

Abstract
The hippocampus plays an integral role in spatial navigation, learning and memory, and is a major site for adult neurogenesis. Critical to these functions is the proper organization of the hippocampus during development. Radial glia are known to regulate hippocampal formation, but their precise function in this process is yet to be defined. We find that in Nuclear Factor I b (Nfib)-deficient mice, a subpopulation of glia from the ammonic neuroepithelium of the hippocampus fail to develop. This results in severe morphological defects, including a failure of the hippocampal fissure, and subsequently the dentate gyrus, to form. As in wild-type mice, immature nestin-positive glia, which encompass all types of radial glia, populate the hippocampus in Nfib-deficient mice at embryonic day 15. However, these fail to mature into GLAST- and GFAP-positive glia, and the supragranular glial bundle is absent. In contrast, the fimbrial glial bundle forms, but alone is insufficient for proper hippocampal morphogenesis. Dentate granule neurons are present in the mutant hippocampus but their migration is aberrant, likely resulting from the lack of the complete radial glial scaffold usually provided by both glial bundles. These data demonstrate a role for Nfib in hippocampal fissure and dentate gyrus formation, and that distinct glial bundles are critical for correct hippocampal morphogenesis.
Introduction
During nervous system development, radial glia regulate boundary demarcation and act as scaffolds for migrating neurons (Götz and Barde, 2005). Early neuroanatomical studies indicated that glia might play a pivotal role in hippocampal morphogenesis (Rickmann et al., 1987; Sievers et al., 1992). These studies implicated two glial bundles in this process, the supragranular bundle, derived from the ammonic neuroepithelium, and the fimbrial bundle, derived from the fimbrial glioepithelium. Both have been postulated to regulate neuronal migration (Altman and Bayer, 1990a,b; Nakahira and Yuasa, 2005). The differentiation of radial glia into glial fibrillary acidic protein (GFAP) positive cells in the hippocampus occurs in mouse from embryonic day 15 (E15) (Fox et al., 2004) and from E17 in the rat (Rickmann et al., 1987). Both the supragranular glial bundle, and the fimbrial glial bundle express GFAP but it is not known whether these glia are derived from similar or distinct progenitors, or how their development is molecularly regulated. Recent work has implicated the transcription factor Emx2 in hippocampal morphogenesis (Oldekamp et al., 2004; Zhao et al., 2006; Skutella et al., 2007), as hippocampal expression of GFAP is reduced at E18.5 in these mutants. However, these studies did not address either earlier glial development, or the importance of glial maturation to hippocampal development.
Members of the Nuclear Factor I (Nfi) transcription factor family (Nfia, Nfib, Nfic and Nfix) are expressed in overlapping patterns during embryogenesis (Gronostajski, 2000; Plachez et al., 2008), and knock-out studies have demonstrated that Nfia, Nfib and Nfix are important during brain development (das Neves et al., 1999; Shu et al., 2003; Steele-Perkins et al., 2005; Driller et al., 2007; Campbell et al., 2008). Mice deficient in Nfib display developmental abnormalities in the hippocampus, including the morphological absence of the dentate gyrus (DG). Importantly, Nfib knock-out mice display defects in formation of the glial wedge, a midline glial structure that regulates formation of the corpus callosum (Steele-Perkins et al., 2005). Furthermore, Nfia and Nfib regulate gliogenesis in the developing spinal cord (Deneen et al., 2006), suggesting that control of glial differentiation throughout development of the nervous system may be critically dependent on the Nfi gene family.
Here we demonstrate that glial maturation in the ammonic neuroepithelium is required for formation of the supragranular bundle. The absence of the supragranular bundle in Nfib-deficient mice leads to the failure of hippocampal fissure formation, culminating in the morphological absence of the DG. However, the fimbrial glial bundle forms normally from the fimbrial glioepithelium in these mice, suggesting that the two glial populations in the developing hippocampus are regulated by distinct molecular determinants. Thus, just as neuronal populations are specified into different subregions of the hippocampus, so too are specific glial populations within this structure. These data provide an understanding of the molecular processes underpinning the neuroanatomical development of the hippocampus by demonstrating that the development of the DG may depend on the formation of the hippocampal fissure.
Materials and Methods
Animals.
Animals used in this study were litters of wild-type C57BL/6J and Nfib mice, which were bred on site at the University of Queensland under ethical approval from the institutional Animal Ethics Committee. The Nfib−/− allele (Steele-Perkins et al., 2005) was backcrossed for >10 generations onto the C57BL/6J background. No hippocampal defects were detected in wild-type or heterozygote animals. Timed-pregnant females were obtained by placing male and female mice together overnight. The following day, females were inspected for the presence of a vaginal plug. If present, this day was designated as E0. Heterozygous Nfib mice were bred to obtain wild-type, heterozygous and homozygous progeny. Embryos were genotyped by PCR as previously described (Steele-Perkins et al., 2005).
Immunohistochemistry.
On the required gestational day, embryos were drop-fixed in 4% paraformaldehyde (PFA; E14 and below) or transcardially perfused with 0.9% saline, followed by 4% PFA (E15 to E18), then postfixed in 4% PFA at 4°C until sectioning. Brains were removed, blocked in 3% noble agar, and then sectioned at 45 μm on a vibratome (Leica). Sections were washed in PBS, blocked in PBS containing 2% normal goat serum (Vector Laboratories) and 0.2% Triton X-100 (Sigma) for 2 h and then incubated in primary antibody diluted in blocking solution overnight. Sections were subsequently washed in PBS, and then incubated in the secondary antibody for 1 h, followed by three PBS washes. They were then incubated in avidin-biotin solution (1:500; Vector Laboratories) for 1 h, after which they were washed again in PBS. Finally, sections were placed in a chromogen solution (2.5% nickel sulfate and 0.02% 3,3′ diaminobenzidine in 0.175 m sodium acetate, activated with 0.01% H2O2) until a dark precipitate had formed. The color reaction was terminated by rinsing in PBS, after which the sections were mounted, dehydrated and coverslipped in DPX mounting medium (Fluka). Primary antibodies used for immunohistochemistry were anti-Tbr1 (rabbit polyclonal, a gift from Robert Hevner, University of Washington, Seattle, WA; 1:100,000); anti-Prox1 (rabbit polyclonal, 1:100,000, Chemicon); anti-Pax6 (rabbit polyclonal, 1:25,000; Chemicon); anti-calretinin (rabbit polyclonal, 1:75,000; SWANT); anti-calbindin (rabbit polyclonal, 1:50,000; SWANT); anti-reelin (G10, mouse monoclonal, a gift from Andre Goffinet, University of Louvain Medical School, Brussels, Belgium; 1:100,000); anti-GFAP (rabbit polyclonal, 1:50,000; DAKO); anti-GLAST (rabbit polyclonal, a gift from Niels Danbolt, University of Oslo, Oslo, Norway; 1:50,000); anti-nestin (mouse monoclonal, 1:1500; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); anti-cleaved caspase 3 (rabbit polyclonal, 1:1000, Cell Signaling Technology); anti-tenascin C (rabbit polyclonal, 1:2000, Chemicon). Secondary antibodies used were biotinylated goat-anti-rabbit IgG and biotinylated donkey-anti-mouse IgG (1:500–1:2000, Vector Laboratories). For all immunohistochemical analyses, three independent brains for both wild-type and Nfib mutants were sectioned and stained. Hippocampal sections from comparable positions along the rostrocaudal axis in wild-type and knock-out samples were imaged using an upright microscope (Zeiss Z1) attached to a digital camera (Zeiss AxioCam HRc), and images were captured using AxioVision software (Zeiss). Immunofluorescence labeling was performed as described above with minor modifications. The primary antibody was incubated overnight at 4°C, and sections were washed and then incubated in secondary antibody, before being washed again and then mounted in PVA/DABCO (Fluka) mounting medium. The primary antibodies used for immunofluorescent labeling were anti-NFIB (rabbit polyclonal, 1:1000; Geneka Biotechnology) and anti-phosphohistone H3 antibody (rabbit polyclonal, 1:1000; Upstate). The secondary antibody used was goat-anti-rabbit IgG AlexaFluor488 (1:1000; Invtrogen). Fluorescence confocal microscopic images of NFIB expression were obtained with a laser scanning microscope (Zeiss 510 Meta). To quantify proliferation in the developing hippocampus, sections were imaged using an upright fluorescence microscope (Zeiss Z1) attached to a digital camera (Zeiss AxioCam HRc) using AxioVision software (Zeiss). Eight to 10 optical sections encompassing the entire 45 μm section were captured with an ApoTome (Zeiss). To calculate the total number of hippocampal phosphohistone H3-positive cells per unit area, the outline of each hippocampus was traced using AxioVision software (Zeiss), and the number of immunolabeled cells in focus in each optical section counted and pooled. (n = 3 for both wild-type and knock-out at all ages).
Immunohistochemistry on paraffin sections.
E17 wild-type and Nfib-deficient brains were perfused as above. PFA solution was removed and replaced with PBS at least 48 h before use. Brains were embedded in paraffin wax and sectioned at a thickness of 6 μm. Antigen retrieval was performed using a 10 mm, pH 6 sodium citrate solution. Immunohistochemistry was performed as described above with minor modifications. Primary antibodies used for immunohistochemistry were: anti-Prox1 (rabbit polyclonal, 1:2000; Chemicon), anti-nestin (rabbit polyclonal, 1:750; Abcam) and anti-β-galactosidase (rabbit polyclonal, 1:1000; MP Biomedicals). A biotinylated goat-anti-rabbit IgG secondary antibody (Vector Laboratories) was used at 1:1000.
Hippocampal cell culture and immunofluorescent staining of cultures.
Cultures containing neurons and glia were prepared from wild-type or Nfib mutant hippocampi at E16. Hippocampi were dissected, trypsinized at 37°C for 5 min, and then triturated using a pipette tip. Trypan-excluding cells were plated at a density of 200,000 cells/13 mm diameter on poly-d-lysine (50 μg/ml)-coated glass coverslips. Cells were grown for 1–7 d in vitro in Neurobasal Medium (Invitrogen) with 10% fetal calf serum, B27 supplement (Invitrogen) and 5 mm glutamine. After this time, cells were washed in PBS and then fixed in 4% PFA for 15 min. Immunocytochemistry was performed by first blocking and permeabilizing cells in PBS containing 0.5% Triton X-100 and 2% normal goat serum. Primary antibody was included for 2 h in blocking buffer according to the following dilutions: anti-NFIB (rabbit polyclonal, 1:1000; Geneka Biotechnology), anti-Tuj1 (mouse monoclonal, 1:1000, Covance), anti-nestin (mouse monoclonal, 1:1500; Developmental Studies Hybridoma Bank) and anti-GFAP (mouse monoclonal, 1:1000; Chemicon). After washes in PBS, cells were incubated in either goat-anti-mouse AlexaFluor488 or goat-anti-rabbit AlexaFluor594 (Invitrogen) at 1:1000 for 1 h. Conjugated anti-phalloidin/AlexaFluor594 (1:50, Invitrogen) was used to stain F-actin. 4′,6-Diamidino-2-phenylindole (DAPI, 1:20,000 in PBS; Sigma) was used as a counterstain (300 nm) and cells were mounted for fluorescence microscopy in PVA/DABCO (Fluka).
Analysis of cultured hippocampal cells.
Dissociated hippocampal cells grown for 24 or 48 h were stained with phalloidin (see above), then imaged with an upright fluorescence microscope (Zeiss Z1) attached to a digital camera (Zeiss AxioCam HRc) using AxioVision software (Zeiss). Only cells that had at least one process >30 μm (the length was arbitrarily selected to ensure viable cells) and that were not in contact with any other cell were included in our analysis. Using ImageJ (NIH, publically available), we measured the following parameters of the cultured hippocampal cells: the length of the longest primary process, the total number of primary processes per cell and the total number of branch points on the primary processes of each cell. A two-tailed unpaired Student's t test was used compare wild-type and knock-out samples. Graphs depict the pooled data from wild-type (n = 5) and knock-out (n = 4) hippocampal cultures (total cells analyzed: wild-type 24 h = 236, 48 h = 143; knock-out 24 h = 168, 48 h = 144). Error bars indicate SEM.
In situ hybridization.
E12 and E18 embryos were collected and fixed as described above (n = 3 for both wild-type and knock-out). In situ hybridization was performed using antisense probes to Wnt2b, Wnt3a, Lhx2, SCIP and KA1 as described previously (Mangale et al., 2008) with minor modifications. The hybridization temperatures were 68°C for Wnt2b and 70°C for the remaining probes. The color reaction solution was NBT/BCIP.
Results
NFIB is expressed in hippocampal neurons and glia
Nfib-deficient mice display hippocampal defects, including the morphological absence of the DG (Fig. 1A–D) (Steele-Perkins et al., 2005). To elucidate the mechanisms responsible for this phenotype we investigated the cell-type specific expression of NFIB during embryogenesis. NFIB is expressed in the hippocampal formation from E13 through postnatal ages, and in the adult (Plachez et al., 2008). At E18, NFIB is highly expressed in the hippocampus (Fig. 1E), and is especially prominent in the ventricular zone (VZ) (Fig. 1F), and the developing DG (Fig. 1G). We observed very low levels of NFIB expression in the fimbrial glioepithelium (Fig. 1H). Immunohistochemistry on dissociated hippocampal cultures demonstrated the coexpression of NFIB with both the neuronal marker TuJ1 (Fig. 1I) and the astrocytic marker GFAP (Fig. 1J), demonstrating that NFIB is expressed in both neurons and glia.

NFIB is expressed in hippocampal neurons and glia. A–D, Hematoxylin staining of coronal sections of E18 wild-type (A, C) and Nfib-deficient (B, D) mouse brains. The DG is morphologically absent in the hippocampus of Nfib-deficient mice (D). The point at which the hippocampal fissure has failed to form in the mutant is indicated with an arrow in D. Panels C and D are higher magnifications of the boxed regions in A and B, respectively. E–H, Coronal section of an E18 wild-type brain (E), showing expression of NFIB protein in the VZ of the hippocampus (arrow in F) and in the DG (arrow in G). Only low levels of NFIB are expressed in the fimbria (arrow in H). I–J, In vitro cultures of wild-type hippocampal cells demonstrating that NFIB colocalizes with both the neuronal marker TuJ1 (I) and the glial marker GFAP (J). Scale bar: A, B, 500 μm; C, D, E, 80 μm; F–H, 25 μm; I, 60 μm; J, 15 μm.
Nfib does not regulate patterning, proliferation or apoptosis in hippocampal development
As NFIB is expressed in the developing hippocampal primordium, and the hippocampus is greatly reduced in Nfib deficient mice, we first examined whether this phenotype was due to an early patterning defect or changes in cell proliferation or cell-death in this region. The cortical hem is a transient medial structure and an important patterning center for early embryonic telencephalic development (Grove and Tole, 1999; Shimogori et al., 2004; Yoshida et al., 2006). Disruption of the cortical hem results in a morphologically aberrant hippocampus (Lee et al., 2000). The cortical hem is an abundant source of Wnt proteins and can be visualized by specific Wnt probes such as Wnt2b and Wnt3a (Grove et al., 1998). Wnt2b and Wnt3a expression appeared normal in the cortical hem of Nfib-deficient mice at E12.5. Strong expression of Lef1 in the cortical neuroepithelium (data not shown) further supported the interpretation that the defect observed in Nfib-deficient mice was not due to patterning defects associated with the hem or the Wnt signaling pathway. Furthermore, expression of Lhx2, a transcription factor required for all cortical derivatives, including the hippocampus, appeared normal in Nfib mutants (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), where it was excluded from the hem (Bulchand et al., 2001). Given the defects within the DG, we next investigated whether this could be due to either a decrease in progenitor proliferation early in development or an increase in apoptosis. We analyzed cellular proliferation (E13-E15) and apoptosis (E14-E18) in the hippocampus and observed no significant differences between wild-type and Nfib-deficient mice (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material). This suggests that neither aberrant proliferation early in development nor excessive apoptosis contributes to the Nfib hippocampal phenotype. As members of the Nfi gene family have previously been implicated in glial development (Shu et al., 2003; Steele-Perkins et al., 2005; Cebolla and Vallejo, 2006; Deneen et al., 2006; Gopalan et al., 2006; Driller et al., 2007), we next investigated glial development in the hippocampus of Nfib-deficient mice.
Nestin is expressed by radial progenitors in Nfib-deficient mice
Radial glial cells are characterized by their bipolar radial processes that extend from their cell bodies in the VZ to the pial and ventricular surfaces of the brain. They differentiate from neuroepithelial progenitor cells and function as a scaffold for migrating cells, and are the major source of neuronal and glial progenitors at E13/14 in mice (Götz and Barde, 2005; Mori et al., 2005). To assess the potential role of Nfib in radial glial development we assessed different stages of glial differentiation using specific markers (Fig. 2A). Nestin is an intermediate filament protein expressed by neuronal and glial progenitors (Lendahl et al., 1990) from E10.5 in the mouse neuroepithelium (Dahlstrand et al., 1995), thereby acting as a marker for these early radial progenitor cells (Yamaguchi et al., 2000). Nestin was expressed in the hippocampus of Nfib-deficient mice from E14 to E18 (Fig. 2B–G), indicating that the radial progenitors are correctly specified in the hippocampus of these animals. We next examined the differentiation of radial progenitors to radial glia.
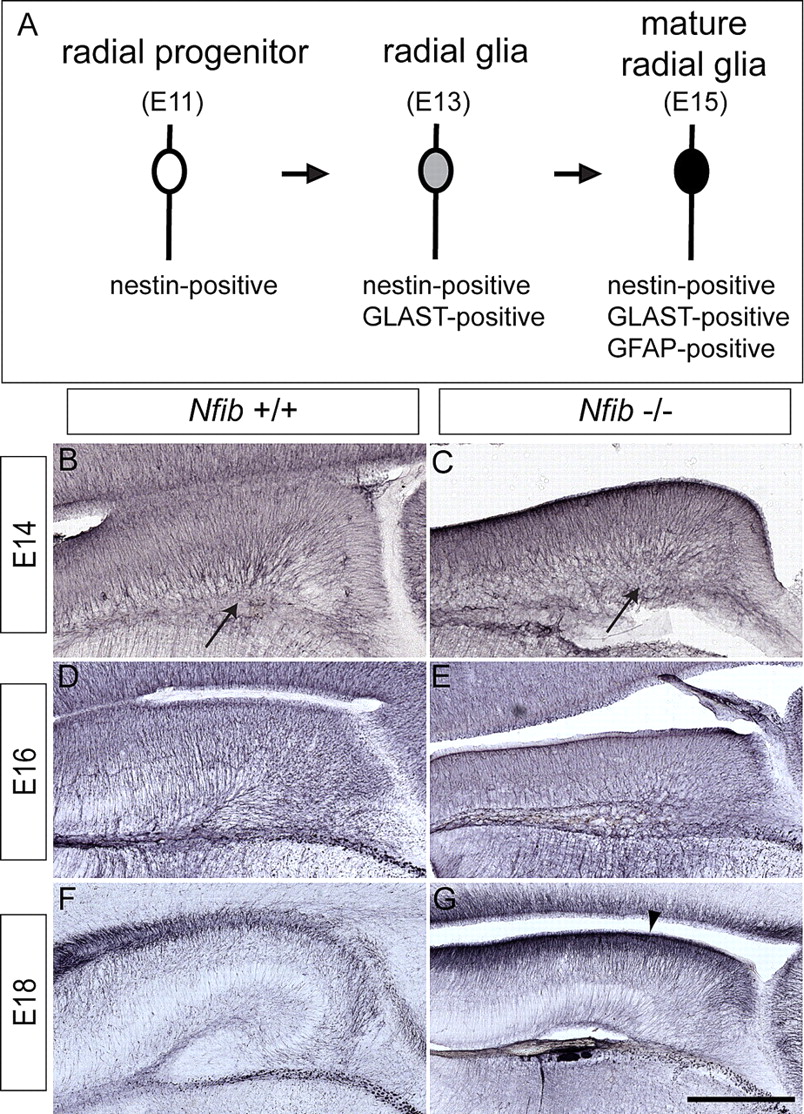
Nestin-positive radial glia are present in Nfib-deficient mice. A, The stages of glial differentiation can be followed through the temporal expression of specific molecules including nestin (E11 onwards), GLAST (E13 onwards) and GFAP (E15 onwards). B–G, Expression of the radial progenitor marker nestin at E14 (B, C), E16 (D, E), and E18 (F, G). Nestin is expressed in wild-type (B, D, F) and Nfib-deficient mice (C, E, G), demonstrating that radial progenitors are specified in the absence of Nfib. Expression of nestin appears higher at E18 in the mutant (arrowhead in G). Arrows in B and C indicate the site of the future hippocampal fissure. Scale bar: B, C, 200 μm; D, E, 125 μm; F, G, 80 μm.
Nfib regulates the development of a subpopulation of radial glia in the hippocampus
Astrocyte-specific glutamate transporter (GLAST) is expressed in radial glial cells that have differentiated from radial progenitors (Shibata et al., 1997). GLAST is a marker for glial differentiation that is expressed from E13/14 in mice, which coincides with the expression of NFIB in the hippocampal anlage. We found that from E14 in wild-type mice, GLAST expression was prominent in the fimbrial region, the basement membrane and in the developing ammonic neuroepithelium (Fig. 3A,C,E). Interestingly, in the ammonic neuroepithelium, GLAST-expressing fibers converge on an area of the marginal zone where the hippocampal fissure will form (Fig. 3A,C). In contrast, expression of GLAST in Nfib-deficient mice was absent from the ammonic neuroepithelium at E14 and E15 (Fig. 3B,D). At E18, low GLAST expression was observed in the ammonic neuroepithelium of the mutant, perhaps indicating a delayed onset of expression (Fig. 3F). Expression of GLAST in the fimbria and basement membrane, however, was normal at all ages studied, indicative of a specific defect in the development of radial glia in the ammonic neuroepithelium of Nfib-deficient mice. Tenascin-C is another marker of radial glial cells in the developing hippocampus (Yuasa, 2001) and its expression was also markedly decreased in the developing ammonic neuroepithelium of Nfib-deficient mice at E15 (Fig. 3H). These data demonstrate that the development of radial glia in the ammonic neuroepithelium of Nfib-deficient mice is aberrant, and, consequently, that distinct molecular pathways regulate the development of the ammonic neuroepithelium and fimbrial glioepithelium.

Aberrant expression of GLAST and tenascin-C in the hippocampus of Nfib-deficient mice. A–H, Expression of the glial markers GLAST (A–F) and tenascin-C (G, H) in coronal sections of wild-type (A, C, E, G) and Nfib-deficient (B, D, F, H) mice. GLAST expression is evident at E14 in the ammonic neuroepithelium of the wild-type hippocampus (A) but, strikingly, is absent from the ammonic neuroepithelium of Nfib null mutants (B), although it is evident in the fimbria (arrowheads in B, D, F). Arrows indicate GLAST-positive fibers converging at the future “anchor-point” of the hippocampal fissure. Asterisks indicate GLAST expression in the meninges. At E18, expression of GLAST in the mutant is present, but at a lower level than in the wild-type (compare E to F). Similarly, expression of tenascin-C in the mutant (H) is markedly lower than in the wild type (G) at E15. Scale bar: A, B, 200 μm; C, D, G, H, 180 μm; E, F, 80 μm.
The supragranular glial bundle is absent, but the fimbrial glial bundle is still present, in Nfib-deficient mice
After the differentiation of radial glia from radial progenitors as shown by GLAST expression, radial glia develop further into more mature glia, with astrocytic characteristics including glial fibrillary acidic protein (GFAP) expression (Noctor et al., 2002). In the wild-type hippocampus at E15, GFAP expression was observed in both the fimbrial glia and the ammonic glia (Fig. 4A). In contrast, in the Nfib knock-out, expression was absent in the ammonic glia (Fig. 4B). Expression was observed in the fimbrial glia, indicating that GFAP expression per se was not delayed in the Nfib mutants. At E16 in wild-type mice, two major glial bundles are present: the fimbrial glial bundle and the supragranular bundle (Fig. 4C). The supragranular bundle can be observed as a fascicle of GFAP-positive processes extending to the pial surface and forming a juncture with the fimbrial bundle (Fig. 4E). In contrast, mature, GFAP-expressing glia do not developin the ammonic neuroepithelium of Nfib-deficient mice, resulting in the absence of the supragranular bundle and a loss of the hippocampal fissure (Fig. 4D,F). The failure of the hippocampal fissure to develop in the mutant is most evident at E18 (Fig. 4G,H). However, GFAP expression was detected in the fimbrial glial bundle across all ages examined in the Nfib knock-out (Fig. 4B,D,H). This confirms that distinct developmental programs control formation of the supragranular bundle and the fimbrial bundle, and raises the hypothesis that the hippocampal fissure is dependent on the formation of the supragranular glial bundle.

The supragranular glial bundle fails to form in Nfib knock-out mice. A–H, Coronal sections of the hippocampus between E15 and E18 in wild-type (A, C, E, G) and Nfib-deficient (B, D, F, H) mice, showing expression of GFAP, a marker for mature radial glia. At E15, expression in the wild type (A) is evident in the fimbrial glioepithelium (arrowhead) and ammonic neuroepithelium (arrow), but is only seen in the fimbrial glioepithelium of the mutant (arrowhead in B). At E16, the disparity in GFAP expression in the ammonic neuroepithelium between the wild type (C) and knock-out (D) becomes more apparent. The boxed regions in C and D are shown at a higher magnification in E and F, respectively. In the wild type (E), both the fimbrial bundle (arrowhead) and the supragranular bundle (arrow) are evident, whereas only the fimbrial bundle is detected in the mutant (arrowhead in F). At E18, GFAP is expressed broadly in the wild-type hippocampus (G). Notably, a low level of GFAP expression is observed in the mutant ammonic neuroepithelium at E18 (arrow in H), indicating that GFAP expression may be delayed or compensated for by another mechanism. Scale bar: A, B, 180 μm; C, D, 125 μm; E, F, 40 μm; G, H, 80 μm.
Altered morphology of nestin-positive cells from Nfib-deficient mice
The decreased expression of GLAST and GFAP in the mutant ammonic neuroepithelium indicates that Nfib regulates the differentiation of nestin-positive progenitors into mature radial glia. As morphological features of glia, such as branching and elongation, are indicative of maturation and are known to be important for neuronal migration during cortical and hippocampal development (Takahashi et al., 1990; Frotscher et al., 2003; Gupta et al., 2003; Elias et al., 2007), we examined the morphology of glial cells from Nfib-deficient hippocampi in more detail using an in vitro culture technique. Cells were harvested from E16 Nfib-deficient and wild-type littermate hippocampi and cultured for 24 or 48 h on poly-d-lysine-coated coverslips. All cells we analyzed were nestin-positive. In wild-type cultures we found that 83 ± 1.91% of cells in the culture expressed both nestin and NFIB (total of 451 cells pooled from 3 independent replicates) (Fig. 5A). We then analyzed the morphological characteristics of cultured wild-type (Fig. 5B) and Nfib-deficient (Fig. 5C) hippocampal cells via phalloidin staining. We found no difference in the length of the longest primary process between Nfib-deficient and wild-type mice (Fig. 5D). However, cells from Nfib-deficient mice extended significantly more processes from the cell body, and these processes were more branched than those from wild-type mice at both 24 and 48 h in vitro (p < 0.0001, Student's t test) (Fig. 5E,F).

Cultured hippocampal radial glia from Nfib-deficient mice display an altered morphology. Dissociated wild-type hippocampal cells were cultured for 24 h in vitro. A, Immunohistochemistry against the radial progenitor marker nestin demonstrated that under culture conditions all cells expressed this protein. Furthermore, 85% were also NFIB-positive (arrow indicates an NFIB-positive cell, arrowhead indicates an NFIB-negative cell). B, C, Hippocampal cells from wild-type (B) and mutant (C) mice were cultured for 24 h. Staining with phalloidin, a marker for filamentous actin, demonstrated that cells lacking Nfib exhibited an aberrant morphology. Analysis of both 24 and 48 h cultures showed that, although the length of the longest process was similar between wild-type (WT) and knock-out (KO) cells (D), cells from Nfib-deficient hippocampi had significantly more processes emanating from the cell body (E) and significantly more process branch points (F) in comparison with wild-type controls. *p < 0.0001, Student's t test. Error bars indicate SEM. G–J, Coronal paraffin sections of wild-type (G, I) and Nfib-deficient brains (H, J) at E17 showing expression of nestin. The boxed regions in G and H are shown at a higher magnification in I and J, respectively. The expression of nestin in knock-outs appears higher than that in controls, and, furthermore, whereas nestin fibers appear straight in the wild type (arrowhead in I), many fibers in the mutant appear wavy (arrow in J). Scale bars: (in C) A, 40 μm; B, C, 10 μm; (in J) G, H, 100 μm; I, J, 15 μm.
A potential explanation for these morphological differences could be that progenitor cells from Nfib-deficient mice are differentiating into neuronal cells. To assess this, we stained cultures with TuJ1, a specific neuronal marker, and found that there was no significant difference in the number of TuJ1-positive cells between wild-type (24 h: 83.79 ± 1.4%, total of 312 cells from 3 independent replicates; 48 h: 81.69 ± 2.19%, total of 291 cells from 3 independent replicates) and Nfib-deficient (24 h: 88.79 ± 2.46%, total of 312 cells from 3 independent replicates; 48 h: 84.36 ± 3.11%, total of 265 cells from 3 independent replicates) cultures. These results suggest that aberrant radial glial maturation, rather than increased neuronal differentiation, underlies the hippocampal phenotype observed in Nfib-deficient mice. To more closely examine in vivo morphology of nestin-positive fibers we analyzed nestin expression in thin paraffin sections at E17. This analysis yielded two major findings. First, nestin-positive fibers in the Nfib mutant appeared wavy compared with controls (Fig. 5G–J). This altered morphology in vivo possibly reflects our in vitro data, as similar morphological differences in radial glia have been observed in the cortex of Pax6-deficient mice (Götz et al., 1998). Second, expression of nestin in the Nfib mutants appeared higher than in the wild-type mice (Fig. 5G–J; see also Fig. 2F,G), perhaps indicative of cells remaining as progenitors in the mutant (Dahlstrand et al., 1995). To investigate this we analyzed proliferation at E18 using the mitotic marker phosphohistone H3 (PH3). Significantly, more PH3-positive cells were per millimeter were located in the ammonic neuroepithelium of Nfib-deficient mice (57.3 ± 2.32) compared with controls (31.5 ± 3.36, p < 0.0001, Student's t test) (Fig. 6A,B) indicative of delayed maturation of radial glia. Furthermore, there were fewer mitotic cells in the dentate migratory stream of Nfib-deficient mice (8.8 ± 0.99) compared with wild-type animals (23.7 ± 2.89, p < 0.0001, Student's t test) (Fig. 6A,B). The decrease in mitotic cells in the dentate migratory stream of Nfib mutant mice may reflect a decrease in the production of dentate granule neurons migrating toward the incipient DG. To investigate this, we examined the development of this population of cells within the hippocampus.

Aberrant proliferation in the hippocampus of Nfi-deficient mice at E18. Phosphohistone H3 (PH3) immunohistochemistry on wild-type (A) and Nfib-deficient mice (B) in the mouse hippocampus at E18. The number of PH3-positive cells in the ventricular zone (VZ) of the hippocampus was higher in Nfib-deficient mice compared with controls, whereas there were fewer PH3-positive cells in the dentate migratory stream (indicated by dashed lines) of the mutant. Scale bars: A, B, 80 μm.
Defective migration of dentate granule cells in Nfib-deficient mice
The absence of dentate granule neurons results in the phenotypic loss of the DG (Zhou et al., 2004). To investigate whether these neurons were affected by the loss of Nfib we analyzed the expression of the transcription factor Prospero-related homeobox 1 (Prox1), a marker of differentiated dentate granule cells in the prenatal and postnatal mouse brain (Pleasure et al., 2000a). From E15.5, Prox1-positive cells migrate from the dentate neuroepithelium adjacent to the fimbria, to the granular layer of the DG (Nakahira and Yuasa, 2005) (Fig. 7 A,B, arrowheads). Dentate granule cells in the wild-type hippocampal formation had begun migrating from the VZ by E16 (Fig. 7A). At E18 the characteristic V-shape of the DG is highlighted by Prox1 staining in the granular layer and hilar region (Fig. 7C). No defect was observed in the generation of Prox1-positive granule cells in the VZ of the Nfib-deficient mice at E16 (Fig. 7B). However, the positioning of the dentate granule cells was disrupted at E18 (Fig. 7D) as they failed to populate the presumptive DG. Using thin paraffin sections we quantified the number of Prox1-positive cells in the hippocampus at E17 and found no significant difference between wild-type (Fig. 7E, 168.7 ± 12.91 cells) and Nfib-deficient (Fig. 7F, 162.3 ± 4.41 cells; p value >0.05) mice indicating dentate granule neuron production is normal, at least prenatally, in the absence of Nfib. However, as the dentate gyrus develops predominantly postnatally further investigation is required to examine the role of Nfib in proliferating precursors within the hilar region that may give rise to additional granule neurons. Nevertheless, the radial glial scaffold is crucial for granule cell migration into the DG (Rickmann et al., 1987; Li and Pleasure, 2005) and it appears that, in the Nfib-deficient mice, the prenatally generated dentate granule cells have only migrated to the farthest extent of the fimbrial glial bundle (compare Figs. 4H and 7D). Both the lack of a mature glial substrate and altered glial morphology may be responsible for the Prox1-positive dentate granule cell migration defect in Nfib-deficient mice, leading to a failure of the DG to form.

Defects in the migration of dentate granule cells in Nfib-deficient mice. A–D, Coronal sections of wild-type (A, C) and Nfib-deficient mice (B, D) at E16 (A, B) and E18 (C, D) labeled with Prox1, a marker for dentate granule cells. Prox1-positive granule cells are born in the dentate neuroepithelium (arrowheads) of both wild-type (A) and Nfib-deficient (B) mice but fail to migrate into the presumptive DG (arrow in C) in mice lacking Nfib (D). Quantification of the number of Prox1-positive cells in 6 μm coronal paraffin sections showed no significant difference between E17 wild-type (E, 168.7 ± 12.91 cells) and Nfib-deficient (F, 162.3 ± 4.41 cells) hippocampal mouse sections. Statistical error indicates SEM. Scale bar: A, B, 125 μm; C, D, 80 μm.E,F 100 μm.
Tbr1-positive pyramidal neurons migrate within the hippocampal formation in Nfib-deficient mice
We next investigated the development of pyramidal neuron populations, as similar DG phenotypes to those observed in Nfib-deficient mice have been reported in mice carrying defects in genes reported to mediate neuronal development. These include Lhx5, cdk5, CXCR4 and LRP6 (Ohshima et al., 1996; Zhao et al., 1999; Lu et al., 2002; Zhou et al., 2004). We first analyzed the development of pyramidal neurons in the hippocampus using Tbr1, a transcription factor expressed by glutamatergic neurons (Englund et al., 2005; Hevner et al., 2006). Expression of Tbr1 protein in the hippocampus of Nfib-deficient mice at E14 was similar to its expression in wild-type littermates (Fig. 8A,B). The migration of the Tbr1-positive cells from the VZ to the presumptive pyramidal cell layer appeared normal, suggesting that the nestin-positive glial scaffold is sufficient for the migration of Tbr1-positive cells, and is unaffected by the loss of Nfib. At E16, lamination is evident in wild-type hippocampus (Fig. 8C, arrowhead), but is not observed until E18 in the mutant mice (Fig. 8F, arrowhead). As Tbr1 is a general marker of projection neurons produced from the pallial neuroepithelium we next examined whether specific hippocampal subfields were generated in the Nfib mutants. Both the CA1-specific marker SCIP (Pou3f1) (Frantz et al., 1994), and the CA3-specific marker, KA1 (Grik4) (Bettler et al., 1990), were expressed in the Nfib mutant. These results indicated that the glutamatergic neurons are born in Nfib-deficient mice, but these regions still appeared smaller in the Nfib mutant compared with wild-type brains. Hence, either there are a reduced number of glutamatergic neurons in these areas, or the migration of the glutamatergic neurons and their lamination within the CA regions is disrupted because of the defects in the maturation of the glial scaffold.

Glutamatergic neurons develop in the hippocampus of Nfi-deficient mice. A–F, Expression of Tbr1, a marker for glutamatergic neurons, in coronal sections of wild-type (A, C, E) and Nfib-deficient (B, D, F) hippocampi at E14 (A, B), E16 (C, D) and E18 (E, F). Tbr1-positive pyramidal neurons are generated and migrate within the hippocampus of Nfib-deficient mice. However, some notable differences between wild types and mutants are apparent. Lamination of the hippocampus is first observed at E16 in the wild type (C, arrows) but only at E18 in the mutant (F, arrows). Furthermore, the migration of Tbr1-positive neurons into the developing DG observed in the wild type (E) is aberrant in the mutant (F). G–J, Expression of the CA1-specfic marker SCIP (G, H) and the CA3-specific marker KA1 (I–J) visualized using in situ hybridization. The specification and differentiation of both CA1 and CA3 hippocampal subfields is apparent in Nfib-deficient mice. Scale bar: A, B, 200 μm; C, D, 125 μm; E–J, 80 μm.
Localization of other neuronal populations in Nfib-deficient mice reflects aberrant glial-derived DG development
We have previously demonstrated that interneurons do not express Nfib (Plachez et al., 2008), and, moreover, it is known that tangential migration of interneurons is not required for the morphological formation of the hippocampus (Pleasure et al., 2000b; Cobos et al., 2005). However, Nfib-mediated glial development may play a role in the localization of these interneurons within the developing hippocampus. Calbindin and calretinin are calcium-binding proteins that are collectively expressed by most interneurons that migrate tangentially from the caudal ganglionic eminence to the DG and CA regions of the hippocampus throughout development (Gulyás et al., 1992; Jiang and Swann, 1997; Förster et al., 2006; Wonders and Anderson, 2006). Calbindin-positive interneurons are observed in a normal pattern within the hippocampus of E16 and E18 wild-type and Nfib-knock-out brains (Fig. 9A–D), suggesting that this population of interneurons is unaffected by the aberrant maturation of ammonic glia. The formation of the DG can be observed in wild-type mice by examining calretinin expression, as these cells line the hippocampal fissure and populate the future hilar region of the DG (Fig. 9E,G). Calretinin-positive neurons migrate correctly into the hippocampal formation of the Nfib mutant at E16 and E18, but are unable to enter the dentate gyrus (Fig. 9F,H).

Migration of interneurons and Cajal-Retzius cells occurs normally in Nfib-deficient mice. A–J, Expression of calbindin (A–D), calretinin (E–H) and reelin (I, J) in coronal sections of wild-type and Nfib-deficient mice. Calbindin-positive interneurons migrate normally into the hippocampus of mice lacking Nfib (B, D). At E16, calretinin expression is evident in the marginal zone of both wild-type (E) and mutant (F) hippocampi. However, the developing hippocampal fissure (arrow in E) is morphologically absent in the mutant. Comparison of wild-type (G) and mutant (H) sections at E18 demonstrates the absence of the DG (arrowhead in G) in mice lacking Nfib. Migration of reelin-positive Cajal-Retzius cells (I, J) into the hippocampus also occurs normally. However, Cajal-Retzius cells fail to migrate dorsally from the marginal zone into the DG by E18 in the mutant (J). Scale bar: A, B, E, F, 125 μm; C, D, G–J, 80 μm.
Calretinin is also a marker of Cajal-Retzius cells, which produce the extracellular molecule reelin (Frotscher, 1998; Bielle et al., 2005). Reelin plays an important developmental role in hippocampus, including regulating the formation of a radial glial scaffold within the DG (Nakajima et al., 1997; Frotscher et al., 2003). We observed no obvious difference in reelin expression in the hippocampus of either Nfib-deficient (Fig. 9J) or wild-type mice (Fig. 9I) at E18. What was striking, however, was that reelin-positive cells were unable to migrate along the hippocampal fissure (Fig. 9J). Furthermore, as reelin-positive hippocampal cells are derived from the cortical hem (García-Moreno et al., 2007) and our analysis indicated cortical hem formation was normal in the Nfib mutant mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), we conclude that the defects observed are not associated with specification of Cajal-Retzius cells or reelin production.
Discussion
Although the morphogenesis of the hippocampal formation has been anatomically characterized (Rickmann et al., 1987; Altman and Bayer, 1990a; Sievers et al., 1992), the molecular control of this process is only now becoming evident (Li and Pleasure, 2007). Especially prominent during the development of this region is the involvement of radial glia. Only a small number of factors have been identified that may be involved in the formation of the GFAP-positive glial scaffold during hippocampal development (Oldekamp et al., 2004; Zhou et al., 2004; Zhao et al., 2006). Here, we demonstrate that the maturation of distinct populations of radial glia are critical for certain aspects of DG formation, particularly the emergence of the hippocampal fissure and the migration of dentate granule cells.
Although there are obvious morphological abnormalities in the hippocampal region throughout development in Nfib-deficient mice, neurogenesis and migration of glutamatergic neurons and interneurons are relatively normal. We found no obvious defects with respect to apoptosis, patterning, or the rates of proliferation during early hippocampal development in Nfib-deficient mice. Moreover, although dentate granule cells only migrate part of the way toward the presumptive DG, as observed using the marker Prox1, they are born in the VZ of mutant mice, as in wild-type controls. The fimbrial bundle forms normally whereas the supragranular bundle is absent in Nfib-deficient mice. These results indicate that the Nfib-deficient mice are a valuable tool for examining the roles of separate glial populations in hippocampal formation without disrupting other developmental processes. Collectively these data also show that the hippocampal fissure is formed predominantly through the maturation of the supragranular bundle of radial glia and the subsequent migration of neurons, and not through another mechanism such as tissue folding, despite this region having a morphologically folded appearance. The data also highlight that different molecular mechanisms are required for the formation of the fimbrial and supragranular glial bundles. Nfib has also been shown to regulate glial maturation of the glial wedge at the midline of the developing cerebral cortex at the cortico-septal boundary (Steele-Perkins et al., 2005). These data suggest the hypothesis that Nfib may regulate the differentiation of glial populations involved in the formation of morphological boundaries that direct cellular migration within the brain. Further investigation of the molecular determinants of specific glial populations in the brain is required to test this hypothesis however.
Members of the Nfi family of transcription factors, namely Nfia, Nfib and Nfix, have been implicated in astrocyte-specific gene expression and glial development (Shu et al., 2003; Steele-Perkins et al., 2005; Cebolla and Vallejo, 2006; Gopalan et al., 2006; Driller et al., 2007; Campbell et al., 2008). Both Nfia and Nfib have been shown to promote gliogenesis and subsequent terminal astrocytic differentiation in the mouse spinal cord, without affecting neurogenesis (Deneen et al., 2006). Nfib-deficient mice display reduced mature glial populations at the cortical midline of the brain as observed using antibodies against GFAP (Steele-Perkins et al., 2005). We show that Nfib affects the differentiation of radial glia, specifically from neuroepithelial progenitors in the mouse ammonic neuroepithelium.
NFIB expression is first detected in the hippocampal primordium at E13 (Plachez et al., 2008) coinciding with the onset of gliogenesis and also neurogenesis in this region in the rodent (Altman and Bayer, 1990b). Radial glia have multiple functions encompassing neurogenesis and boundary and patterning specification, as well as acting as migratory scaffolds for neurons (Götz and Barde, 2005). The glial scaffold in the DG may serve to fulfill all these functions, namely demarcating the area in which the DG will develop, establishing a radial migratory route for incoming granule cells and thus playing a major role in the morphological formation of the DG (Rickmann et al., 1987; Sievers et al., 1992; Li and Pleasure, 2005). In wild-type mice, the GFAP-expressing supragranular bundle connects the ventricular surface to the site of the developing fissure on the pial surface, and our results suggest that this glial bundle plays a role as a substrate and/or structural determinant in the development of the DG (Fig. 10). The site on the pial surface that acts as an anchor-point for the supragranular bundle is also the target scaffold for the fimbrial bundle. Both glial bundles are necessary to complete the scaffold required for dentate granule cell migration. In the Nfib-deficient mice there was a severe lack of glial maturation in cells originating from the ammonic neuroepithelium and as a result no supragranular bundle was formed (Fig. 10B). The fimbrial bundle, which originates from the fimbrial glioepithelium, forms in the Nfib-deficient mice, and thus represents an apparently distinct radial glial population to those that form the supragranular glial bundle. As glial wedge and indusium griseum glia development are also disrupted in Nfib-deficient mice (Steele-Perkins et al., 2005), the fimbrial glia may represent a glial population with molecular control distinct from the rest of the pallium. Importantly, the fimbrial bundle alone is not sufficient to overcome the loss of the supragranular bundle in the formation of the DG and for the complete migration of the dentate granule cells. These results demonstrate that the morphological development of the DG requires radial glia from two distinct neuroepithelial origins and that the molecular control of radial glial differentiation is not uniform across the hippocampal primordium.

Summary of hippocampal defects in Nfib knock-out mice. A, B, Differentiating radial glial cells are depicted in the hippocampus of wild-type (A) or Nfib-deficient mice (B). At E18 in the wild-type hippocampus, the GFAP-expressing supragranular and fimbrial glial bundles have formed and the DG is developing its characteristic V-shape (A). Incomplete glial development occurs in Nfib-deficient mice, resulting in the formation of the fimbrial glial bundle but not the supragranular glial bundle, thus severely disrupting hippocampal development (B). These results demonstrate that the correct morphogenesis of the DG requires both GFAP-expressing glial bundles.
In addition to our data, previous studies have shown that Nfi genes can regulate the expression of GFAP in vitro through direct binding to the GFAP promoter (Cebolla and Vallejo, 2006). Furthermore, we identified Nfi binding sites in the promoter regions of GLAST and tenascin-C (data not shown). Hence the absence of these markers alone could not definitively be used to study glial differentiation. To examine the morphological development of the radial glia from the ammonic neuroepithelium, we performed in vitro experiments using nestin to label the radial glial progenitors. Our in vitro data suggest that Nfib regulates primary glial process formation and branching, thus affecting the morphological differentiation of radial glial cells. This is further supported by our observations of irregular nestin-positive fibers in the Nfib mutant mice in vivo. Whether the regulation of glial morphological development is controlled directly by NFIB or indirectly through NFIB's regulation of GFAP, GLAST and tenascin-C remains to be elucidated. However, the results provide an explanation for the secondary effects on neuronal migration and the formation of the DG, because increased neuronal and glial branching has been shown to affect migration (Gupta et al., 2003; Koizumi et al., 2006; Elias et al., 2007), possibly because of branch destabilization and adhesion (Elias et al., 2007).
Factors controlling progenitor cell fate and differentiation have been shown to have widespread effects including hippocampal development. For example, forkhead box G1 (Foxg1) functions to repress early-born cell fate, especially Cajal-Retzius (CR) cells from the cortical hem and the pallial-subpallial boundary, and Foxg1 mutant mice show gross telencephalic abnormalities (Hanashima et al., 2004, 2007; Muzio and Mallamaci, 2005). Foxg1, expressed very early in the telencephalon, most likely regulates cell fate upstream or independent of Nfib, as we did not detect any abnormality of the cortical hem of Nfib-deficient mice. Mice deficient in the transcription factors Emx2 and Pax6 have a similar hippocampal phenotype to the Nfib-deficient mice (Pellegrini et al., 1996; Tole et al., 2000; Oldekamp et al., 2004; Zhao et al., 2006). For instance, each mutant lacks a discernible hippocampal fissure. Furthermore, Emx2 mutants display impaired GFAP expression and have defective Prox1-positive granule cell migration (Oldekamp et al., 2004; Zhao et al., 2006). As Emx2 is expressed earlier than Nfib in the mouse brain (Simeone et al., 1992) it may be that Nfib is a downstream target of Emx2. In support of this possibility, regulation of an Nfi family member, Nfia, by Emx2 has been suggested from microarray analysis of cortically derived neurospheres (Gangemi et al., 2006). Emx2 and Pax6 are involved in the regulation of multipotent progenitor cells in the VZ of the cortex (Götz et al., 1998; Heins et al., 2001; Quinn et al., 2007). The loss of either factor results in premature neuronal differentiation of progenitor cells, thereby depleting precursor cells and dramatically reducing the size of the cortex and DG (Pellegrini et al., 1996; Quinn et al., 2007). Consequently, when Nfib is expressed and would normally instruct progenitors to differentiate into GLAST- and subsequently GFAP-positive cells in these mutants, there are few undifferentiated progenitors remaining. We investigated whether Nfib deficiency resulted in increased neurogenesis in vitro but found no evidence for this. Although this result does not preclude Nfib/Emx2/Pax6 acting in the same pathway, it does suggest that the glial deficiency in the Nfib-knock-out mice is not due to precocious neurogenesis. It remains to be determined whether Emx2 or Pax6 form part of the same signaling network as Nfib. Significantly, Nfib-deficient mice do not have gross defects in neurogenesis and are, therefore, the first mutants allowing investigation into glial-specific hippocampal phenotypes.
The subgranular zone of the DG is a major site for the birth of new neurons from glial progenitors in mammals (Kaplan and Hinds, 1977; Eckenhoff and Rakic, 1988; van Praag et al., 2002). This specialized site is formed prenatally and postnatally but the details thereof have been scarce, especially with respect to the glial progenitors and their role in adult neurogenesis. We have demonstrated the importance of glial development in DG formation by using Nfib-deficient mice that allow us to study the specific loss of glial maturation in the ammonic neuroepithelium. NFIB is expressed in the subgranular zone of the adult hippocampus (Plachez et al., 2008) and hence further studies, using conditional knock-out technology, are needed to investigate the role of Nfib in adult neurogenesis and gliogenesis. Our data have underscored the importance of specific glial populations in the morphogenetic development of the hippocampus, especially the hippocampal fissure and dentate gyrus.
Footnotes
-
This work was funded by a National Health and Medical Research Council project grant (L.J.R.) and a Clive and Vera Ramaciotti grant (M.P.). The following authors are supported by National Health and Medical Research Council fellowships: L.J.R. (Senior Research Fellowship), M.P. (Howard Florey Centenary Fellowship), R.M. (CJ Martin Fellowship). S.M. is supported by a University of Queensland F.G Meade PhD Scholarship. We thank Lynette Knowles, John Baisden, Jane Ellis, Daphne Kusters, Sarah Croft, and Chantelle Reid for technical assistance, Rowan Tweedale for critical analysis of this manuscript, and Robert Hevner (University of Washington), Niels Danbolt (University of Oslo) and Andre Goffinet (University of Louvain Medical School) for providing antibodies. We thank Steve Kinsman and Aurora Anderson for initial observations and early discussions of the phenotype.
- Correspondence should be addressed to Linda J. Richards, The University of Queensland, Queensland Brain Institute, Brisbane QLD 4072, Australia. richards{at}uq.edu.au





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}