondingly, CF–EPSCs with small amplitudes and slow kinetics were found specifically in GluRδ2ΔT mice, whereas the populations of CF–EPSCs with fast kinetics were comparable between wild-type and mutant mice. Previous studies suggested that similar atypical CF–EPSCs with small amplitudes and slow kinetics observed in GluRδ2 null mutant mice are ascribed to ectopic CF innervation to neighboring PCs (Hashimoto et al., 2001; Ichikawa et al., 2002). Thus, CF–EPSCs with small amplitudes and slow kinetics in GluRδ2ΔT mice are also likely to correspond to ectopic CF synapses at distal dendrites.
The distal extension of the CF innervation territory was also reported in GluRδ2 and cerebellin 1 precursor protein Cbln1 null mutant mice (Ichikawa et al., 2002; Hirai et al., 2005). Because nearly half of the spines were not in contact with PF terminals in these mutant mice, a one-sided decrease in the number of PF–PC synapses appeared to result in the expansion of CF innervation territory through the heterosynaptic competition between PF and CF synapses. In contrast to these null mutant mice, PF–PC synaptic connections were intact and PF synaptic transmission appeared to be normal in GluRδ2ΔT mice. Thus, our results suggest that the expansion of CF innervation territory does not necessarily require a decrease in the number of PF–PC synapses or the appearance of free spines. Instead, the distal extension and ectopic innervation of CF axon terminals in GluRδ2ΔT mice should be ascribed to the impairment of the signaling through the C terminal T site of GluRδ2 because GluRδ2 is localized at PF–PC synapses but not at CF synapses (Takayama et al., 1996; Landsend et al., 1997). Thus, these results suggest that the regulation of CF innervation territory by signaling through the T site of GluRδ2 at PF–PC synapses underlies the heterosynaptic competition between PF and CF in PC. Morphologically, the presence of CF synapses seems to exclude nearby PF–PC contact (Llinás and Walton, 1998). After blocking electrical activity by tetrodotoxin, CF terminal arbors retracted from the proximal dendrites and the CF innervation territory decreased (Bravin et al., 1999; Morando et al., 2001). PF inputs, normally distributed on the spines of the distal dendrite, formed ectopic synapses on the proximal dendrite under the condition of electrical activity block. Decrease in CF innervation territory and ectopic formation of PF synapses at proximal dendrites were also observed in mutant mice lacking the VDCCα1A subunit (Miyazaki et al., 2004). Functionally active CFs exerted repressive action on the dendritic domain surrounding their synapses through the ionotropic AMPA receptor as a kind of lateral inhibition (Cesa and Strata, 2005; Cesa et al., 2007). These observations suggest that, at proximal dendrites, PF synapses are repressed by the activity of CFs that generate strong excitation and Ca2+ influx to PC dendrites. To compete against this mechanism, signaling through the T site of GluRδ2, most likely by protein–protein interactions, may be required for restricting the CF synapse territory to proximal dendrites. The topographical distributions of different classes of synaptic inputs are well exemplified in several neurons, including granule cells in the dentate gyrus, interneurons in the hippocampus, and thalamocortical relay cells (Freund and Buzsáki, 1996; Erisir et al., 1997; Förster et al., 2006). Extracellular matrix molecules play an important role in the proper lamination specificity of entorhinal fiber growth into the outer molecular layer in the dentate gyrus (Förster et al., 2001; Zhao et al., 2003). In the cerebellum, the targeting of GABAergic synapses on axon initial segment of PCs is regulated by the ankyrin-based subcellular gradient of neurofascin186, an L1 family Ig cell adhesion molecule (Ango et al., 2004).
In early development, PCs are innervated by multiple CFs. Then a single CF input is selected, allowed to mature, and strengthened, whereas surplus CFs are eliminated by the end of the third postnatal week in mice (Crépel, 1982; Hashimoto and Kano, 2005). In mGluR1 null mutant mice, most PCs remained to be multiply innervated by CFs with fast kinetics in the adulthood (Hashimoto et al., 2001), suggesting the failure of CF elimination from proximal dendrites. Similarly, the multiple innervation of PCs by CFs with fast kinetics was sustained in GluRδ2 null mutant mice. In contrast, the ratios of single to multiple innervations by CFs with fast kinetics were similar between wild-type and GluRδ2ΔT mice (Fig. 10D). Thus, surplus CFs appeared to have been successfully eliminated from proximal dendrites in GluRδ2ΔT mice.
The present study with GluRδ2ΔT mice provides evidence that diverse GluRδ2 functions are mediated by the distinct domains of GluRδ2. The C-terminal T site of GluRδ2 is essential for LTD induction at PF–PC synapses and the proper restriction of the CF territory but is dispensable for PF–PC synaptic connection and the elimination of surplus CF synapses at proximal dendrites during development.
Footnotes
-
This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and Solution-Oriented Research for Science and Technology, the Japan Science and Technology Corporation. We thank Yoshinori Mikami, Dr. Hiroshi Sagara, and Joel Ju for help in preliminary experiments, Dr. Tomonori Takeuchi and Rie Natsume for help in mutant mouse generation, and Yuki Takahashi for mouse genotyping.
- Correspondence should be addressed to Dr. Masayoshi Mishina, Department of Molecular Neurobiology and Pharmacology, Graduate School of Medicine, University of Tokyo, Tokyo 113-0033, Japan. mishina{at}m.u-tokyo.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}