

Abstract
The inability of retinal ganglion cells (RGCs) to regenerate damaged axons through the optic nerve has dire consequences for victims of traumatic nerve injury and certain neurodegenerative diseases. Several strategies have been shown to induce appreciable regeneration in vivo, but the regrowth of axons through the entire optic nerve and on into the brain remains a major challenge. We show here that the induction of a controlled inflammatory response in the eye, when combined with elevation of intracellular cAMP and deletion of the gene encoding pten (phosphatase and tensin homolog), enables RGCs to regenerate axons the full length of the optic nerve in mature mice; approximately half of these axons cross the chiasm, and a rare subset (∼1%) manages to enter the thalamus. Consistent with our previous findings, the axon-promoting effects of inflammation were shown to require the macrophage-derived growth factor Oncomodulin (Ocm). Elevation of cAMP increased the ability of Ocm to bind to its receptors in the inner retina and augmented inflammation-induced regeneration twofold. Inflammation combined with elevated cAMP and PTEN deletion increased activation of the phosphatidylinositol 3-kinase and mitogen-activated protein kinase signaling pathways and augmented regeneration ∼10-fold over the level induced by either pten deletion or Zymosan alone. Thus, treatments that synergistically alter the intrinsic growth state of RGCs produce unprecedented levels of axon regeneration in the optic nerve, a CNS pathway long believed to be incapable of supporting such growth.
Introduction
Like most pathways in the mature CNS, the optic nerve cannot regenerate if injured, leaving victims of traumatic nerve injury or degenerative diseases such as glaucoma with life-long visual losses. This situation can be partially reversed by enhancing the intrinsic growth state of retinal ganglion cells (RGCs), the projection neurons of the eye. Induction of a controlled inflammatory reaction in the eye causes injured RGCs to upregulate the expression of GAP-43 and other genes related to axon growth and to regenerate lengthy axons beyond the site of nerve injury (Berry et al., 1996; Leon et al., 2000; Yin et al., 2003; Fischer et al., 2004b). This growth is stimulated by the macrophage-derived growth factor Oncomodulin (Ocm), acting in concert with other factors (Yin et al., 2006, 2009). Strategies that counteract cell-extrinsic inhibitors of axon growth promote only modest regeneration on their own (Chierzi et al., 1999; Lehmann et al., 1999) but greatly enhance regeneration once the intrinsic growth program of RGCs has been activated (Fischer et al., 2004a,b). An alternative way to enhance the intrinsic growth state of RGCs is by deleting the gene that encodes PTEN (phosphatase and tensin homolog) or SOCS-3 (suppressor of cytokine signaling-3), proteins that suppress signaling through the phosphatidylinositol 3-kinase (PI3K)–Akt and Jak–STAT (signal transducer and activator of transcription) pathways, respectively (Park et al., 2008; Smith et al., 2009). Increasing intracellular cAMP ([cAMP]i), although stimulating appreciable regeneration in other parts of the nervous system (Neumann et al., 2002; Qiu et al., 2002; Hannila and Filbin, 2008), has only a small effect in promoting axon regeneration through the optic nerve (Monsul et al., 2004; Yin et al., 2006) but can augment the effects of intraocular inflammation (Müller et al., 2009). It is currently unknown whether intraocular inflammation, inactivating suppressors of cell signaling, and elevating intracellular cAMP levels activate complementary mechanisms that would lead to markedly stronger regeneration when combined, or whether they activate common intracellular signaling pathways and would therefore show little additivity. Our results show that intraocular inflammation, deletion of the PTEN gene, and elevation of intracellular cAMP exert complementary effects that together enable RGCs to regenerate axons all the way from just behind the eye to the diencephalon in a few weeks. These effects involve increased binding of Ocm to its cognate receptor and activation of complementary cell-signaling pathways. To our knowledge, these results represent the best documented instance of long-distance axon regeneration in the optic nerve to date.
Materials and Methods
Animals.
Studies were performed at Children's Hospital (Boston, MA) with the approval of the Institutional Animal Care and Use Committee. Experiments used male mice with a conditional deletion of exon 5 in the pten gene (PTENflx/flx), which encodes the phosphatase domain of the protein (Groszer et al., 2001), wild-type mice of the same strain (C57BL/6J), or 8-to 10-week-old male Fisher rats. Long-term regeneration experiments (6 weeks) used mice that were 8 weeks old at the time of viral infection. All other mouse experiments used animals that were 4 weeks old when injected with viruses (and 6 weeks old at the time of optic nerve injury).
Surgery.
RGC-selective deletion of the pten gene was accomplished via Cre–lox recombination, taking advantage of the tropism of adeno-associated virus seroform 2 (AAV2) for RGCs (Cheng et al., 2002; Martin et al., 2002). AAV2 expressing Cre recombinase [AAV2–Cre, 3 μl (Vector Laboratories); titer, 1 × 1012 GC/ml] was injected intraocularly 14 d before optic nerve crush, with care taken to avoid injuring the lens. Recombination was verified in a Rosa–lox–STOP–lox–PLAP reporter line of mice (gift from Dr. F. Wang, Duke University, Durham, NC). AAV2–green fluorescent protein (GFP) (3 μl; Vector Laboratories) was injected intraocularly as a control virus. Optic nerve crush surgery and intraocular injections were performed under general anesthesia as described previously (Yin et al., 2009). Reagents injected intraocularly included Zymosan (1.5–12.5 μg/μl, sterilized before use; Sigma); 8-(4-chlorophenylthio) (CPT)–cAMP (4–500 μm; Sigma), a membrane-permeable, nonhydrolyzable cAMP analog; Rp-cAMPs (100 μm; Sigma), a protein kinase A antagonist; recombinant Oncomodulin (rOcm) (50 ng/eye); P1, a 24 aa peptide that competes with Ocm for receptor occupancy and acts as a potent antagonist; or Pα, a control peptide based on a region of α-parvalbumin that is partially homologous to P1 (Yin et al., 2009) (both injected at 2.3 μg/μl in 1 μl of 0.1% DMSO–saline).
Quantitation of axon regeneration and RGC survival.
In most of the studies, mice were killed with an overdose of anesthesia 14 d after optic nerve injury and were perfused with saline and 4% paraformaldehyde (PFA). These mice were 8 weeks old when killed. Optic nerves and eyes were dissected and postfixed in PFA. Nerves were impregnated with 10% and then 30% sucrose, embedded in OCT Tissue Tek Medium (Sakura Finetek), frozen, cut in the longitudinal plane at 14 μm, and mounted on coated slides. Regenerating axons were visualized by staining with a sheep antibody to GAP-43 followed by a fluorescently labeled secondary antibody. Axons were counted manually in at least eight longitudinal sections per case at prespecified distances from the injury site, and these numbers were converted into the number of regenerating axons at various distances as described previously (Leon et al., 2000).
To study axon regeneration over longer times and distances, mice received intraocular injections of the B subunit of cholera toxin (CTB) (3 μl, 1%; List Biological) 4 d before being killed, enabling us to verify that axons visualized within the chiasm and thalamus had originated in the retina. These animals survived a total of 6 weeks after optic nerve surgery and were 16 weeks old at the completion of the study. In addition to examining the optic nerves and eyes, we prepared 14 μm frozen sections in the horizontal plane through the optic chiasm and 50 μm coronal sections through the diencephalon. These sections were double immunostained for GAP-43, as above, and for CTB using a primary antibody prepared in rabbit (1:500; GenWay), followed by differentially labeled fluorescent secondary antibodies (1:500; Invitrogen). A monoclonal antibody to the neuronal marker neuronal-specific nuclear protein (NeuN) (MAB377; Millipore Bioscience Research Reagents) was used to visualize the boundaries of thalamic structures, including the lateral geniculate nucleus (LGN). In horizontal sections through the optic chiasm, CTB-labeled axons crossing an imaginary horizontal line passing through the center of the chiasm were counted in all sections under fluorescent microscopy and summed (see Fig. 6H). For the LGN, we summed all CTB-labeled axons visualized within the plane of section in all sections through the dorsal and ventral portions of the nucleus contralateral to the injured optic nerve. We then averaged the total number of axons in each area across the five cases of long-distance regeneration that were suitable for quantitative analysis. In addition to these cases, there were three others in which we examined long-distance regeneration qualitatively. RGC survival was evaluated in flat-mounted retinas immunostained with a rabbit antibody to βΙΙΙ-tubulin (1:500; Abcam), followed by a secondary antibody made in goat and conjugated to Alexa Fluor 594, taking advantage of the selective expression of βΙΙΙ-tubulin in RGCs (Cui et al., 2003). Images of eight prespecified areas, 2 mm from the optic disc were captured under fluorescent illumination (400×; E800; Nikon). βΙΙΙ-Tubulin-positive cells were counted using NIH ImageJ software (Wayne Rasband, National Institutes of Health, Bethesda, MD). Cell densities were averaged across all eight areas, and data were represented as means ± SEM based on five to eight mice per group.
Ocm binding assays in vivo.
In separate groups of animals, we investigated the ability of Ocm to diffuse from the vitreous and bind to the inner retina in vivo. These studies were performed either 1 d after injecting rOcm, with or without CPT–cAMP, into the vitreous, or at 1 or 7 d after injecting Zymosan with or without CPT–cAMP, Rp-cAMP, P1, or Pα. Cryostat sections through the retina were immunostained with TUJ1, a monoclonal antibody to βIII-tubulin, and an affinity-purified rabbit polyclonal antibody to Ocm (Yin et al., 2009), followed by an Alexa Fluor 594-labeled secondary antibody to mouse IgG and an Alexa Fluor 488-labeled antibody to rabbit IgG. As a negative control, we used the same anti-Ocm antibody preabsorbed with recombinant Ocm. As noted previously, preadsorption removes most, but not all, Ocm-specific IgGs (Yin et al., 2009). The inner plexiform layer (IPL) of the retina was identified by 4′,6′-diamidino-2-phenylindole (DAPI) and TUJ1 staining. The mean intensity of Ocm in the IPL was measured in three regions per case in at least four cases per condition using NIH ImageJ software.
Western blots.
Retinas were obtained from normal rats or from rats injected intraocularly with Zymosan (12.5 μg/μl), alone or with CPT–cAMP (50 μm) and/or the Ocm-blocking peptide P1 or the control peptide Pα (same concentrations as in mouse studies). Retinas were dissected without fixation 1 d later and homogenized in Eppendorf tubes with PBS, pH 7.4, containing a mixture of protease inhibitors. Soluble and particulate protein fractions were separated by centrifugation (13,000 rpm, 15 min, 4°C), pellets were washed once with the same buffer and resedimented, and proteins were extracted in buffer containing 1% Triton X-100. Ocm was detected in the soluble and particulate fractions using Western blots as described (Yin et al., 2009). Briefly, proteins were separated by SDS-PAGE, transferred electrophoretically to polyvinylidene difluoride membranes, blocked for 2 h in buffer containing 5% milk, and then incubated with a mouse monoclonal antibody to Ocm (1:200; gift from Michael Henzl, University of Missouri, Columbia, MO) in 5% BSA, 4°C overnight. After several washes, an HRP-conjugated secondary antibody made in sheep against mouse IgG (GE Healthcare) was applied at 1:2000 in 2% milk for 1 h. Signals were visualized with ECL-Plus (GE Healthcare).
Signal transduction pathways.
Using immunohistochemistry, we investigated changes in the activation of three signal-transduction molecules, mitogen-activated protein kinase (MAPK), Akt, and S6 kinase (S6K), along with overall levels of these proteins. These studies were performed in PTENflx/flx mice injected with either AAV2–Cre or AAV2–GFP as described above. After allowing 2 weeks for transgene expression and gene deletion, mice sustained a crush injury of the optic nerve, followed by intraocular injections of Zymosan (12.5 μg/μl), CPT–cAMP (50 μm), both, or neither. Eyes were removed 3 d later and immersed in 4% PFA overnight, transferred to 30% sucrose for another day, embedded in OCT medium (Sakura Finetek), frozen, and sectioned on a cryostat (14 μm). Sections were double immunostained with the TUJ1 antibody and a rabbit antibody against active (phospho-) MAPK (1:200), active (phospho-) Akt (1:150), or activated S6K (1:200) (all from Cell Signaling Technology). Other sections were stained with antibodies to recognize the overall abundance of the three signaling molecules, i.e., a mouse antibody to total MAPK (1:200), or rabbit antibodies to Akt (1:400) or S6K (1:200) (all from Cell Signaling Technology), followed by a goat anti-mouse or anti-rabbit IgG antibody conjugated to either Alexa Fluor 488 or 594. As negative controls, we omitted the primary antibodies. RGCs and the IPL were identified by TUJ1 and DAPI staining. The mean staining intensity in RGCs was calculated using NIH ImageJ software based on 10 cells per section, two sections per case, and three to four cases per condition. Staining intensity in the IPL was determined using the methods described above to measure Ocm levels. Individual mean values were corrected by staining levels in the corresponding negative controls and then averaged across each group. We present both raw data and data normalized by the total level of the relevant proteins (background subtracted).
Statistics.
Data are represented as means ± SEM. Statistical significance was determined using unpaired Student's t tests. Statistical analysis was performed by ANOVA with Bonferroni's and Dunnett's post hoc tests for multiple comparisons.
Results
cAMP enhances regeneration in an Ocm-dependent manner
cAMP elevation was shown recently to enhance inflammation-induced regeneration in the optic nerve (Müller et al., 2009). The authors of that study proposed that ciliary neurotrophic factor (CNTF) is the principal mediator of inflammation-induced regeneration and that cAMP acts by enhancing the effects of CNTF. Our studies, in contrast, point to Ocm as the primary mediator of inflammation-induced regeneration (Yin et al., 2006, 2009) but leave open the possibility that cAMP could act by increasing the effects of CNTF or other factors. A second issue is that, at high concentrations, cAMP is detrimental to axon regeneration (Monsul et al., 2004; Yin et al., 2006), and the optimal concentration of cAMP to enhance the effects of intraocular inflammation is unknown. We therefore performed preliminary studies to determine the optimal concentration of cAMP to induce regeneration in mice. As expected, even in the absence of exogenous cAMP, intraocular Zymosan (12.5 μg/ml, sterilized) greatly increased the amount of regeneration seen 2 weeks after optic nerve crush (Fig. 1A,B,E: p < 0.001 at 0.5–1 mm). Zymosan was far more effective than lens injury (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) (p < 0.05). CPT–cAMP increased Zymosan-induced regeneration continuously up to concentrations of 50–100 μm (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). CPT–cAMP did not stimulate long-distance regeneration on its own (Fig. 1C,E) but, at 50–100 μm, increased the effects of intraocular inflammation approximately twofold (Fig. 1D,E) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) (p < 0.001 at 0.5 mm, p < 0.02 at 1 mm, NS at 2 mm and beyond). In terms of cell viability, Zymosan doubled the number of RGCs that remained alive 2 weeks after optic nerve injury, and this remained unchanged in the presence of CPT–cAMP at concentrations up to 50 μm (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). However, higher concentrations diminished RGC survival (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Parallel studies in the rat (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material) similarly show that outgrowth stimulated by lens injury, an alternative way to promote intraocular inflammation, is maximally augmented by 20–100 μm CPT–cAMP, with higher concentrations having a negative effect.

cAMP enhances inflammation-induced axon regeneration in the mouse optic nerve. A–D, Longitudinal sections through the adult mouse optic nerve (ON) showing GAP-43-positive axons distal to the injury site (asterisks) 2 weeks after optic nerve crush. A, Absence of regeneration after surgery alone. B, C, Increased regenerating after intraocular injection of Zymosan (Zymo) (B; 12.5 mg/ml) but not CPT–cAMP alone (C; 50 μm). D, Extensive regeneration induced by Zymosan plus CPT–cAMP. Scale bar, 200 μm. E, Quantitation of axon growth at indicated distances beyond the crush site. ***p < 0.001 compared with optic nerve crush alone. †††p < 0.001 compared with cAMP treatment. #p < 0.05, ###p < 0.001 compared with Zymosan treatment alone. Data in all figures represent group means ± SEM.
Excessive inflammation diminishes RGC survival in rats (Yin et al., 2003), and we therefore performed additional pilot studies to determine the optimal concentration of Zymosan for the mouse studies. Maintaining CPT–cAMP concentration at 50 μm, axon regeneration rose continuously with increasing concentrations of Zymosan, reaching a plateau at concentrations at 6.3–12.5 μg/μl. RGC survival was similar across all concentrations (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Based on these results, we used Zymosan at 12.5 μg/μl and CPT–cAMP at 50 μm for the subsequent studies.
To investigate whether the ability of cAMP to enhance inflammation-induced regeneration depends on Ocm, we used P1, a peptide that competes with Ocm for receptor occupancy (Yin et al., 2009). P1 completely eliminated the axon-promoting effects of Zymosan (Fig. 2A,B,E; compare with Fig. 1E) and also eliminated the enhancement of regeneration seen when Zymosan was combined with CPT–cAMP (Fig. 2E) (p < 0.001). The control peptide Pα had no effect in either case (Fig. 2A,C). These results indicate that the ability of cAMP to enhance inflammation-induced regeneration depends on Ocm and imply that no other factors are present that can promote regeneration in the absence of Ocm, even when [cAMP]i is elevated. Although P1 suppressed the axon-promoting effects of inflammation, it did not suppress the ability of inflammation to enhance RGC survival (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Like P1, Rp-cAMP also suppressed Zymosan-induced regeneration without altering RGC survival (Fig. 2E,F: p < 0.05) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).

cAMP enhances regeneration in an Oncomodulin-dependent manner. A–D, Longitudinal sections through the adult mouse optic nerve (ON) showing GAP-43-positive axons distal to the injury site (asterisk) 2 weeks after optic nerve crush. All animals received intravitreal injections of Zymosan (Zymo) immediately after optic nerve surgery. Regeneration in the absence of exogenous cAMP is unaffected by the control peptide Pα (A) but is blocked by the Ocm antagonist P1 (B). The enhanced regeneration seen in the presence of exogenous cAMP is unaffected by Pα (C) but is eliminated by P1 (D). Rp-cAMP diminishes Zymosan-induced axonal regeneration (E). Scale bar, 200 μm. F, Quantitation of axon regeneration in the presence or absence of CPT–cAMP, Rp-cAMP, and peptides. †††p < 0.001 compared with Pα injections. #p < 0.05 compared with Zymosan injections (decrease).
cAMP enables Ocm to bind to the inner retina
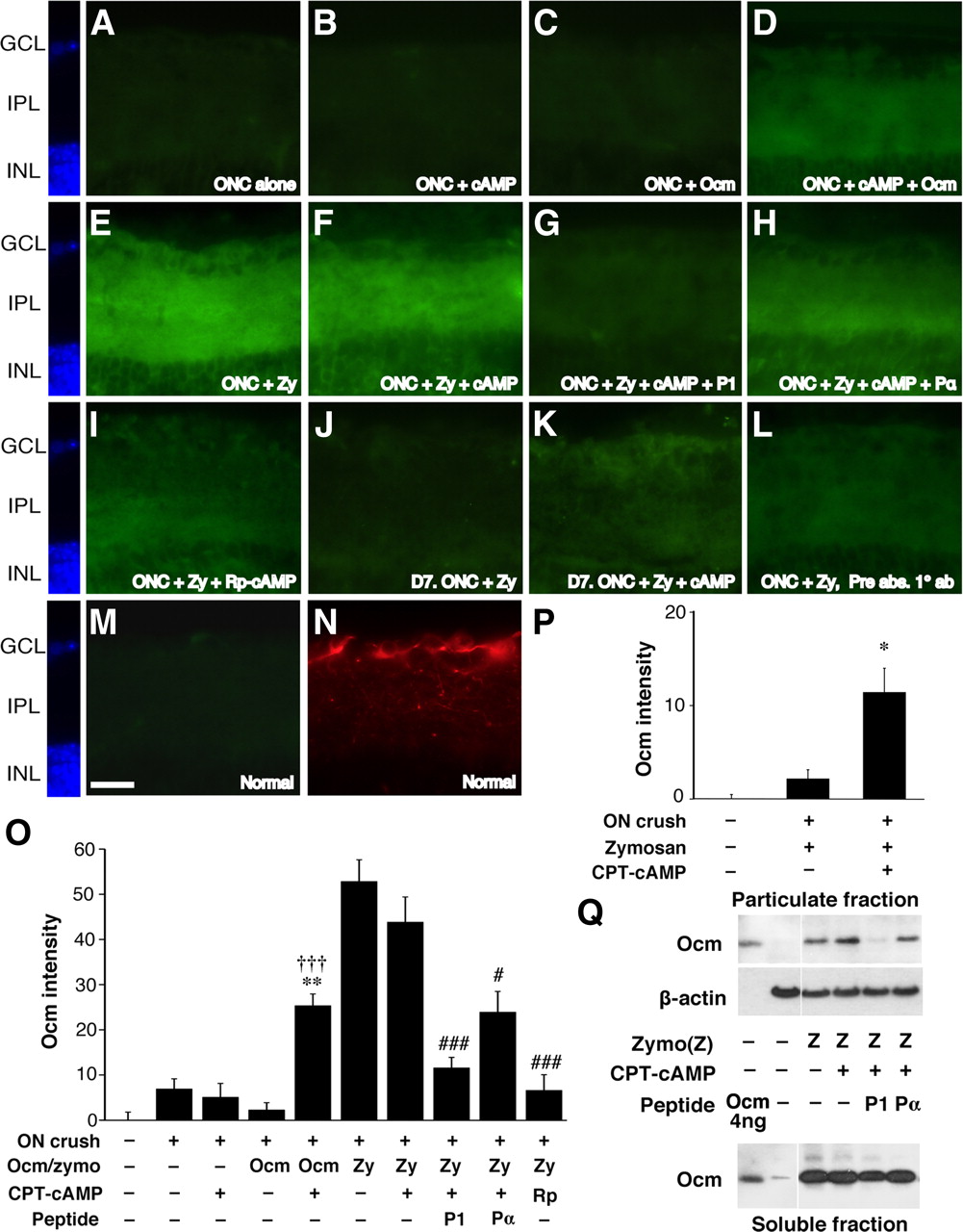
Studies in culture show that the ability of Ocm to promote outgrowth requires elevation of [cAMP]i and correlates with the cAMP-dependent appearance of an Ocm receptor on the cell surface (Yin et al., 2006). To investigate whether cAMP controls the ability of Ocm to bind to its receptor(s) in vivo, we examined levels of Ocm in the IPL of the retina after injecting rOcm or Zymosan, with or without CPT–cAMP or the protein kinase A antagonist Rp-cAMP, into the vitreous. Ocm was undetectable in the retinas of intact mice (Fig. 3M) and in the retinas of mice 1 d after optic nerve crush, even when either rOcm (50 ng) or CPT–cAMP (50 μm) was injected into the vitreous (Fig. 3A–C). However, injecting rOcm together with CPT–cAMP resulted in a marked elevation of intraretinal Ocm (Fig. 3D,M: p < 0.01 and p < 0.001 for Ocm plus CPT–cAMP compared with CPT–cAMP or Ocm alone, respectively). Intravitreal Zymosan (12.5 μg/μl) led to even greater increases in retinal Ocm (Fig. 3E,M). These findings show that the binding of Ocm in the retina is cAMP dependent and suggest that intraocular inflammation results in the secretion of high levels of Ocm as well as factors that lead to an increase in [cAMP]i. Based on the intensity of Ocm binding obtained after injecting 50 ng of rOcm plus CPT–cAMP, it would appear that intraocular inflammation releases ≥100 ng of Ocm into the eye. The addition of CPT–cAMP did not elevate Ocm immunostaining in the retina beyond the level seen after injecting Zymosan alone (Fig. 3F,M). The specificity of Ocm binding in the retina was verified by the fact that it was fully displaced by P1 (Fig. 3G,M). The control peptide Pα had a partial effect but still left appreciable levels of Ocm bound to the retina (Fig. 3H,M). Ocm intensity after Zymosan injection was also strongly diminished by Rp-cAMP (Fig. 3I,M: p < 0.001). These latter results further demonstrate the specificity and cAMP dependence of Ocm binding in the retina.

cAMP regulates Ocm binding in the inner retina. A–N, Sections through the inner retina of mice with optic nerve crush (ONC; A–L) or normal mice (M, N) immunostained to detect Ocm (A–I, M) or βIII tubulin (N) 1 or 7 d after injecting various agents into the posterior chamber of the eye. A–D, Binding of recombinant Ocm in the retina 1 d after optic nerve crush. Ocm is undetectable in the absence of exogenous Ocm or when Ocm or CPT–cAMP are injected alone, but becomes apparent when the latter two are coinjected. E–I, Binding of native Ocm after intraocular injections of Zymosan. Zymosan strongly elevates Ocm immunostaining (E); this is not enhanced by the addition of CPT–cAMP (F). Ocm binding is suppressed by peptide P1 (G) and by Rp-cAMPs (I); the control peptide Pα has a partial effect (H). J, K, Ocm binding 7 d after Zymosan injection is enhanced by CPT–cAMP. L, Reduced immunostaining after preadsorption of the primary antibody with rOcm. M, N, Normal retina shows no detectable Ocm (M). RGCs and their dendrites in the IPL are visualized with an antibody to βIII-tubulin (N). Scale bar: A–N, 25 μm. O, Quantitation of Ocm intensity in the inner plexiform layer of the retina. **p < 0.01 compared with CPT–cAMP alone. †††p < 0.001 compared with Ocm alone. ###p < 0.001 compared with Zymosan plus CPT–cAMP. ON, Optic nerve; Zy, Zymosan. P, Quantitation of Ocm intensity in the inner plexiform layer 7 d after optic nerve (ON) crush. *p < 0.05 compared with Zymosan treatment alone. Q, Ocm binding in the rat retina. One day after injecting Zymosan (Zymo or Z) with or without other agents as indicated, retinas were dissected, homogenized, and fractionated into particulate and soluble fractions, and the proteins were separated by SDS-PAGE. Western blotting used a monoclonal antibody to visualize Ocm and a β-actin antibody to evaluate protein loading. Within the particulate fraction, Ocm levels rose above background after the injection of Zymosan, with or without CPT–cAMP, into the vitreous. P1 but not Pα diminished Ocm binding in the particulate fraction but did not affect levels in the soluble protein fraction. GCL, Ganglion cell layer; INL, inner nuclear layer.
The above findings show that intraocular inflammation causes Ocm binding in the inner retina to saturate, at least after 1 d, raising the question of how exogenous cAMP enhances inflammation-induced regeneration. One possibility is that the factor(s) that cause [cAMP]i to increase in RGCs after inflammation decline rapidly and that exogenous cAMP prolongs Ocm binding. To investigate this possibility, we examined Ocm levels in the retina 7 d after injecting Zymosan in the presence or absence of CPT–cAMP. When Zymosan was injected alone, Ocm levels declined to baseline after 7 d. However, when Zymosan was injected with CPT–cAMP, Ocm levels remained higher (Fig. 3J,K,N: p < 0.05). Thus, although CPT–cAMP does not increase the initial binding of Ocm to the retina, it prolongs this binding.
The cAMP dependence and specificity of Ocm binding in the retina was also investigated by subcellular fractionation and Western blotting. These studies were performed in rats because the monoclonal anti-Ocm antibody used for Western blotting gives a clearer signal for rat Ocm than for the mouse protein (Y.Y., unpublished observations) and because of the greater ease of working with retinas from rats versus mice. To differentiate bound Ocm from total, retinas were homogenized and separated into particulate and soluble fractions by centrifugation. Proteins were then separated by SDS-PAGE, transferred to a membrane, and probed with antibodies to Ocm. Intravitreal Zymosan injections increased Ocm levels in the particulate fraction of the rat retina, and the addition of CPT–cAMP increased levels even further. The P1 peptide displaced Ocm from the bound fraction of the retina but left Ocm levels in the soluble fraction high; Pα did not appreciably alter levels of bound or soluble Ocm.
Intraocular inflammation, cAMP, and PTEN deletion act synergistically to augment regeneration
As noted in the Introduction, conditional deletion of PTEN in RGCs promotes axon regeneration through the injured mouse optic nerve (Park et al., 2008). This finding implies that PI3K activation is sufficient to induce appreciable regeneration. Complementary studies using inhibitors have also suggested that the PI3K–Akt signal-transduction pathway may play a role in Ocm-induced axon regeneration in RGCs (Yin et al., 2006). However, it is not known whether enhancement of PI3K signaling by PTEN deletion is functionally equivalent to intraocular inflammation. If it were, we would predict that inflammation/cAMP and PTEN deletion combined might not result in higher levels of regeneration than either treatment alone. Conversely, if Ocm stimulation and PTEN deletion act through complementary pathways, the combination of inflammation plus cAMP and PTEN deletion might be expected to give stronger regeneration than either one alone. To investigate this question, we used mice with a conditional deletion of the PTEN gene (PTENflx/flx) and injected these intraocularly with either AAV2 expressing Cre recombinase (AAV2–Cre) to delete the pten gene in RGCs or a control virus expressing green fluorescent protein (AAV2–GFP). Using a line of reporter mice that express PLAP after Cre–lox recombination, we found that AAV2–Cre injections infected 80–90% of RGCs, as reported previously (Park et al., 2008). AAV2–GFP led to a similar level of infection, 81% of RGCs (data not shown). After allowing 2 weeks for transgene expression to reach high levels, mice underwent optic nerve surgery and were injected intraocularly with Zymosan, CPT–cAMP, neither, or both.
PTEN deletion, induced by infecting RGCs of PTENflx/flx mice with AAV2–Cre, led to robust regeneration (Fig. 4A,I). The same strain of mice injected with the control virus AAV2–GFP showed almost none (Fig. 4I). Regeneration induced by PTEN deletion was unaffected by the P1 peptide (Fig. 4J), indicating, first, that this growth is independent of Ocm, and second, that the inhibitory effects of P1 seen earlier are specific to inflammation-induced growth and interference with Ocm signaling. Growth induced by PTEN deletion was not enhanced by the addition of CPT–cAMP (50 μm) (Fig. 4B,I). Thus, although cAMP elevation enhances regeneration associated with intraocular inflammation, it does not exert a general effect on regeneration stimulated by other means.

Synergistic effects of intraocular inflammation, cAMP, and pten gene deletion. PTEN was deleted in RGCs by injecting AAV2–Cre into the eyes of PTENflx/flx mice 2 weeks before optic nerve surgery (ONC). A–D, Longitudinal sections through the optic nerve showing GAP-43-positive axons 2 weeks after optic nerve injury. PTEN deletion caused appreciable regeneration (A) that was not enhanced by CPT–cAMP (B) and only slightly enhanced by intraocular inflammation (C). The combination of PTEN deletion plus Zymosan (Zymo) plus CPT–cAMP resulted in far greater regeneration than any other treatments (D). E–H, Retinal whole mounts immunostained with TUJ1 to visualize RGCs. E, Normal retina. F, Loss of RGCs 2 weeks after optic nerve crush in PTENflx/flx mouse injected with the control virus (AAV2–GFP). G, H, Preservation of RGCs 2 weeks after optic nerve crush in a PTENflx/flx mouse injected with AAV2–Cre to delete PTEN (G). Survival is not enhanced further by intraocular inflammation plus cAMP (H). Scale bar: A–D, 200 μm; E–H, 50 μm. I, Quantitation of axon regeneration after 2 weeks. *p < 0.05, **p < 0.01, ***p < 0.001 compared with PTEN deletion alone. †p < 0.05, ††p < 0.01, †††p < 0.001 compared with PTEN deletion plus Zymosan. #p < 0.05, ##p < 0.01 compared with PTEN deletion plus Zymosan plus CPT–cAMP. J, P1 peptide does not diminish regeneration induced by PTEN deletion. K, Quantitation of RGC survival. PTEN deletion more than tripled the number of RGCs surviving 2 weeks after optic nerve damage compared with PTENflx/flx mice injected with control virus. Survival was not enhanced further by intraocular inflammation and/or CPT–cAMP. ***p < 0.001 compared with nerve-damaged controls.
PTEN deletion combined with intraocular inflammation resulted in somewhat higher levels of regeneration than PTEN deletion alone (Fig. 4C,I: p < 0.05 at 1 and 3 mm, p < 0.01 at 2 mm). However, when PTEN deletion was combined with Zymosan plus CPT–cAMP, the effects were dramatic. At short distances from the injury site, the three-way combination more than doubled the amount of regeneration seen after PTEN deletion alone, and the difference became greater and greater at increasing distances. At 3 mm and beyond, PTEN deletion combined with inflammation plus CPT–cAMP increased the number of regenerating axons >10-fold compared with PTEN deletion alone (Fig. 4D,I: differences significant at p < 0.001 at 0.5–2 mm, p < 0.01 at 3 mm) and threefold compared with Zymosan plus CPT–cAMP (Fig. 4D,I: different significant at p < 0.001 at 0.5 and 1.0 mm, p < 0.0 1 at 2 mm, p < 0.05 at 3 mm). The addition of P1 to the combination of PTEN deletion, Zymosan, and CPT–cAMP decreased the number of regenerating axons to the level seen with PTEN deletion alone (Fig. 4I). Thus, the increase observed in the presence of all three treatments is dependent on Ocm.
As reported previously (Park et al., 2008), PTEN deletion enhanced RGC survival after optic nerve crush. Survival was not enhanced further by intraocular inflammation, with or without CPT–cAMP (Fig. 4K). Thus, the augmented regeneration seen when PTEN deletion was combined with intraocular inflammation plus CPT–cAMP is unrelated to changes in RGC survival.
The dramatic enhancement of regeneration seen when PTEN deletion was combined with intraocular inflammation plus CPT–cAMP suggests that this combination may activate key cell-signaling pathways to a greater degree than the single treatments. We investigated this question using immunocytochemistry to detect changes in the activation of p42/p44 MAPK (extracellular signal-regulated kinase), Akt, and S6K in RGCs. S6K is a regulator of protein translation and can be activated by either the PI3K–Akt or MAPK pathway (Carracedo and Pandolfi, 2008; Chalhoub and Baker, 2009). The principal results of this study are shown in Figure 5, and supplemental Figure 4 (available at www.jneurosci.org as supplemental material) shows all conditions tested, along with results obtained when labeling intensities for the activated forms of the kinases were normalized by their abundance. In several instances, changes were more apparent within the IPL of the retina, which contains the dendrites of RGCs and impingent axons, than within RGC somata. Within the IPL, MAPK phosphorylation was augmented by either PTEN deletion (p < 0.01) or by inflammation plus CPT–cAMP (p < 0.05), and the increase became even greater when all three were combined (p < 0.01 compared with optic nerve crush alone in mice expressing PTEN in RGCs; p < 0.05 comparing mice with intraocular inflammation plus CPT–cAMP in RGCs lacking PTEN vs having PTEN present). Increased MAPK activation was also apparent within RGCs after PTEN deletion and intraocular inflammation/cAMP. PTEN deletion also increased Akt phosphorylation, as expected, although this effect was more pronounced within the IPL than within RGCs. Intraocular inflammation/CPT–cAMP did not enhance Akt activation or further augment the effect of PTEN deletion. The phosphorylation of S6K followed the same pattern as Akt within RGCs but showed strong changes within the IPL when PTEN deletion was combined with intraocular inflammation plus CPT–cAMP (p < 0.001). Thus, combining PTEN deletion with inflammation plus CPT–cAMP activated both the PI3K and MAPK signaling pathways; the pattern of S6K activation suggests that it is influenced by both pathways.

Activation of cell-signaling pathways. A, Sections through the inner retina 3 d after optic nerve crush (ONC) alone or combined with intraocular Zymosan (Zymo) plus CPT–cAMP. PTENflx/flx mice were injected 2 weeks earlier with either AAV2–GFP, allowing RGCs to continue expressing PTEN (PTEN+), or AAV2–Cre to excise the pten gene in RGCs (PTEN−). Sections were stained with antibodies to the phosphorylated (activated) forms of MAP kinase (pMAPK), Akt (pAkt), or S6K (pS6K) as indicated, followed by fluorescent secondary antibodies. Scale bar, 25 μm. GCL, Ganglion cell layer. B, Quantitation of fluorescence in βIII-tubulin-positive RGCs and in the IPL. *p < 0.05, **p < 0.01, ***p < 0.001 compared with PTEN-positive mice with optic nerve crush (ONC) alone. †p < 0.05, †††p < 0.001 compared with PTEN-positive mice injected with Zymosan (Zymo) plus CPT–cAMP after optic nerve crush.
We attempted to normalize levels of the activated forms of the kinases by the overall levels of the proteins based on immunostaining with antibodies that do not differentiate the phosphorylated and dephosphorylated forms of the proteins (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). This analysis revealed that some of the treatments altered the overall levels of the kinases. In the case of Akt, normalization altered the pattern of phosphorylation changes from that seen when measuring kinase activity alone (supplemental Fig. 4E vs B, available at www.jneurosci.org as supplemental material). However, normalization did not alter the pattern of phosphorylation changes seen for MAPK (supplemental Fig. 4D vs A, available at www.jneurosci.org as supplemental material) or S6K (supplemental Fig. 4F vs C, available at www.jneurosci.org as supplemental material).
Retino-thalamic regeneration
In light of the extensive regeneration described above, we investigated whether axons would be able to extend into the diencephalon after longer survival periods. These studies were performed in mice that were 8 weeks old at the time of AAV2 injections and 10 weeks old at the time of optic nerve surgery. Because Ocm levels in the retina decrease to background levels 7 d after Zymosan injections (Fig. 3K,L), mice received a second Zymosan injection at half the original concentration 2 weeks after the first, together with CPT–cAMP (50 μm) to maintain Ocm binding. To provide an alternative way of visualizing regenerating axons and to verify that these axons originate in the retina, mice received an intraocular injection of CTB as an anterograde tracer 4 d before being killed.
Combinatorial treatment enabled numerous GAP-43-positive axons to extend the full length of the optic nerve after 6 weeks (Fig. 6A). The great majority of these axons were double labeled for GAP-43 and CTB (Fig. 6B–E), verifying that they were of retinal origin and actively growing. The number of lengthy axons increased markedly from 2 to 6 weeks (Fig. 6F: p < 0.001). Many of these entered the optic chiasm and continued growing (Fig. 6G). In general, the most lateral CTB-labeled axons entering the chiasm appeared to extend ipsilaterally when exiting (Fig. 6G, arrows), whereas the most medial ones turned at the chiasm and grew into the contralateral optic nerve, failing to turn in the knee of Wilbrand (Fig. 6G, arrowheads). Some axons continued on into the contralateral optic tract and could be visualized in the contralateral LGN (Fig. 6H–K). Based on CTB immunostaining, we quantified the number of regenerating axons in the optic nerve just proximal to the chiasm (4.0 and 4.5 mm from the injury site), at the middle of the chiasm, and within the contralateral LGN. On average, the number of regenerating axons in the chiasm and LGN were 41 and 1%, respectively, of the numbers seen 4.5 mm down the optic nerve (Fig. 6M). In addition to the five long-term regeneration cases that were suitable for quantitation, we examined three others at a qualitative level. Of a total of eight cases, three had regenerating axons within the LGN. In terms of cell survival, although some cell death occurred between 2 and 6 weeks, RGC survival remained elevated (mean number of TUJ1-positive cells per square millimeter declined from 54% of the original number at 2 weeks to 39% after 6 weeks) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In summary, intraocular inflammation combined with CPT–cAMP and PTEN deletion enabled some axons to regenerate from just behind the eye through the entire optic nerve, across the optic chiasm, and into their central target area within 6 weeks.

Synergistic interactions lead to long-distance regeneration after 6 weeks. A, B, Regenerating axons extend the full length of the optic nerve in mice lacking the pten gene in RGCs and exposed to intraocular inflammation plus CPT–cAMP. Mice received intraocular injections of CTB 4 d before being prepared for histology. Nerves were double immunostained for GAP-43 (A) and CTB (B). C, D, Enlarged images of the areas enclosed within the white boxes in A and B, respectively. E, Merged image of C and D. F, Quantitation of axon regeneration 3.5 mm from the crush site. The number of lengthy axons increased almost 10-fold between weeks 2 and 6 (***p < 0.001), and most of these axons were double labeled for GAP-43 and CTB. G, Regenerating axons in the optic chiasm. Arrows point to regenerating axons that extend into the thalamus; arrowheads show axons growing into the contralateral optic nerve (ON). H–K, Extension of CTB-labeled axons into the thalamus. Sections were double stained to detect CTB in regenerating axons (red) and NeuN to visualize neurons (green). H and J show double staining, whereas I and K show the axons alone at high contrast. Some axons can be seen in the contralateral optic tract (H, I) and ventral lateral geniculate nucleus (J, K). Arrowheads indicate CTB-labeled axons. L, Schematic drawing through the thalamus showing positions of labeled axons. M, Quantitation of CTB-labeled axons in different segments of the visual pathway. Data are based on four or five cases. dLGN, Dorsal lateral geniculate nucleus; vLGN, ventral lateral geniculate nucleus; OPT, optic tract; Ch, chiasm; CP, cerebral peduncle; Zymo, Zymosan. Scale bars: A, B, 200 μm; C–E, H–K, 50 μm.
Discussion
Although previous studies have identified several strategies to stimulate axon regeneration in the mature optic nerve, extensive regeneration from the eye to the brain has remained a major challenge. The present study was motivated by the hypothesis that several of the known axon-promoting strategies activate distinct mechanisms with additive or even synergistic effects. Our results show that intraocular inflammation, cAMP elevation, and PTEN deletion exert complementary effects that, when combined, result in unprecedented levels of regeneration.
The role of Ocm
In agreement with previous studies (Yin et al., 2006, 2009), our results show that Ocm plays a central role in inflammation-induced regeneration. Injuring the lens or injecting Zymosan into the eye leads to an influx of macrophages that produce high levels of Ocm (Yin et al., 2009). In the presence of appropriate cofactors, Ocm binds to a high-affinity receptor on RGCs and stimulates extensive axon outgrowth in culture and in vivo (Yin et al., 2006). P1, a small peptide that competes with Ocm for receptor occupancy, was shown previously to suppress most of the regeneration induced by intraocular inflammation in rats (Yin et al., 2009). In the present study, P1 eliminated inflammation-induced regeneration in mice but did not diminish the effects of PTEN deletion. In all, these data show that Ocm is necessary and, in the presence of appropriate cofactors, sufficient to account for inflammation-induced regeneration in the optic nerve. Others have argued that CNTF mediates these effects (Müller et al., 2009), but this seems highly unlikely in view of the loss of regeneration caused by blocking Ocm and by many studies showing that CNTF exerts relatively little axon-promoting effects on RGCs in vivo in the absence of intraocular inflammation (Leon et al., 2000; Weise et al., 2000; Pernet and Di Polo, 2006; Cen et al., 2007; Lingor et al., 2008; Smith et al., 2009) (but see Leaver et al., 2006).
Role of cAMP
Studies in cell culture show that cAMP enables Ocm to bind to a cell-surface receptor on RGCs (Yin et al., 2006), probably by stimulating receptor translocation (Meyer-Franke et al., 1998). The present results show that this also occurs in vivo. Recombinant Ocm injected directly into the vitreous could be detected 1 d later in histological sections through the retina only when cAMP had been elevated. Intraocular inflammation resulted in nearly twice as much retinal Ocm after 1 d as injecting rOcm with a cAMP analog, and this binding was diminished by the protein kinase A antagonist Rp-cAMPs. The cAMP dependence of Ocm binding suggests that, in addition to producing high levels of Ocm, intraocular inflammation produces factors that cause [cAMP]i to increase in RGCs. The specificity of Ocm binding was further demonstrated by its loss in the presence of the P1 peptide. Pα, a control peptide with 50% sequence identity to P1 (and 58% homology), partially blocked Ocm binding in the retina after intraocular inflammation but did not suppress inflammation-induced regeneration. This discrepancy may be explained by our previous finding that Ocm can exert its full biological effect even when occupying a fraction of the available receptors (KD = 28 nm) (Yin et al., 2006).
Exogenous cAMP doubled the amount of regeneration induced by inflammation. Besides promoting the translocation of growth factor receptors, elevation of [cAMP]i can also improve regeneration by rendering growth cones unresponsive to cell-extrinsic inhibitors of growth (Ming et al., 1997; Song et al., 1998) and by increasing the expression of growth-promoting proteins, including Arginase I (Cai et al., 2002). One downstream product of Arginase I, spermidine, stimulates some axon regeneration in the optic nerve on its own (Deng et al., 2009). However, the primary effect of cAMP in the present study appears to be mediated via Ocm. CPT–cAMP had little effect on its own and did not enhance regeneration induced by PTEN deletion, as might be expected if its primary effect were to promote regeneration in general. Second, the ability of CPT–cAMP to enhance regeneration was abrogated by the P1 peptide, indicating that its effect requires Ocm. CPT–cAMP prolonged the binding of endogenous Ocm to its receptor in vivo, providing a plausible mechanism for its effect on regeneration.
PTEN gene deletion
Intraocular inflammation, cAMP elevation, and PTEN gene deletion showed a dramatic synergy when combined, increasing the number of axons reaching the distal optic nerve by a factor of 10 compared with any of the treatments in isolation. PTEN suppresses signaling through the PI3K pathway, and, as anticipated (Cully et al., 2006), its deletion increased Akt phosphorylation in RGCs and RGC viability. In culture, although the axon-promoting effects of Ocm are unaffected by inhibiting PI3K, MAPK, or Janus kinase alone, they are fully inhibited by blocking a combination of these pathways (Yin et al., 2006). Combining PTEN deletion with intraocular inflammation and cAMP elevation did not enhance Akt phosphorylation or RGC survival beyond the level seen with PTEN deletion alone. Together, these observations imply that the role of the PI3K pathway in Ocm-stimulated growth is constitutive rather than regulatory. They also indicate that the effect of the combinatorial treatment on axon regeneration is not a consequence of increased cell survival. MAPK showed increased activation within the IPL, which contains RGC dendrites and axons that project onto these, in response to both inflammation plus cAMP and PTEN deletion, and even stronger activation when all three were combined. The regulation of MAPK signaling by PTEN, although not previously demonstrated in the nervous system, has been shown to occur elsewhere (Yart et al., 2001). Finally, in conformity with the established role of PTEN in suppressing the mammalian target of rapamycin (mTOR) pathway, we found that PTEN deletion increased the phosphorylation of S6K, a downstream target of mTOR and a key regulator of protein translation. Inflammation plus CPT–cAMP did not enhance S6K activation further within RGC somata but did so within the IPL. The latter observation is consistent with the dual regulation of S6K by the PI3K and MAPK pathways (Carracedo and Pandolfi, 2008). At this stage, we do not know why some changes appear to be stronger in the IPL than in RGC somata, but this could reflect a saturation of the signal in RGC somata or that some changes occur selectively in RGC dendrites or even in the axons that synapse on them.
Intraocular inflammation increases mRNA levels for GAP-43, SPRR1A, and other genes related to axon growth (Fischer et al., 2004b), and thus the synergy between intraocular inflammation, cAMP elevation, and PTEN deletion is likely to reflect a combined increase in transcription and translation. Synergistic effects on optic nerve regeneration have also been obtained by combining intraocular inflammation with methods that counteract cell-extrinsic inhibitors of growth (Fischer et al., 2004b). Studies elsewhere in the nervous system have demonstrated a synergy between trophic factors and either cAMP or agents that counteract cell-extrinsic inhibitors of growth (Schnell et al., 1994; Guest et al., 1997; Lu et al., 2004; Pearse et al., 2004; Verma et al., 2008; Kadoya et al., 2009).
By 6 weeks, combinatorial treatment enabled many axons to regenerate the full length of the optic nerve and through the optic chiasm. Strong regenerative growth across the chiasm has also been seen when axons are injured just proximal to this structure and RGCs are stimulated to grow by intraocular peripheral nerve implants (Berry et al., 1999). In our studies, some axons continued on into the diencephalon, and, in three of eight cases, a few reached the contralateral LGN. Looking ahead to the future, it is not yet known whether regenerating axons would be able to form topographically organized maps if they could be stimulated to innervate the correct target areas in sufficient numbers, but this possibility is supported by retino-tectal stripe assays (Bähr and Wizenmann, 1996; Wizenmann and Bähr, 1998) and by studies showing persistent gradients of Eph receptors and ephrins (Knöll et al., 2001; Rodger et al., 2001; Symonds et al., 2007) within the retina and superior colliculus after optic nerve injury. Although a few studies have reported full-length axon regeneration in the optic nerve, they did not rigorously demonstrate that the axons seen in central target areas had been damaged in the first place (Fischer et al., 2001) or that physiological responses recorded in central target areas were associated with regenerated axons (Eitan et al., 1994). The present results represent the first clear evidence that mature mice can regenerate axons from just behind the eye, through the entire optic nerve, across the chiasm, and on into the thalamus. Obtaining significantly greater levels of regeneration is likely to require the development of methods to maintain RGCs in an active growth state over an extended period.
Footnotes
-
This work was supported by National Eye Institute Grants 2 R01 EY05690-29A1 and 3 R01 EY05690-29A1S1 and the Miriam and Sheldon Adelson Medical Research Foundation. We thank Dr. Zhigang He and members of his laboratory (Dr. Kevin Park and Lauren Connolly) for providing PTENflx/flx mice, assistance with breeding, and validation of viral infection efficiency, Dr. Mustafa Sahin for providing several antibodies, and Dr. Lihong Bu and the Developmental Disabilities Research Center (National Institutes of Health Grant H P30 HD018655 for assistance with imaging.
- Correspondence should be addressed to Dr. Larry Benowitz, CLSB 13071, Children's Hospital, 115 Longwood Avenue, Boston, MA 02115. larry.benowitz{at}childrens.harvard.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}