

Abstract
NMDA receptor (NMDAR)-dependent strengthening of neurotransmitter release has been widely observed, including in layer 5 (L5) pyramidal cells of the visual cortex, and is attributed to the axonal expression of NMDARs. However, we failed to detect NMDAR-mediated depolarizations or Ca2+ entry in L5 pyramidal cell axons when focally stimulated with NMDAR agonists. This suggests that NMDARs are excluded from the axon. In contrast, local GABAA receptor activation alters axonal excitability, indicating that exclusion of ligand-gated ion channels from the axon is not absolute. Because NMDARs are restricted to the dendrite, NMDARs must signal to the axon by an indirect mechanism to alter release. Although subthreshold somatic depolarizations were found to spread electrotonically hundreds of micrometers through the axon, the resulting axonal potential was insufficient to open voltage-sensitive Ca2+ channels. Therefore, if NMDAR-mediated facilitation of release is cell autonomous, it may depend on voltage signaling but apparently is independent of changes in basal Ca2+. Alternatively, this facilitation may be even less direct, requiring a cascade of events that are merely triggered by NMDAR activation.
Introduction
The expression of ligand-gated ion channels on axons near presynaptic sites of release provides a direct signaling pathway to locally modulate the strength of synaptic transmission between neurons independent of the somatodendritic compartment (MacDermott et al., 1999; Engelman and MacDermott, 2004). The regulatory action of presynaptic receptor signaling often affects Ca2+ influx at the site of release (Zucker and Regehr, 2002). Of particular interest, then, are axonal receptors that are Ca2+ permeable, such as NMDA receptors (NMDARs), because their influence on neurotransmitter release could be independent of any secondary or indirect signaling mechanism (Corlew et al., 2008). In fact, NMDAR activation alters transmitter release in a number of neuron types arguing for an axonal localization (Corlew et al., 2008). Yet, a search for direct NMDAR-mediated Ca2+ entry in cerebellar stellate cell axons failed to reveal functional channels (Christie and Jahr, 2008) despite NMDAR-dependent facilitation of release (Glitsch and Marty, 1999; Duguid and Smart, 2004; Glitsch 2008). Instead, dendritic NMDARs were found to depolarize the axon and allow Ca2+ influx through voltage-sensitive Ca2+ channels (VSCCs), consistent with an emerging view that somatodendritic electrical signaling alters axonal release (Alle and Geiger, 2006; Shu et al., 2006; Kole et al., 2007). Whether NMDARs can be directly detected in axons of other cell types has not been explored.
In contrast to GABAergic stellate cell interneurons in the cerebellum, pyramidal cells in neocortical layer 5 (L5) of visual cortex release glutamate, are primary projection neurons, and have extensive axonal arbors spanning multiple cortical layers. It has been argued that NMDAR-dependent strengthening of spontaneous and action potential-evoked neurotransmission in L5 pyramidal cells must reflect the close spatial association of NMDARs and sites of release on the axon (Sjöström et al., 2003; Corlew et al., 2007), because NMDAR-mediated Ca2+ entry and diffusion are highly constrained within a cell (Mainen et al., 1999a; Sabatini et al., 2002). However, without direct observation of functional NMDARs on L5 pyramidal cell axons, it remains possible that the regulatory functions attributed to axonal receptors are instead mediated by dendritic NMDARs.
In this report, we investigated the distribution of NMDARs in L5 pyramidal cells of the visual cortex using a variety of methods. In contrast to observations of NMDAR-mediated activity in dendrites, we find no evidence for the expression of functional NMDARs on axons. Focal stimulation of pyramidal cell axons with an NMDAR agonist failed to depolarize the axon, alter axonal excitability, or evoke NMDAR-mediated Ca2+ influx. Alteration of axonal excitability resulting from local GABAA receptor (GABAAR)-mediated activity suggests that not all ligand-gated ion channels are excluded from the axon. These data imply that dendritic NMDARs must signal, through an unknown mechanism independent of basal Ca2+, to presynaptic varicosities, thereby altering release.
Materials and Methods
Slice preparation and electrophysiology.
Cortical slices were prepared from Sprague Dawley rats (postnatal day 13–19) based on the method of Markram et al. (1997). Following isoflurane anesthesia, rats were decapitated, and the brain was removed by dissection. The sagittal surface of the right cerebral hemisphere was glued onto a cutting block angled at 30° such that during slicing the blade of the vibroslicer (VT12005; Leica Instruments) traversed from the rostral border of the neocortex toward the caudal border and down toward the midline. Slices (250 μm) were cut in an ice-cold solution containing (in mm) 87 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 10 glucose, and 75 sucrose. Slices were then transferred to a chamber containing artificial cerebrospinal solution (ACS) (in mm) 119 NaCl, 26.2 NaHCO3, 2.5 KCl, 1 NaH2PO4, 0.1 MgCl2, 3 CaCl2, and 11 glucose and incubated at 34°C for 30 min. Thereafter, slices were maintained at 22–25°C. The same solutions and procedures were used for slices prepared from the cerebellar vermis (see Fig. 6; Christie and Jahr, 2008). All solutions were bubbled with 95% O2 and 5% CO2. Animal handling and procedures were performed in accordance with Oregon Health and Science University Institutional Animal Care and Use Committee protocols.
Whole-cell recordings were obtained from large L5 pyramidal cells in visual cortex identified with gradient contrast infrared optics (Dodt et al., 2002) and two-photon fluorescence microscopy. On the basis of the somatic position within L5, distinct morphology, and characteristic bursting behavior, most cells were likely thick-tufted L5B pyramidal cells (Larkman and Mason, 1990; Mason and Larkman, 1990). Pyramidal cell axons were directly stimulated with an extracellular pipette filled with ACS (2–20 V, 20–100 μs) that was positioned within several micrometers of the process of interest. Iontophoretic pipettes (impedance >100 MΩ) were filled with either a 1 m solution of l-aspartate, pH 7.2, or GABA, pH 5.0. A retention current of approximately −1 nA was applied to prevent passive leakage of l-aspartate and ∼4 nA for GABA. Simultaneous two-photon fluorescence and scanning-infrared imaging (Mainen et al., 1999b) were used to position both the extracellular and iontophoretic pipettes near axons and dendrites. For axonal excitability testing, the iontophoretic pipette was positioned within ∼15 μm of the stimulating pipette.
Patch pipettes with resistances of 3–5 MΩ were used for somatic whole-cell recording. For axonal recordings, resistances were >5 MΩ. The internal solution contained (in mm) 142 potassium gluconate, 2 KCl, 10 HEPES, 4 MgCl2, 4 NaATP, 0.5 NaGTP, 10 potassium BAPTA, and 0.05–0.07 Alexa 594 (Molecular Probes) for NMDAR recordings, except for Ca2+-imaging experiments in which Fluo-5F (200 μm; Molecular Probes) replaced potassium BAPTA. For GABAAR recording, KCl (140 mm) replaced potassium gluconate, and potassium BAPTA was lowered to 5 mm. An internal solution consisting of (in mm) 114 CsMeSO3, 10 HEPES, 14 sodium phosphocreatine, 4 NaATP, 0.4 NaGTP, 0.05 Alexa 594, and 0.2 Fluo5F was used for cerebellar granule cell recording. Electrophysiological potentials and currents were recorded with a Multiclamp 700B amplifier (Molecular Devices). Electrode series resistance was compensated by bridge balance in current-clamp and was uncompensated during voltage-clamp recording. Analog signals were filtered at 3–10 kHz and digitized at 10–20 kHz. Data were collected using custom software (J. S. Diamond, National Institute of Neurological Disorders and Stroke, Bethesda, MD) written in IgorPro (Wavemetrics).
Pharmacological agents, including NMDA, GABA, 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), and TTX, were obtained from Tocris Cookson, picrotoxin from Sigma, and the peptide toxin ω-conotoxin MVIIC from Peptides International. Cytochrome c (Sigma) was added (0.25 mg/ml) to solutions containing peptides to prevent surface adsorption in the perfusion system.
Two-photon imaging.
Fluorescence was monitored with a custom two-photon laser-scanning microscope using an Olympus objective (60×; 0.9 NA) and a Ti:sapphire laser (Coherent). The laser was tuned to 810 nm, except for photolysis experiments when 780–790 nm was used. Line scanning occurred at 500 Hz. Fluorescence in both epifluorescence and transfluorescence pathways was split into green and red pathways using a 565 nm dichroic and 525/50 and 620/60 bandpass filter glass (Chroma and Semrock) and collected in photomultiplier tubes (H8224; Hamamatsu). ScanImage software (Pologruto et al., 2003) was used for data acquisition. Fluorescence changes were quantified as increases in green fluorescence divided by red fluorescence (ΔG/R) (Sabatini et al., 2002). Peak amplitude measurements of evoked Ca2+ transients (except for action potentials) reflect the average ΔG/R of a 125–200 ms epoch centered on the peak. For most axonal line scan measurements, alternating trials in which no stimulation occurred were used to determine the background fluorescence levels in cells. When a peak in Ca2+ fluorescence was not apparent in the axon, the average amplitude was computed from an epoch corresponding to the peak occurring during a dendrite-evoked response in the same cell. Axonal distance values (i.e., distance from soma) in the text and figures reflect measurements made by tracing axonal trajectories from maximum projection images and therefore are underestimates.
Data analysis.
Microsoft Excel and InStat (GraphPad Software) were used for statistical analysis. ANOVA (Bonferroni post hoc procedures) and t tests were used as appropriate. A value of p < 0.05 was considered significant. Reported values are mean ± SEM. In figures, asterisks denote statistical significance.
Results
NMDARs and axonal excitability
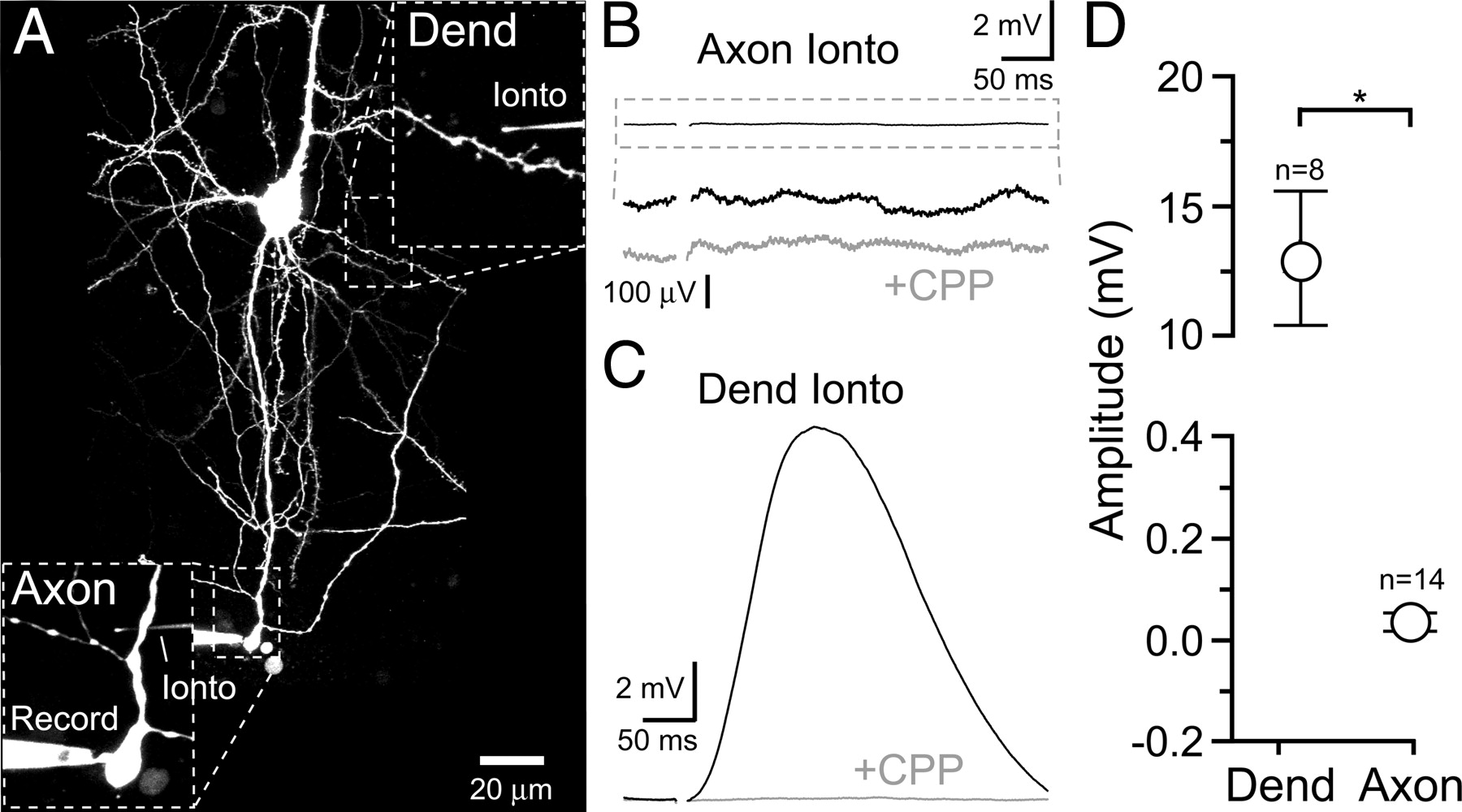
To determine if functional NMDARs are expressed on axons of L5 pyramidal cells in visual cortex, we tested for NMDAR-dependent increases in axonal excitability. Antidromic action potentials were directly evoked in a lateral axon branch from an L5 pyramidal via an extracellular stimulating electrode. Cells were filled with the morphological indicator Alexa 594 (50–70 μm) and imaged by two-photon microscopy so that the stimulating electrode could be precisely positioned near the axon of interest (Fig. 1A). The resulting action potentials were detected in the soma during whole-cell current clamp recording as full amplitude spikes. The stimulus intensity was set for a high rate of action potential failure, averaging close to 75% (range, 60–84%) (Fig. 1B). In alternate trials, iontophoresis of l-aspartate (−50 to −200 nA; 5–40 ms) preceded the electrical stimulus by 100–300 ms and was used in an attempt to locally activate axonal NMDARs near the stimulating electrode (distance within ∼15 μm). Iontophoresis was targeted to bead-like axon varicosities that are hot spots of VSCC activity and therefore likely en passant sites of release (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) because synaptic release of glutamate is thought to activate nearby NMDARs (Sjöström et al., 2003). Bath conditions were favorable for NMDAR opening (100 μm Mg2+; 10 μm d-serine), whereas Ca2+-dependent synaptic transmission and AMPA receptor-driven excitation were blocked to isolate axonal NMDARs and enhance the sensitivity of the assay (1 μm ω-conotoxin MVIIC; 10 μm NBQX). If NMDARs were expressed on the axon then an l-aspartate-evoked NMDAR depolarization would be expected to lower the stimulus threshold for action potential initiation. However, pairing l-aspartate iontophoresis with axonal stimulation did not reduce the action potential failure rate (Fig. 1, B and D; >40 trials in each condition). Iontophoresis of l-aspartate onto dendrites before and following axonal application elicited NMDAR-mediated potentials (12.5 ± 0.7 mV; n = 8; R-CPP, 2.9 ± 1.8% of control), confirming the reliability of iontophoresis within single-cell recordings and that a sufficient concentration of l-aspartate had been reached to evoke NMDAR-mediated responses (Fig. 1C).

l-Aspartate iontophoresis does not alter axonal excitability. A, Two-photon fluorescence image of an L5 pyramidal cell filled via patch pipette with Alexa 594. Magnified views show the position of the dendrite, axon, and the extracellular stimulating and iontophoretic pipettes. B, On the left, two superimposed somatic records show that direct electrical stimulation of the axon could evoke an antidromic action potential or result in a failure (flat line). Below, Raster display of 25 sweeps is shown. On the right, somatic responses to axonal electrical stimulation paired with l-aspartate iontophoresis. The electrical artifact that occurs during iontophoresis was blanked. C, Dendritic iontophoresis of l-aspartate evoked a depolarization that was blocked by the NMDAR antagonist R-CPP (20 μm). D, Axonal l-aspartate iontophoresis did not evoke an NMDAR-dependent alteration in the antidromic action potential failure rate. E, Pairing axonal electrical stimulation with small (3.1 mV) and moderate (6.4 mV) somatic depolarizations (middle and right, respectively) reduced failures. Four superimposed sweeps are shown in each condition. F, Somatic depolarization significantly decreased the failure rate of antidromic action potential stimulation. ASP, l-Asparate; Cont, control; Dend, dendrite; Depol, depolarization; Ionto, iontophoresis; Stim, stimulation.
The detection of axonal NMDAR expression by excitability testing requires sensitivity to small depolarizations. The sensitivity range of the excitability test was determined by pairing subthreshold depolarizations evoked by direct current injection in the soma (10–70 pA; 150–200 ms before axonal stimulation) with antidromic action potential stimulation in the axon (Fig. 1E). Somatic depolarizations were clearly capable of penetrating into the axon and enhancing excitability as evidenced by a significant reduction in action potential failure rate following pairing (Fig. 1, E and F; average distance from soma to axonal stimulation site 225 ± 26 μm; n = 11). Small somatic depolarizations (2.1 ± 0.1 mV; n = 11) were effective in reducing failures, whereas larger depolarizations (4.2 ± 0.3 mV; n = 11) nearly eliminated failures altogether. This demonstrates that minute changes in membrane potential near the site of antidromic action potential initiation are clearly reported by the spike failure rate. The lack of an NMDAR-dependent alteration in axonal excitability suggests the absence of axonal NMDARs.
GABAARs and axonal excitability
Given the lack of NMDAR-mediated excitability in axons, we tested whether activation of ligand-gated ion channels on L5 pyramidal cell axons, in general, could alter excitability. Direct GABAAR-mediated channel recordings from hippocampal mossy fiber boutons, calyceal terminals of the brainstem, and posterior pituitary nerve terminals have unequivocally demonstrated an axonal expression pattern (Zhang and Jackson, 1995a; Turecek and Trussell, 2002; Alle and Geiger, 2007). Therefore, GABAARs are likely axonal targets for excitability testing in L5 pyramidal cells. Antidromic action potentials were evoked in lateral axon branches by extracellular stimulation and were alternatively paired with iontophoresis of GABA (200 nA, 5–35 ms) to activate GABAARs (Fig. 2A). AMPA, NMDA, and GABABRs were blocked (10 μm NBQX; 20 μm R-CPP; 0.1 μm CPG 55845) and the reversal potential for GABAARs was set to ∼0 mV to mimic depolarizing NMDARs by including 140 mm Cl− in the internal solution. As shown by dendritic iontophoresis (Fig. 2B), GABA-evoked depolarizations (14.1 ± 1.6 mV; n = 6) were fully blocked by the GABAAR antagonist picrotoxin (100 μm; 4.9 ± 2.3% of control; n = 3).

GABA iontophoresis increases axonal excitability and evokes a GABAAR-mediated depolarization. A, On the left, direct electrical stimulation of the axon evoked an antidromic action potential or a failure (flat line). On the right, axonal GABA iontophoresis preceded electrical stimulation. Raster displays of 25 sweeps in each condition are shown below. B, Iontophoresis of GABA onto the dendrite evoked a depolarization that was blocked by the GABAAR antagonist picrotoxin (100 μm). C, Pairing electrical stimulation with GABA iontophoresis significantly decreased the likelihood of antidromic action potential failure. D, A small depolarization detected in the soma was evoked by axonal GABA iontophoresis (indicated by arrow). The trace is an average of 49 sweeps. For clarity, only action potential failure trials were included in the average at the bottom. E, The GABA iontophoresis-dependent decrease in antidromic action potential failures was related to the amplitude of the evoked depolarization in the axon. F, GABA iontophoresis onto a proximal dendrite (22 μm from the soma) evoked a larger depolarization than a more distal process (98 μm from the soma). Withdrawing the iontophoretic electrode 24 μm from the initial distal site failed to evoke a depolarization as shown at higher gain at the bottom. Ampl, Amplitude; Cont, control; Ionto, iontophoresis; Stim, stimulation.
In contrast to l-aspartate iontophoresis, pairing GABA iontophoresis with antidromic axonal stimulation significantly decreased spike failures (Fig. 2, A and C), indicating that axonal GABAAR-mediated depolarization lowered the stimulus threshold for spike initiation. Consistent with this idea, small GABAAR-mediated depolarizations evoked by axonal GABA iontophoresis were often detected in the soma (Fig. 2D; 0.3 ± 0.1 mV; n = 7). The amplitude of evoked depolarizations varied across axonal locations and was well correlated with the change in failure rate indicating a clear relationship between these two parameters (Fig. 2E).
We considered the possibility that GABAAR-mediated depolarizations elicited by axonal GABA iontophoresis were inadvertently evoked in the dendrite. However, directly targeting GABA iontophoresis onto distal dendrites, the dendritic processes nearest to axons, evoked small depolarizations (Fig. 2F; 1.7 ± 0.6 mV; n = 5; proximal stimulation 15.6 ± 0.8 mV, n = 3; p < 0.05), suggesting a low expression density of GABAARs. Second, withdrawing the iontophoretic pipette a short distance from the distal dendrite (from 10 ± 1 to 31 ± 2 μm) failed to evoke GABAAR-mediated responses (Fig. 2F; 0.0 ± 0.0 mV; n = 5), revealing the spatial extent of GABA diffusion (<30 μm). The average distance from the location of axonal iontophoresis to the nearest dendrite (60 ± 12 μm) was beyond this limit. Last, Ca2+-dependent transmission was blocked (200 μm cadmium) so that electrical stimulation of neighboring axons by the extracellular electrode should not result in release of GABA onto the dendrites of the test cell. Therefore, depolarizations evoked by axonal iontophoresis reflect GABAAR-mediated potentials that propagate from the axon into the cell body similar to antidromic propagation of subthreshold potentials into axons from the calyx of Held (Paradiso and Wu, 2009).
Direct axonal recording and NMDARs
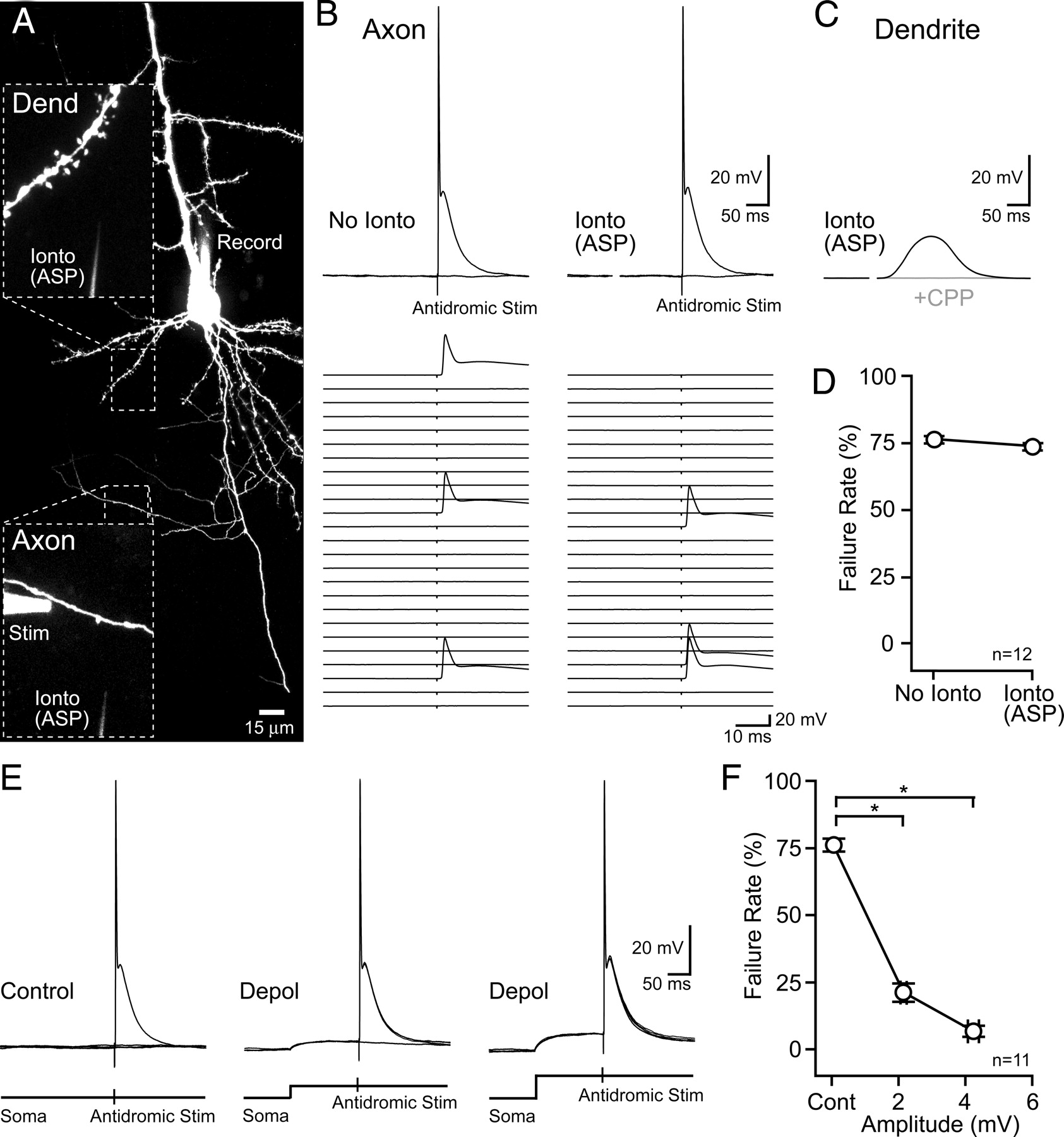
In light of the observation of somatic depolarization resulting from activation of axonal GABAARs, we tested whether similar somatic depolarizations could be evoked by axonal NMDARs (10 μm NBQX, 10 μm d-serine). However, axonal iontophoresis of l-aspartate failed to evoke detectable depolarizations during somatic recording (0.0 ± 0.1 mV; n = 12). To mitigate axonal filtering of the responses we made whole-cell recordings from swellings of severed primary axons (“blebs”; Shu et al., 2006; Kole et al., 2007) and iontophoresed l-aspartate onto nearby lateral axon branches (Fig. 3A). The distance from the bleb and l-aspartate stimulation site was <32 μm (average, 17 ± 3 μm; n = 13). Similar to somatic recording, we found no evidence for NMDAR-mediated axonal depolarization. Trials with l-aspartate iontophoresis were no different than iontophoretic trials in the presence of R-CPP (Fig. 3, B and D; 0.1 ± 0.0, n = 15; and 0.1 ± 0.0 mV, n = 9; control and R-CPP, respectively; p = 0.13). Dendritic l-aspartate iontophoresis before and following axonal iontophoresis (188 ± 36 μm distal to the axonal recording site, n = 9) evoked large depolarizations (Fig. 3, B and D), confirming the efficacy of l-aspartate discharge from the pipette and the responsiveness of the axonal recording configuration.
Direct axonal recordings fail to reveal l-aspartate-evoked depolarization by NMDARs. A, Image of an L5 pyramidal cell patched and filled with Alexa 594 via an axon bleb. Magnified views show the location of the iontophoretic pipette relative to the axon and dendrite. B, l-Aspartate iontophoresis onto the axon failed to evoke an NMDAR-mediated depolarization. The amplified record is shown compared with iontophoretic trials in R-CPP (20 μm). C, Dendritic l-aspartate iontophoresis evoked a depolarization that was blocked by R-CPP (20 μm). D, Comparison of dendritic and axonal stimulation with l-aspartate iontophoresis. Dend, Dendrite; Ionto, iontophoresis.
Fluorescence measurement of NMDAR-mediated Ca2+ entry
In whole-cell recordings from most pyramidal-type neurons, single-NMDAR-channel currents cannot be resolved because of background electrical noise. In contrast, two-photon microscopy can be used to monitor Ca2+ influx through single NMDAR channels in hippocampal CA1 pyramidal cells (Nimchinsky et al., 2004). To enhance the sensitivity of detecting axonal NMDAR channels, we used two-photon microscopy to measure NMDAR-mediated Ca2+ influx. L5 pyramidal cells were filled via the patch pipette with the green Ca2+ indicator Fluo5F (200 μm) and the red morphological dye Alexa594 (Fig. 4A). In voltage-clamped cells (100 μm Mg2+; 10 μm d-serine; 10 μm NBQX; 0.5 μm TTX), iontophoresis of l-aspartate onto dendrites evoked Ca2+ transients in dendrites near the iontophoretic pipette (Fig. 4, B and D). Block by R-CPP (20 μm) confirmed that Ca2+ response was mediated by NMDARs (2.1 ± 0.6% of control; n = 4).

Axonal iontophoresis of l-aspartate does not evoke NMDAR-mediated Ca2+ transients. A, L5 pyramidal cell image shows the dendritic position of the iontophoretic pipette and the line scan for Ca2+ measurement. B, Dendritic l-aspartate iontophoresis evoked a Ca2+ transient that was blocked by R-CPP (20 μm). C, Ca2+ entry in an axon varicosity (inset) was not evoked by l-aspartate iontophoresis. The amplified average record is shown on the right compared with the average response without iontophoresis and to the iontophoretic response in R-CPP (20 μm). An action potential, evoked by somatic current injection before TTX application, revealed that the recoding site was a hot spot for Ca2+ entry. D, Summary for data acquired in the presence or absence of extracellular Mg2+. AP, Action potential; Dend, dendrite; Ionto, iontophoresis.
In contrast to l-aspartate iontophoresis-evoked Ca2+ transients in dendrites, axonal iontophoresis failed to evoke Ca2+ transients in axons. Trials with l-aspartate iontophoresis were indistinguishable from those in R-CPP or those without iontophoresis (Fig. 4, C and D). Even in the absence of extracellular Mg2+, l-aspartate iontophoresis failed to cause axonal Ca2+ entry (Fig. 4D). In this condition (0 mm Mg2+), spontaneous epileptiform activity in the slice was suppressed by tonically activating GABAARs (1 μm muscimol). Ca2+ measurements were obtained along the entire axonal arbor of L5 pyramidal cells (L6-L1; distance from the soma, 71–776 μm; n = 47 varicosities), suggesting that NMDAR expression did not depend on axonal location.
To test for NMDAR expression in larger regions of axons than afforded by line scans, we bath-applied NMDA (4 μm) while imaging with frame scans (100 μm Mg2+; 10 μm d-serine; 10 μm NBQX; 0.5 μm TTX). Frame scans were taken at 0.05 Hz to monitor Ca2+ entry in short segments of axons that always included at least one varicosity. Consistent with observations using l-aspartate iontophoretic stimulation, axonal segments (>150 μm from the soma) did not reveal increases in Ca2+ during NMDA application (Fig. 5A; ΔG/R, 0.0004 ± 0.0001 and 0.0002 ± 0.0001; n = 14 axon segments; control and NMDA, respectively; p = 0.17). Similar results were obtained in the absence of extracellular Mg2+ (ΔG/R 0.0040 ± 0.0006; and 0.0053 ± 0.0007, n = 13 axon segments; control and NMDA, respectively; p = 0.19). NMDAR-dependent toxicity and epileptiform activity was diminished in this condition (0 mm Mg2+) by voltage-clamping the cell (Vh, −60 mV) and including muscimol (1 μm) in the bath. In dendrites, NMDA (4 μm; 100 μm Mg2+) evoked large Ca2+ elevations (ΔG/R, 0.0029 ± 0.0007 and 0.1114 ± 0.0111; n = 3; p < 0.05; control and NMDA, respectively) and depolarized the cell (25.4 ± 1.0 mV; n = 20), indicating that low concentrations of NMDA potently activated NMDARs.

Bath-applied NMDA does not evoke Ca2+ entry in axons. A, Image of an L5 pyramidal cell axon showing the axon segment, including a varicosity, used to monitor Ca2+ (white dotted box) and a series of frame scans taken throughout the experiment. There was no observable increase in Ca2+ during NMDA application (4 μm; 100 μm Mg2+). B, In the same cell, a sequence of frame scans from the dendrite shows that NMDA evoked a large increase in Ca2+.
Microscope sensitivity
To determine whether our microscope can detect Ca2+ transients expected from low-density NMDAR expression, we monitored intracellular Ca2+ levels during single NMDAR-mediated channel openings. Because of the difficulty in resolving single-channel electrical activity in L5 pyramidal cells, we recorded from granule cells in cerebellar slices (Fig. 6A) whose compact size and exceptionally high input resistance is amenable for discriminating small signals (Silver et al., 1992). In the absence of extracellular Mg2+ (10 μm d-serine; 10 μm NBQX; 100 μm picrotoxin; 0.5 μm TTX), clearly resolvable single-NMDAR-channel currents infrequently occurred spontaneously likely due to a low ambient concentration of glutamate in the slice (Cavelier and Attwell, 2005; Herman and Jahr, 2007; Yamashita et al., 2009; data not shown). Two-photon laser uncaging of 4-methoxy-7-nitroindolinyl (MNI)-glutamate (2–5 mm) was used to weakly stimulate NMDARs in a location-specific manner by continuous line scanning over a distal dendrite (0.5–1 s) at a wavelength near threshold for MNI-glutamate photolysis (780–790 nm). Emitted fluorescence in both red (Alex594; 40 μm) and green channels (Fluo5F; 200 μm) was continuously collected during scanning to monitor for NMDAR-mediated Ca2+ entry. Moderate laser stimulation (∼5 mW) evoked small inward currents that were composed of barrages of single-channel activity and elicited Ca2+ influx in the dendrite in which photolysis occurred (Fig. 6B). Inward currents and Ca2+ influx were blocked by R-CPP confirming that they were NMDAR-mediated (−0.888 ± 0.077 and 0.002 ± 0.026 pA/s; control and R-CPP current integrals, respectively; peak ΔG/R, 0.214 ± 0.065 and −0.001 ± 0.002; control and R-CPP, respectively; n = 3; p < 0.05). When laser power was lowered so that channel openings occurred infrequently, clear Ca2+ transients were observed in numerous trials (five stimulus locations) that were unambiguously time-locked to single-channel currents (Fig. 6C). This demonstrates that our microscope is capable of detecting Ca2+ influx through single NMDAR channels. Therefore, if an axonal NMDAR was activated on an L5 pyramidal cell axon, it is very likely that we would observe corresponding NMDA-mediated Ca2+ influx.

Ca2+ entry through single NMDAR channels. A, Representative image of a cerebellar granule cell filled with Alexa 594 and Fluo5F. B, MNI-glutamate (2–5 mm) was uncaged by a line scan across a dendrite (duration indicated by black bar). An NMDAR-mediated inward current and Ca2+ influx were evoked in the dendrite in which laser scanning occurred. The traces are the averages of 7 and 16 sweeps in control and R-CPP, respectively. Dend, Dendrite. C, Three examples of single-NMDAR-channel currents and simultaneously recorded Ca2+ entry evoked by MNI-glutamate photolysis. Each example trace was acquired from a separate imaging location (2 cells). Note that the sensitivity of the photomultiplier tubes was tuned down in B so that the scaling comparisons to C are not linear.
High-frequency spiking and NMDAR-mediated Ca2+ entry
High-frequency spiking is reported to facilitate glutamate release from L5 pyramidal cell axons in a cell-autonomous, NMDAR-dependent manner (Sjöström et al., 2003). In this scenario, spike-evoked glutamate release binds to axonal NMDARs, and subsequent spikes relieve NMDAR channel block by Mg2+, allowing Ca2+ influx. Therefore, action potential-evoked Ca2+ transients were monitored in L5 pyramidal cell axon varicosities during burst firing (six spikes; 20–30 Hz) for an NMDAR-mediated Ca2+ component (100 μm Mg2+). Block of NMDARs by R-CPP (20 μm) did not alter the amplitude of the first two action potential-evoked Ca2+ transients in the spike burst (Fig. 7A; R-CPP 103.0 ± 4.4% and 103.7 ± 2.9% of control; first and second action potential, respectively; n = 7; p = 0.88), suggesting that NMDARs did not contribute to the initial Ca2+ transient. Comparisons were not made on subsequent spikes because the indicator (Fluo5F; 200 μm) was beyond the linear reporting range for Ca2+ binding. To examine the entire spike train, cells were filled with the lower-affinity Ca2+ indicator Fluo4FF (200 μm) to avoid saturation during repetitive firing (Fig. 7, B and D). However, action potential-evoked Ca2+ transients often displayed rundown during the course of an experiment so R-CPP comparisons were made following normalization of the first action potential-evoked Ca2+ transient. There were no significant alterations in spike-evoked Ca2+ following R-CPP application (ANOVA; p = 0.96) confirming that NMDARs did not contribute to Ca2+ entry in axon varicosities during high-frequency spiking (Fig. 7, C and D).
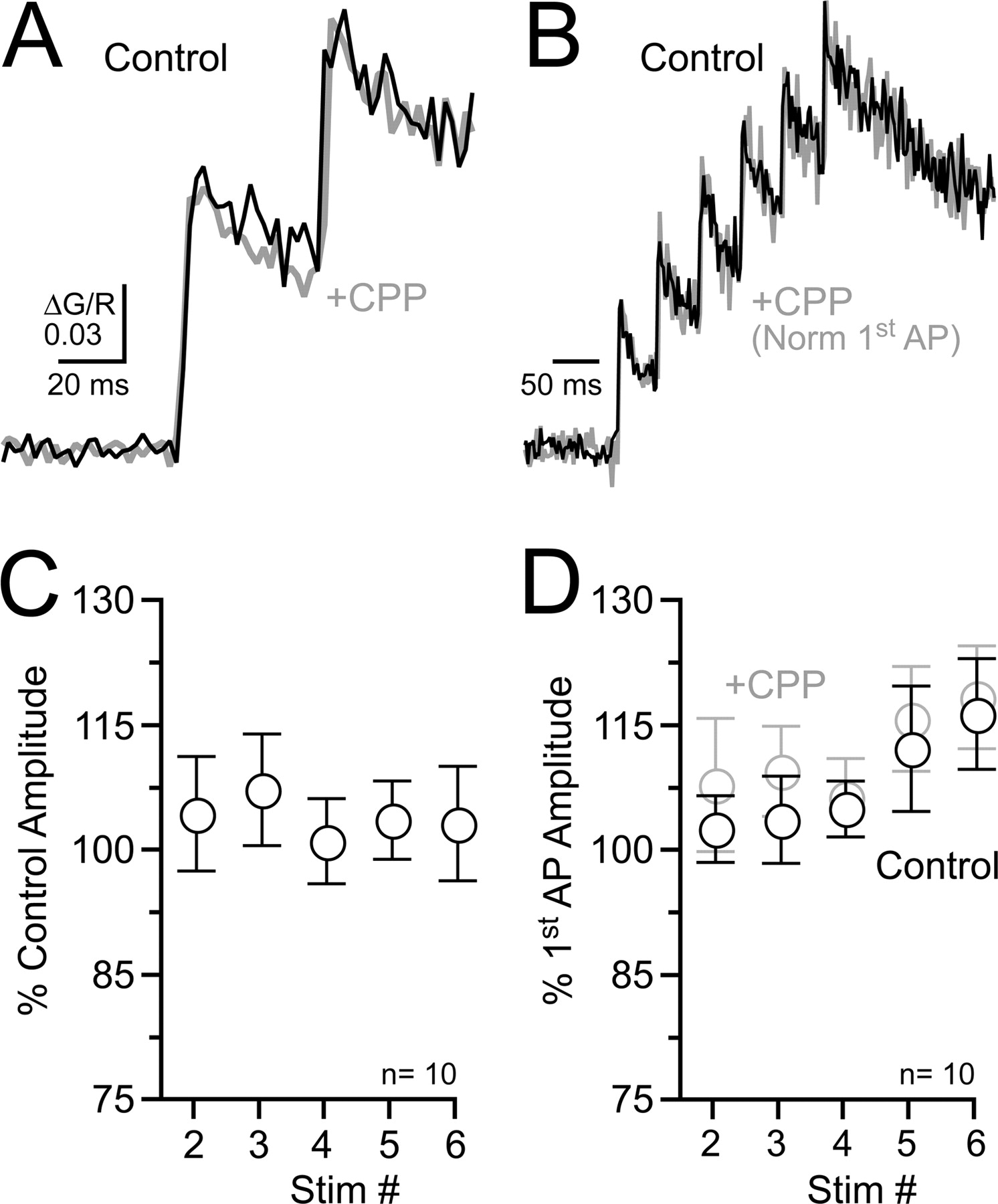
High-frequency spiking does not evoke NMDAR-mediated Ca2+ entry in axons. A, In an L5 pyramidal cell axon varicosity, the Ca2+ transient evoked by the first two action potentials during high-frequency firing (25 Hz) was unaltered by the NMDAR antagonist R-CPP (20 μm). The cell was filled with the moderate-affinity Ca2+ indicator Fluo5F (200 μm). B, Ca2+ transients evoked by an action potential train (25 Hz) recorded in an axon varicosity with the low-affinity Ca2+ indicator Fluo4FF (200 μm) in control and in R-CPP (20 μm). For comparison, responses were normalized to the first action potential peak. C, Summary shows that R-CPP had no effect on action potential-evoked Ca2+ transients (Fluo4FF). For this analysis, action potential peak comparisons were made after normalizing the amplitudes of the first spike. D, The amplitude of each action potential-evoked Ca2+ transient (Fluo4FF) within a burst compared with the size of the first spike in each condition. AP, Action potential; Stim, stimulation.
Somatodendritic electrical signaling into axons
In cerebellar stellate cells, NMDAR-mediated depolarizations subthreshold for action potential initiation spread electrotonically from the dendrite into the axon opening VSCCs in varicosities (Christie and Jahr, 2008). This axonal Ca2+ transient may mediate NMDAR-dependent alterations of release (Glitsch and Marty, 1999; Duguid and Smart, 2004; Glitsch, 2008). To determine whether passive electrical signaling is a common mechanism by which dendritic NMDARs open axonal VSCCs, we simulated NMDAR-like depolarizations by direct somatic current injection into L5 pyramidal cells (average amplitude, 21.0 ± 0.8 mV; half-width 324 ± 12 ms, n = 17) and monitored for Ca2+ in lateral axon varicosities (Fig. 8, A1 and A2). Unlike results in stellate cells, subthreshold depolarizations from the soma failed to evoke measurable Ca2+ transients in L5 axons (Fig. 8, A3 and B; ΔG/R; 0.0004 ± 0.0001 and 0.0003 ± 0.0001, n = 17 varicosities; control and subthreshold depolarization trials, respectively; p = 0.59). Action potential-evoked Ca2+ transients confirmed that the selected sites were hot spots of Ca2+ entry (Fig. 8A1–A3).

Subthreshold somatic depolarizations fail to evoke Ca2+ entry in axon varicosities. A1, An action potential and a subthreshold depolarization elicited by current injection in a somatic whole-cell recording. A2, Position of the line scan used for Ca2+ imaging on a lateral axon varicosity. A3, Subthreshold depolarization failed to evoke a Ca2+ transient in the axon varicosity. An action potential confirmed the varicosity was a hot spot of Ca2+ entry. The amplified records show the response to subthreshold depolarization compared with a control condition in which no stimulation occurred. B, Subthreshold depolarizations failed to evoke Ca2+ entry in lateral axonal arbors of pyramidal cells. AP, Action potential; Stim, stimulation; Subthres Depol, subthreshold depolarization.
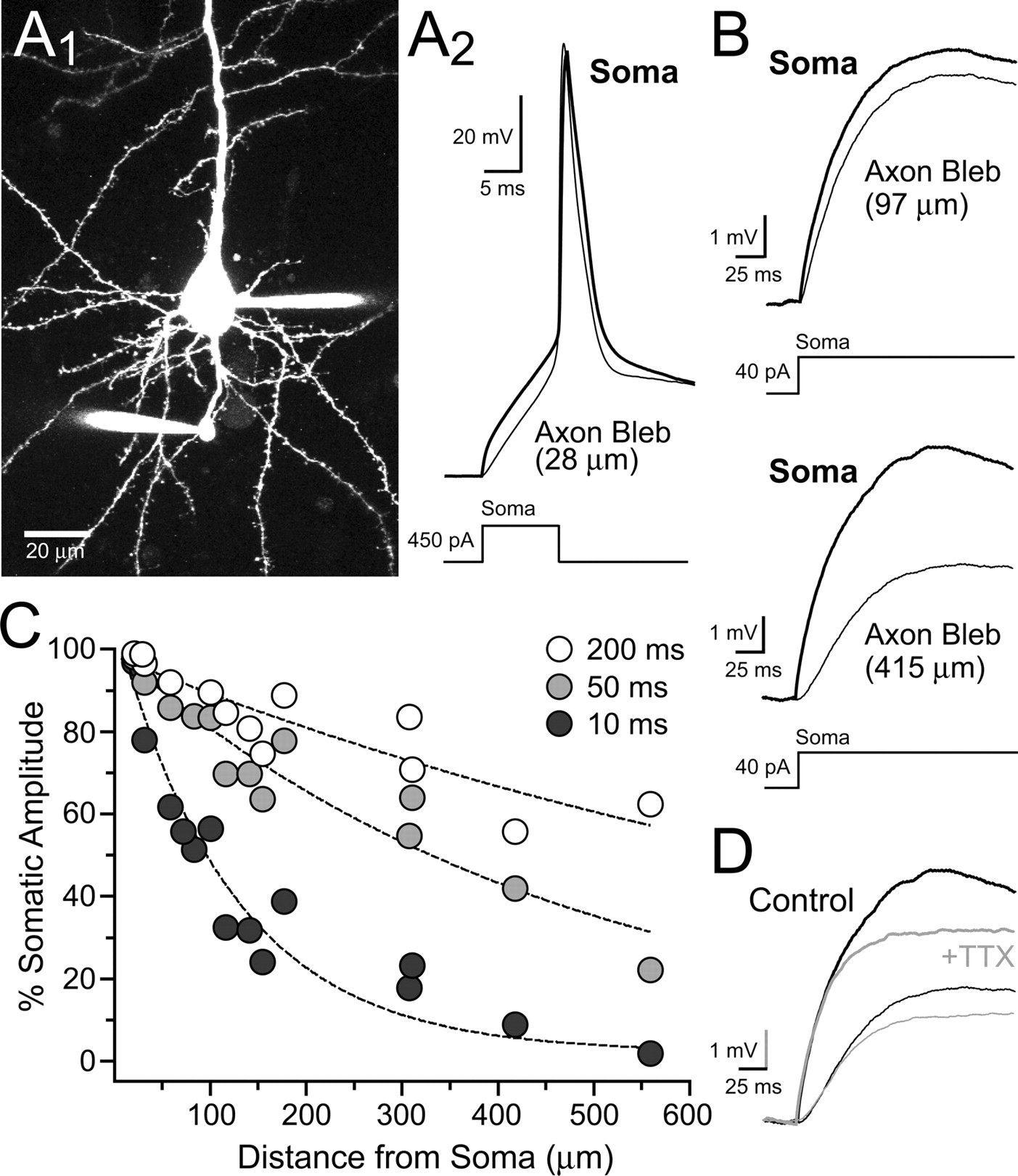
The lack of subthreshold depolarization-evoked Ca2+ transients in L5 pyramidal cell axon varicosities might suggest that somatic depolarizations are greatly attenuated by axonal cable filtering. Therefore, we made simultaneous whole-cell recordings from both the soma and axon blebs to directly determine the extent of axonal filtering (Fig. 9, A1 and A2). Somatic depolarizations (≥200 ms) were well transferred to the cut-end of the axon with modest attenuation at proximal distances and moderate attenuation at distances over several hundred micrometers (Fig. 9, B and C) similar to prior reports (Shu et al., 2006; Kole et al., 2007). Shorter duration potentials were attenuated to a greater extent (Fig. 9C) as expected from frequency-dependent cable filtering. Blocking sodium channels with TTX (0.5 μm) slightly diminished the amplitude of the somatic potential; however, the axonal potential was diminished to the same extent (Fig. 9D; 84.2 ± 2.6% and 86.0 ± 2.9% of control, n = 4; soma and axon, respectively; p = 0.66) ruling out active propagation. Taken together, these results suggest that dendritic NMDAR-mediated potentials spread far into the axon arbor, but the resulting axonal potentials are too small to activate VSCCs expressed in varicosities. Therefore, NMDAR-mediated facilitation of release does not appear to depend on Ca2+ entry elicited by subthreshold depolarization.

Subthreshold somatic depolarizations passively spread far into the axon arbor. A1, Image shows a simultaneous whole-cell recording from both the soma and axon bleb (28 μm from the cell body) of an L5 pyramidal cell. A2, An action potential, evoked by somatic current injection, recorded in the soma and axon bleb. B, Somatic subthreshold depolarizations elicited by current injection recorded in both the soma and a proximal (top) or distal (bottom) axon bleb. The recordings were made from two separate cells. C, Summary shows the both the frequency and distance dependence of axonal attenuation of somatic depolarizations. The data are plotted as the ratio of amplitude measurements from the somatic and axonal potential at three time points (10, 50, and 200 ms) during a prolonged somatic depolarization. Length constants for curves: 10 ms, τ = 122 μm; 50 ms, τ = 462 μm; 200 ms, τ = 1001 μm. D, TTX (0.5 μm) blocked the somatic and axonal potential to the same extent.
Discussion
Despite observations of NMDAR-dependent facilitation of release (Sjöström et al., 2003; Corlew et al., 2007), we find no evidence for functional expression of NMDARs on L5 pyramidal cell axons. Assays that specifically probe axons for NMDAR-mediated excitation or direct Ca2+ influx failed to uncover NMDAR activity. In contrast, axonal GABA iontophoresis evoked GABAAR-mediated depolarizations that decreased the threshold for antidromic spiking, suggesting that not all ligand-gated ion channels are excluded from axons. Therefore, NMDAR-mediated alteration of release does not depend on axonal receptors but, instead, on NMDARs expressed on the soma, dendrites, or surrounding cells.
NMDARs are enriched in postsynaptic membranes (Fagg and Matus, 1984; Monaghan and Cotman; 1986), including L5 pyramidal cell dendrites and spines, and contribute to synaptic excitability and Ca2+ influx. Observations of NMDAR immunoreactivity in some axonal processes of the neocortex (Aoki et al., 1994; Charton et al., 1999; Corlew et al., 2007) suggest that NMDARs have a more extensive distribution and purpose in cortical neurons. Similar observations have been made in spinal cord, cerebellum, hippocampus and amygdala (Corlew et al., 2008). However, our search failed to detect functional NMDARs on L5 pyramidal cell axons raising the possibility that immunopositive NMDARs are inoperable or unavailable for binding extracellular agonist. Consistent with this notion, the majority of axonal NR1-immunoreaction product in L5 visual cortex is intracellular and associated with synaptic vesicles rather than on or near the plasma membrane (Aoki et al., 1994). In addition, the physiological effect of presynaptic NMDARs is lost by postnatal day 23 in mice, and yet one-half of the immunoreactivity for the NR1 subunit remains at postnatal day 27 (Corlew et al., 2007).
The most sensitive method for detecting functional NMDARs in L5 pyramidal cells is optical monitoring of Ca2+ influx through NMDARs. This allows resolution of single-channel openings (Sabatini and Svoboda, 2000; Nimchinsky et al., 2004). Therefore, it is unlikely that axonal Ca2+ transients evoked from sparse or low-density NMDAR expression were below detection threshold and escaped observation. Although NMDAR desensitization will decrease sensitivity, neuronal responses to NMDAR agonists do not fully desensitize in the presence of glycine-site agonists (Mayer et al. 1989). Unlike conventional NR1-NR2 heteromeric channels, NMDARs that incorporate NR3 subunits have a greatly diminished permeability for Ca2+ (Matsuda et al., 2002; Sasaki et al., 2002). NR3A mRNA transcript has been observed in adult neocortex (Ciaberra et al., 1995), but whether putative NMDAR channels in axons incorporate NR3 is not known. Excitability testing and direct recording in L5 pyramidal cell axons failed to reveal NMDAR-mediated depolarization suggesting NR3-containing NMDARs, if expressed in L5 pyramidal cells, are also excluded from the axon. Even if we assume that small axonal depolarizations are below the detection limit of our recording methods (bleb recording and excitability testing), without Ca2+ influx, such small depolarizations would be very unlikely to alter release.
One limit of our approach is that a relatively small number of axonal sites could be surveyed (several per cell) compared with the total number of release sites on an L5 pyramidal cell axon. If NMDARs were selectively expressed at release sites in a target cell-specific manner then it is possible that random sampling could miss these sites. In L4 pyramidal cell axons of the somatosensory cortex, physiological evidence suggests that NMDARs are selectively expressed at excitatory release sites onto L2/3 cells but not at synapses onto neighboring L4 cells (Brasier and Feldman, 2008). Similar target cell-specific expression of both axonal metabotropic and ionotropic glutamate receptors has been observed in the hippocampus (Scanziani et al., 1998; Sun and Dobrunz, 2006). However, L5 to L5 synapses are strongly facilitated by action potential-evoked NMDAR activity suggesting that if present on the axon, most L5 to L5 pyramidal cell synapses would have associated presynaptic NMDARs (Sjöström et al., 2003).
NMDARs clearly modulate spontaneous and action-potential-evoked transmission in pyramidal cell neurons throughout the neocortex (Berretta and Jones, 1996; Sjöström et al., 2003; Yang et al., 2006; Corlew et al., 2007; Brasier and Feldman, 2008). Because NMDARs are excluded from L5 pyramidal cell axons, dendritic NMDARs must signal to the axon and thereby alter the likelihood of release. In dentate granule cells in the hippocampus and L5 pyramidal cells in cortex, electrotonic passage of somatodendritic depolarization into presynaptic boutons facilitates action potential-evoked release (Alle and Geiger, 2006; Shu et al., 2006; Kole et al., 2007). Similarly, in stellate cells, NMDAR-mediated depolarizations originating in the dendrite pass into the axon and open VSCCs at presumed sites of release (Christie and Jahr, 2008). Although not directly tested, this axonal Ca2+ transient may be the basis for NMDAR-dependent alterations in the strength and frequency of evoked and spontaneous release (Glitsch and Marty, 1999). Although we and others (Shu et al., 2006; Kole et al., 2007) have shown that somatodendritic subthreshold potentials spread hundreds of micrometers into L5 axons, these potentials are insufficient to open VSCCs in varicosities. This indicates that low-threshold VSCCs contribute little to axonal Ca2+ influx. It may be that longer depolarizations, those mimicking cortical up-states, would cause appreciable Ca2+ entry. Therefore, NMDAR-mediated facilitation of spontaneous and evoked release (Sjöström et al., 2003; Corlew et al., 2007) must be independent of alterations in basal Ca2+ in L5 pyramidal cell varicosities. However, subthreshold depolarization-mediated facilitation of release in L5 pyramidal cells can be blocked by presynaptic dialysis with Ca2+ chelators (Shu et al., 2006), raising the possibility that regulation of Ca2+ is important in axonal electrical signaling.
Facilitation elicited by somatodendritic electrical signaling in dentate granule cells is either weakly or completely independent of basal or action potential-evoked Ca2+. Despite suggestions of modeling studies using VSCC recordings from mossy fiber boutons (Li et al., 2007), direct evidence for subthreshold depolarization-mediated Ca2+ entry is absent and Ca2+ chelation does little to alter facilitation mediated by somatodendritic electrical signaling (Alle and Geiger, 2006; Scott et al., 2008). This suggests that multiple mechanisms can be triggered by somatodendritic depolarization to alter axonal release. For example, axonal depolarization can shape the action potential at the site of release by controlling the availability of voltage-sensitive sodium and potassium channels (Zhang and Jackson, 1995b; Kole et al., 2007). In addition, depolarization may directly alter the release machinery (Zhang and Zhou, 2002; Scott et al., 2008). Last, it is unknown if tonic activation of dendritic NMDARs by ambient glutamate (Cavelier and Attwell, 2005; Herman and Jahr, 2007; Yamashita et al., 2009) engages second-messenger signaling cascades that reach the axon and alter release.
Thus far, we have failed to detect functional NMDAR expression in axons of two widely divergent neuronal types, L5 pyramidal cells of visual cortex and stellate cell interneurons of the cerebellum (Christie and Jahr, 2008). The restriction of NMDAR expression to somatodendrites in both stellate cells and L5 pyramidal cells contradicts the concept that axonal NMDARs are general elements in the repertoire of presynaptic ligand-gated receptors. However, the exclusion of NMDARs from axons may be an exception among ligand-gated ion channels. In contrast to NMDARs, we directly observed axonal GABAAR-mediated potentials in L5 pyramidal cells. Axonal expression of GABAARs has been conclusively demonstrated in hippocampus, brainstem and posterior pituitary nerve terminals (Zhang and Jackson, 1995a; Turecek and Trussell, 2002; Alle and Geiger, 2007) and GABAAR-mediated activity has long been known to modulate transmitter release (Frank and Fuortes, 1957; Dudel and Kuffler, 1961). AMPA, kainate, and glycine receptors have also been shown to modulate synaptic transmission and are likely expressed on the axon (Engelman and MacDermott, 2004), although direct axonal observations of these channel types are often lacking.
By excluding NMDARs from the axon, neurons could constrain excessive Ca2+ entry at presynaptic boutons and prevent direct NMDAR-evoked release (Chen et al., 2000; Halabisky et al., 2000) or excitotoxicity following glutamate release near the axon. The prolonged open-time of NMDARs (∼100 ms; Lester et al., 1990) produces extensive Ca2+ influx requiring morphological specialization and endogenous buffers and pumps to tightly limit diffusion in small neuronal structures (Sabatini et al., 2002). The strength of action potential-evoked release is extraordinarily sensitive to ambient or residual Ca2+ in the bouton (Felmy et al. 2003) and can be greatly facilitated by small increases in membrane potential that allow VSCC opening and Ca2+ entry (Awatramani et al., 2005). Therefore, voltage signaling by dendritic NMDARs or axonal ligand-gated channels, such as GABAARs, may be a more effective and less toxic mechanism to modulate release than direct Ca2+ influx through axonal NMDARs.
Footnotes
-
This work was supported by National Institute of Health Grants NS40056 and MH074989 (to C.E.J.). We thank Vanessa Bender, Delia Chiu, Melissa Herman, and Jason Pugh for their helpful discussions and comments on this manuscript.
- Correspondence should be addressed to Dr. Jason M. Christie, Vollum Institute, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, OR 97239. christij{at}ohsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}