

Abstract
The Kir4.1 channel is crucial for the maintenance of the resting membrane potential of glial cells, and it is believed to play a main role in the homeostasis of extracellular potassium. To understand its importance in these two phenomena, we have measured in vivo the variations of extracellular potassium concentration ([K+]o) (with potassium-sensitive microelectrodes) and membrane potential of glial cells (with sharp electrodes) during stimulations in wild-type (WT) mice and glial-conditional knock-out (cKO) Kir4.1 mice. The conditional knockout was driven by the human glial fibrillary acidic protein promoter, gfa2. Experiments were performed in the hippocampus of anesthetized mice (postnatal days 17–24). Low level stimulation (<20 stimuli, 10 Hz) induced a moderated increase of [K+]o (<2 mm increase) in both WT and cKO mice. However, cKO mice exhibited slower recovery of [K+]o levels. With long-lasting stimulation (300 stimuli, 10 Hz), [K+]o in WT and cKO mice displayed characteristic ceiling level (>2 mm increase) and recovery undershoot, with a more pronounced and prolonged undershoot in cKO mice. In addition, cKO glial cells were more depolarized, and, in contrast to those from WT mice, their membrane potential did not follow the stimulation-induced [K+]o changes, reflecting the loss of their high potassium permeability. Our in vivo results support the role of Kir4.1 in setting the membrane potential of glial cells and its contribution to the glial potassium permeability. In addition, our data confirm the necessity of the Kir4.1 channel for an efficient uptake of K+ by glial cells.
Introduction
Glial cells are highly specialized cells in the nervous system. In addition to their implication in the modulation of neuronal transmission and functional hyperemia (Volterra and Meldolesi, 2005; Haydon and Carmignoto, 2006), glial cells are also involved in ionic homeostasis. In particular, glial cells are essential for potassium buffering: they maintain an adequate extracellular potassium concentration ([K+]o) for neuronal transmission and excitability, and uptake excessive extracellular potassium to prevent neuronal hyperexcitability. Several mechanisms are believed to underlie the [K+]o homeostasis, among which particular emphasis was attributed to glial potassium conductances, glial and neuronal Na+/K+-ATPase activity, and Na+/K+/2Cl− cotransporters.
The concept of potassium buffering stems from the pioneering work of Kuffler's group, who first demonstrated that slow depolarizations of glial cells induced by nerve stimulations were attributable to the high potassium permeability of glial cells. According to their spatial buffering hypothesis, glial cells uptake excessive extracellular potassium ions, then potassium diffuses within glial syncytium toward domains where the concentration gradient is favorable for potassium extrusion (Orkand et al., 1966). Pharmacological studies suggest that Kir channels have a predominant role in K+ buffering (Ballanyi et al., 1987; Karwoski et al., 1989; Oakley et al., 1992), with special emphasis on the glia-specific Kir4.1 channel, which is the main glial inward conductance (for review, see Olsen and Sontheimer, 2008). By now, numerous in vitro studies have demonstrated the importance of Kir4.1 in the high potassium permeability of glial cells, thus setting the glial membrane potential (Kofuji et al., 2000; Neusch et al., 2001, 2006; Olsen et al., 2006; Djukic et al., 2007; Kucheryavykh et al., 2007). All these studies converge to implicate the Kir4.1 channel as the main mediator of glial potassium buffering. However, the relative involvement of this mechanism in the extracellular potassium uptake is still largely debated, because alternative pathways have been identified (Xiong and Stringer, 1999; D'Ambrosio et al., 2002; Wallraff et al., 2006).
Here we present an in vivo study involving [K+]o recordings with simultaneous glial cell impalements, performed in the hippocampus of anesthetized juvenile (P17–P24) wild-type (WT) and conditional Kir4.1 knock-out (Kir4.1 cKO) mice. The Kir4.1 cKO mice, generated in Dr. Ken McCarthy's laboratory, feature a floxed Kir4.1 gene and the Cre recombinase driven by the human glial fibrillary acidic protein promoter (gfa2; Djukic et al., 2007). Our in vivo results support the previously published in situ study establishing the role of the Kir4.1 channel in setting a hyperpolarized membrane potential in glial cells and in mediating their high potassium permeability (Djukic et al., 2007). Furthermore, the in vivo [K+]o recordings disclosed the relative contribution of glial Kir4.1 channel-mediated K+ uptake in the extracellular potassium buffering. We show that Kir4.1 depletion causes slower recovery of moderated [K+]o increases induced by stimulation.
Materials and Methods
Conditional knock-out mice generation and identification
The Kir4.1 cKO mice (Djukic et al., 2007) were generated in the laboratory of Dr. Ken McCarthy (University of North Carolina, Chapel Hill, NC). The conditional knockout of Kir4.1, directed to astrocytes, was accomplished by using the human glial fibrillary acidic protein (hGFAP) promoter, gfa2. The Cre/loxP system was used because it takes advantage of the bacteriophage enzyme Cre recombinase capable of excising a DNA fragment surrounded by its recognition sequences or loxP sites (Le and Sauer, 2000). A 2.2 kb fragment of the hGFAP promoter was used to drive Cre expression in astrocytes and the subsequent excision of the floxed Kir4.1 gene. Two lines of mice were generated and bred to homozygosity: a recombinant Kir4.1 floxed (Kir4.1f/f) line and a transgenic hGFAP-Cre line named B6-Tg(GFAP-Cre)1Kdmc line (Casper and McCarthy, 2006). The depletion was effective in Kir4.1f/f/hGFAP-Cre mice once the gene of the Cre-recombinase “Cre” under the promoter of the human GFAP was expressed. The hGFAP gene and hence the Cre-recombinase start to be expressed in precursor cells at embryonic day 14 (Malatesta et al., 2003). Additionally, immunostaining and Western blotting have demonstrated the complete absence of Kir4.1 at postnatal day (P) 20 (Djukic et al., 2007). Because it has been shown that removal of one copy of the Kir4.1 gene does not influence animal behavior or the channel distribution and function (Djukic et al., 2007), data collected from wild-type and heterozygous animals was combined in the present study. The Kir4.1 cKO mice were identified on the basis of behavioral criteria. They were clearly distinguishable from their littermates after P16 and appeared smaller and dehydrated, with wobbly movements, pronounced body tremor, lethargy, and ataxia (Djukic et al., 2007). In addition, Kir4.1 cKO animals did not display any startle reflex to abrupt auditory stimuli (such as finger snapping), in support of previous studies showing that Kir4.1 depletion led to deafness (Rozengurt et al., 2003).
Animal preparation
All experiments were performed between postnatal days 17 and 24. The surgical procedure started with the intraperitoneal administration of ketamine-xylazine anesthesia (2.25 and 0.15 mg/kg, respectively). Supplemental doses of anesthesia were applied on appearance of the withdrawal reflex in response to leg pinching. In addition, the electroencephalogram (EEG) and heart rate were monitored to assess depth of anesthesia. A suitable anesthesia was considered when the EEG pattern was dominated by ample and slow (mainly ∼1 Hz) waves and the heart rate was regular and <110 beats/min. Once anesthetized, skin of the scalp was removed and animals were placed in a stereotaxic frame. The craniotomy exposed the parietal cortex. Stimulating electrodes were aimed at the hippocampus (1 mm depth, 1 and 2 mm AP; 1 mm lateral, all coordinates with respect to bregma). Pipettes for intracellular/field potential recordings, as well as K+-sensitive electrodes, were progressively lowered into the hippocampus until single stimulation induced increased extracellular K+. The depth at which this occurred was ∼1.3 mm (±0.2 mm) for WT mice and 1.1 mm (±0.2 mm) for cKO mice, the latter presenting thinner structures overlying the hippocampus (Djukic et al., 2007). As detailed in a following section, the precise localization of these electrodes was confirmed in all animals postmortem by fast-green injections (see Fig. 2A). Thus, our recordings were performed in the dorsal part of the septal hippocampus (oriens, pyramidal layer, and statum radiatum). The stimulations consisted of 0.2 ms electrical pulses of 1.5 mA maximal intensity. At the end of the experiments, the animals received a lethal dose of ketamine-xylazine. All experimental procedures were performed according to the guidelines of the National Institutes of Health and were also approved by the committee for animal care of Laval University.
In vivo electrophysiology and extracellular K+ recording
Intracellular recordings were obtained with glass micropipettes (tip diameter <0.5 μm, in situ impedance 30–50 MΩ) filled with a 3 m solution of potassium acetate. The signal was passed through a high-impedance amplifier with active bridge circuitry (Neurodata). The EEG was recorded with sliver electrodes at the surface of the cortex (with the reference electrode placed under the skin of the neck). These potentials were bandpass filtered between 1 Hz and 3 kHz.
The K+-sensitive microelectrodes were made according to the procedure described in other studies (Amzica and Steriade, 2000; Amzica et al., 2002). We used double-barrel pipettes (tip diameter of ∼1–3 μm each) in which the K+-sensitive microelectrodes were pretreated with dimethylchlorosilane, dried at 120°C for 2 h, and the tip was filled with the K+ ionophore I-cocktail B (Fluka). The rest of the K+-sensitive barrel was filled with KCl (0.2 m), whereas the other barrel was filled with NaCl (0.2 m) for the recording of DC extracellular field potentials. The calibration of K+-sensitive microelectrodes was done by stepping the electrodes through solutions of different K+ concentrations (2.5, 4.5, 6.5, and 22.5 mm). The solutions contained the following (in mm): 119.78 NaCl, 22.02 NaHCO3, 10 glucose, 1.25 NaH2PO4, 4.09 MgSO4, 2.65 CaCl2, pH 7.4. The K+ concentration was adjusted between 1 and 22.5 mm with KCl. The relationship between concentration and voltage was derived in accordance with the Nicolsky–Eisenmann equation (Ammann, 1986). Electrodes used in our experiments were kept only if displaying voltage responses of at least 50 mV/decade increase in K+ concentration. The potential contamination of the K+ signals through capacitive coupling was eliminated by subtracting the DC field potential signal measured with the second barrel from the signal measured with the ion-sensitive barrel. The resulting signal was linearized and transposed into concentration values using the parameters extracted from the logarithmic fitting of the calibration points. The headstage amplifier for K+-sensitive microelectrode was modified with an ultra ultra low input current (<25 fA) amplifier (National Semiconductors). All signals were digitally converted (20 kHz sampling rate) for off-line analysis (Igor Pro software).
Analysis
Exponential fitting.
To perform the analysis of [K+]o recovery after stimulations, we used an exponential fitting function described as follows:
 InvTau (1/τ) was used for statistical analysis. [K+]o recovery after 5, 10, and 20 stimulations was evaluated from the average of 10 trials for each of these stimulation types delivered every 50 s. The fitting was done on the 10 trial average for each mouse.
InvTau (1/τ) was used for statistical analysis. [K+]o recovery after 5, 10, and 20 stimulations was evaluated from the average of 10 trials for each of these stimulation types delivered every 50 s. The fitting was done on the 10 trial average for each mouse.
Cross-correlation.
The degree of synchronization between glial membrane potential and [K+]o fluctuations during stimulations was quantified by means of cross-correlations, as defined for time series (Bendat and Piersol, 1980).
Power spectra.
The level of network activity was evaluated by means of fast Fourier transforms (FFTs) of the local field potentials from the hippocampus. This represents an indirect measure of the neuronal excitability. From each recording, we chose 2 periods of 400 s free of artifacts, one preceding and the other following stimulation. Each period was divided into 200 consecutive windows of 2 s each. FFT was calculated from each window over the 1–50 Hz range, and the 200 ensuing spectra were averaged, producing the power spectrum of the 400 s period. Results were plotted against on a logarithmic scale.
Fast-green staining
To localize the placement of the recording tip, we filled the field electrode with fast-green dissolved in 0.2 m NaCl. The iontophoretic delivery of fast-green was achieved by injecting −15 μA current for 15–20 min. At the end of the experiments, intracardiac perfusion was performed with 4% paraformaldehyde and brains were removed and kept at 4°C in 4% paraformaldehyde. One day before cutting, brains were cryoprotected with 30% sucrose. Coronal sections (50 μm) were cut with a vibratome, washed in 0.1 m phosphate buffer, and treated with cresyl violet.
Statistical evaluation
Data are expressed as mean values ± SE. Repeated-measures ANOVA was performed for examination of data with N > 2 (Fisher's PLSD post hoc test). Normal distribution and variance homogeneity were always first examined with Shapiro–Wilks and Bartlett's test, respectively. Nonparametric Mann–Whitney U test was performed for samples too small to verify the normal distribution or when a normal distribution could not be assumed. Differences were considered significant at p < 0.05. We used Statview and Statgraphics software.
Results
Depolarization of Kir4.1 cKO glial cells
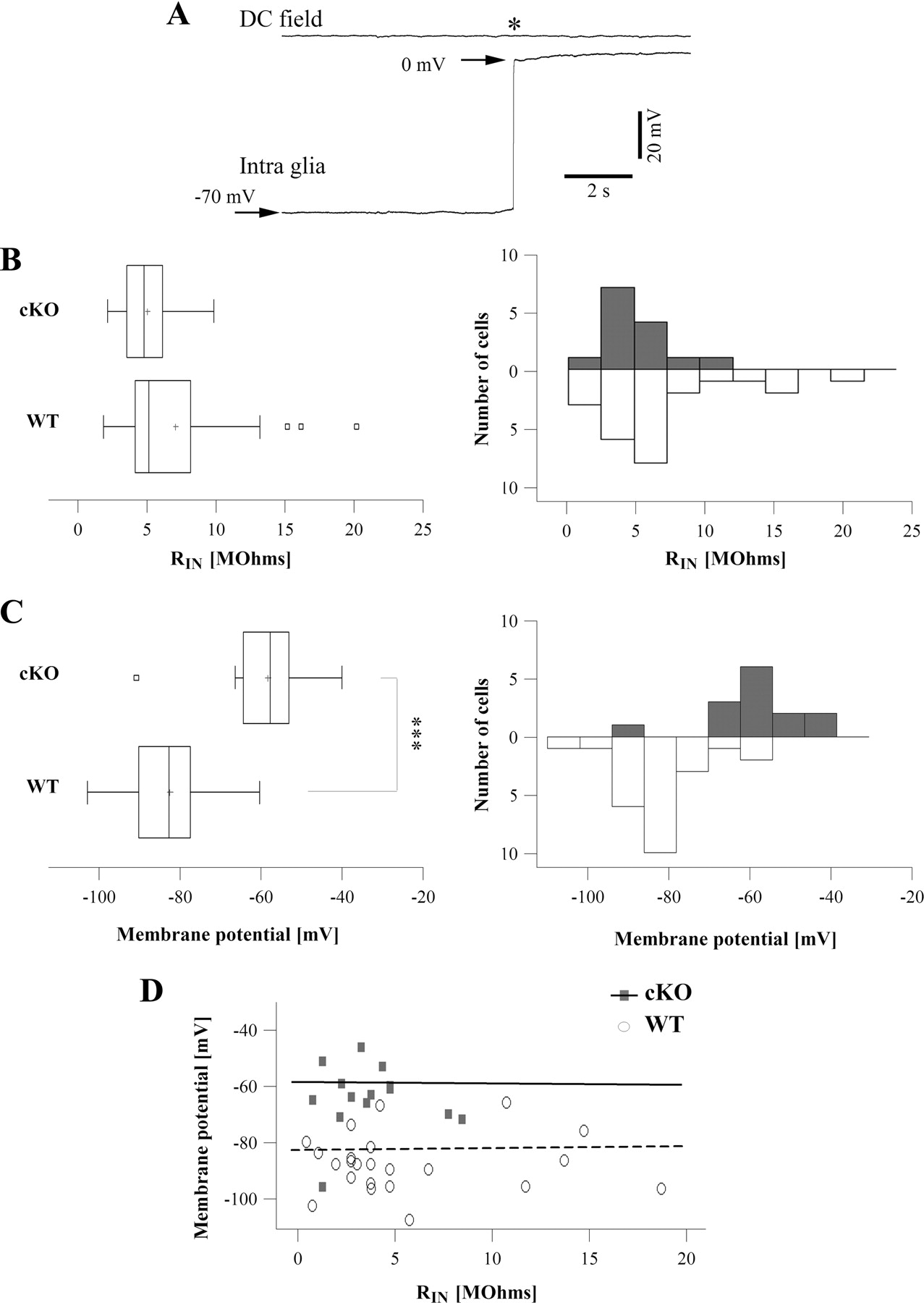
To understand the in vivo functional impact of Kir4.1 channels on the electrophysiological membrane properties of glial cells, we performed intracellular recordings with sharp electrodes (current-clamp) in the hippocampus of anesthetized mice. Recordings considered for analysis started with a sudden drop of the membrane potential upon impalement, followed by a stable resting level lasting for tens of minutes without hyperpolarizing current compensation. No action potentials were observed during any of the following situations: spontaneous activity, penetration of the cell, withdrawal from the cell, or intracellular injection of depolarizing current. The resting membrane potential and the input resistance of the membrane (RIN) were examined. The membrane resistance was tested by means of hyperpolarizing current pulses injected through the recording microelectrode (80 ms duration, 0.5 nA intensity). Glial cells from WT mice displayed a membrane potential of −81.82 ± 2.07 mV (Fig. 1C; n = 24), close to the Nernst equilibrium potential for potassium (Ek), reflecting the presence of high resting K+ conductance. A representative intraglial recording is depicted in Figure 1A. Interestingly, the Kir4.1 gene depletion in glial cells was associated with a strong depolarization of >20 mV; glial cells from cKO mice had a membrane potential of −58.07 ± 3.08 mV (Fig. 1C) (n = 14). This value was significantly different from WT (Mann–Whitney U test, p < 0.001). Despite a slight decrease in RIN for cKO glial cells, no significant difference was found between the two groups (WT RIN = 6.92 ± 0.97 MΩ, n = 24; cKO RIN = 4.89 ± 0.6 MΩ, n = 14; Mann–Whitney U test, p = 0.28) (Fig. 1B). In both groups, there was no significant correlation between membrane potential and RIN (the correlation coefficients for WT and cKO are, respectively, 0.03 and −0.007) (Fig. 1D).

Kir4.1 cKO glial cells are more depolarized than WT glia. A, Example of an in vivo intraglial and DC field recordings in a WT mouse under anesthesia. The star marks the moment of electrode withdrawal from the cell. B, Box plot (left) and distribution (right) of Kir4.1 cKO (n = 14 cells; gray histogram) and WT RIN (n = 24 cells; white histogram). No significant difference was found (Mann–Whitney U test, p = 0.28). The central box covers the middle 50% of each sample; the sides of the box are the lower and the upper quartiles, and the vertical line drawn through the box is the median. The whiskers extend out to the lower and upper values of the samples. Outliers are represented as individual squares beyond the whiskers. The mean is presented with a cross. C, Box plot (left) and distribution (right) of Kir4.1 cKO (n = 14 cells; gray histogram) and WT (n = 24 cells; white histogram) membrane potential. cKO glial cells are significantly more depolarized than WT glial cells (Mann–Whitney U test, p < 0.001). D, Absence of correlation between membrane potential and RIN in both groups. Dots represent individual values, lines are linear fits of the former.
Modified K+ clearance in Kir4.1 cKO mice
Kir4.1 depletion leads to slower K+ clearance
Short-lasting hippocampal stimulations at 10 Hz induced a fast [K+]o increase (∼0.1 mm/electrical pulse), clearly surpassing the basal variations of [K+]o recorded in the hippocampus during spontaneous activity under ketamine-xylazine anesthesia (Fig. 2B). Twelve WT mice and 10 cKO mice were used for this paradigm. Recordings were performed in the dorsal part of the septal hippocampus (Fig. 2A) without any differences of localization between groups. Trains of 5, 10, and 20 stimulations at 10 Hz induced [K+]o increases of 0.62 ± 0.084, 0.91 ± 0.13 mm, and 1.31 ± 0.17 mm, respectively (cumulative averages, independent of the group; n = 22 animals). The differences between [K+]o increases resulting from the three stimulation paradigms were significant (repeated-measures ANOVA, p < 0.001). Although the average peak amplitude of the [K+]o increase was slightly smaller in cKO versus WT groups, this difference was not statistically significant (ANOVA, p = 0.51; WT mice, n = 12; cKO mice, n = 10).

Kir4.1 cKO mice display impairment of extracellular potassium clearance. A, Hippocampal fast-green injection in one of the animals allowing the localization of the K+ recording electrodes. B, Representative recordings of [K+]o responses induced by trains of stimulation in WT and Kir4.1 cKO mice. The stimulations are indicated below the trace with rectangles. The inset depicts an average response to a train of 10 stimuli from one animal (black trace) and the exponential curve fitting of the former (red trace). C, The grand average trace of the exponential fittings after 5, 10, and 20 stimulations for WT (n = 12; black traces) and cKO (n = 10; red traces) groups. Circles indicate the respective time constants. D, Grand average of power spectra of local field potentials from the two groups of mice (WT, black trace; cKO, red trace; n = 5 in each group). The two power spectra are statistically distinct (p < 0.05) for the whole calculated range, with an additional difference within the 6–13 Hz frequency band (between dotted vertical lines) case in which the p values were <0.01 (Mann–Whitney U test).
After reaching its maximum, [K+]o slowly recovered in an exponential fashion regardless of the group. The decay time (15–25 s) depended on the number of stimulations used. Because the main goal of this study was to evaluate the implication of the Kir4.1 channel in the clearance of the [K+]o increase, we calculated the time constant from the exponential fit of the averaged [K+]o traces (Fig. 2C). This provides a measure that is independent of the amplitude of the signal and corresponds to the time needed for a 67% decay. Repeated-measures ANOVA was performed on the inverse of time constant (1/τ) to ensure the condition of the distribution (Shapiro–Wilks, p = 0.65) needed for this analysis. This led to the conclusion that time constants were significantly slower for the Kir4.1 cKO group (WT mice, n = 12, cKO mice, n = 10; repeated-measures ANOVA, p < 0.05). No interaction between the “group” factor and the three different stimulation paradigms was significant (p = 0.92). The post hoc test revealed a difference for each paradigm of stimulation (Fisher's PLSD post hoc test, p < 0.05 for 5 and 10 stimulations, p < 0.02 for 20 stimulations). For 5 stimulations, the associated averaged time constant was 4.17 ± 0.96 s for WT mice and 6.33 ± 1.5 s for cKO mice, for 10 stimulations it was 4.48 ± 0.73 s for WT mice and 6.75 ± 1.06 s for cKO mice, and for 20 stimulations it was 4.81 ± 0.63 s for WT mice and 6.86 ± 0.70 s for mice.
To obtain an estimate of the neuronal excitability of the WT and cKO mice, we calculated the spectral content of local field potentials (Fig. 2D). The grand average of power spectra from WT (Fig. 2D, black trace) and cKO (Fig. 2D, red trace) mice showed that, at the same dose of anesthesia, oscillatory frequencies in the field potential activity were similarly distributed throughout the calculated range (1–50 Hz). This suggests that the global network activity in the hippocampus was generally similar in the two groups of mice, with the exception of the frequency range between the vertical dotted lines (6–13 Hz), where the activity of WT mice displayed a peak that was not visible in cKO recordings. Moreover, in all cases (n = 5 for each group), the amplitude of the power spectrum was always higher in WT than in cKO mice. This resulted in a clearly more ample average power spectrum for WT mice (Fig. 2D). The difference between the two curves was statistically different (Mann–Whitney, p < 0.05) for each frequency value. Moreover, for the frequency range between the two vertical dotted lines (6–13 Hz), the p values were <0.01. We also calculated for each animal separated power spectra for field potentials recorded before and after electric stimulations and did not obtain any significant difference (data not shown). The lower amplitude of the cKO power spectra may be explained by the reduced tissue volume present in the brain of these animals (Djukic et al., 2007).
Kir4.1 cKO mice display a pronounced [K+]o undershoot after long-lasting stimulation
The clearance of [K+]o after long-lasting stimulation was studied in five WT and five cKO mice. Prolonged stimulations usually induced a high [K+]o increase reaching a ceiling level, in accordance with previous reports both in vivo and in vitro (Heinemann and Lux, 1977; Krnjevic et al., 1982; D'Ambrosio et al., 2002). In our conditions, the [K+]o ceiling level amplitude was 2.78 ± 0.3 mm above the control level (cumulative average, independent of the group, n = 10 animals). Although cKO mice displayed a slightly lower amplitude of the plateau than the WT mice, no statistical difference between WT and cKO mice was detected (Mann–Whitney p = 0.53) (see Fig. 4B3), supporting the results of D'Ambrosio (D'Ambrosio et al., 2002). Figure 3 shows an example of the [K+]o behavior during long-lasting stimulations at 10 Hz in the hippocampus of a WT mouse. The [K+]o ceiling level was reached after at least 200 stimulations. The subsequent return of [K+]o to control levels was accompanied by an undershoot following >100 stimulations (Fig. 3). This undershoot could result from the activation of ATP pumps following large K+ increases (see Discussion) and is associated with decreased neuronal discharge (Heinemann and Lux, 1977). Since 300 stimulations at 10 Hz gave rise to an overt undershoot, we used this paradigm to compare WT and Kir4.1 cKO mice.

In vivo [K+]o undershoot after long-lasting stimulations. Representative responses of the [K+]o variations induced by long trains of stimulations (100, 200, 300, 600 stimuli at 10 Hz) in a WT mouse. Note the appearance of an undershoot (long K+ decrease below the basal level once stimulations stopped) with the increase of the duration of the stimulation. A clear undershoot starts to be visible with 300 stimulations. Top trace shows a representative EEG recording before and after long-lasting stimulations. In the central box, average of the first 10 (1) and last 10 (2) stimulations in the stimulation train. In the EEG recording, the epoch with stimuli has been skipped. In the [K+]o traces, stimulation artifacts were artificially removed to illustrate the evolution of the [K+]o dynamic.
The time constants of the initial K+ return toward baseline after long lasting stimulations were not statistically different between WT and cKO groups (Mann-Whiney; p = 0.83) (Fig. 4B1,B2). However, the [K+]o undershoot observed in the Kir4.1 cKO was different from the WT (Fig. 4A,B). In the hippocampus of WT mice, the maximum amplitude of the undershoot was obtained at a latency of 86 ± 8.49 s with respect to the end of the stimulation (n = 5). Its average magnitude was 0.21 ± 0.075 mm below the control level. The final [K+]o recovery was reached 155 ± 27.95 s after the end of the stimulation. cKO mice displayed a more pronounced undershoot: the maximum amplitude was more than twice higher than in WT (0.49 ± 0.06 mm; Mann–Whitney p < 0.05) (Fig. 4B5), and the [K+]o took longer to recover (277 ± 14.57 s; Mann–Whitney p < 0.05) (Fig. 4B6). This led to a three times larger overall area of the undershoot in the Kir4.1 cKO mice compared with the WT (Fig. 4B4). The maximum undershoot amplitude in cKO mice was reached 82.6 ± 8.72 s after the end of stimulation.

Long-lasting stimulation induces a larger [K+]o undershoot in Kir4.1 cKO mice. A, Comparison between representative [K+]o response to 300 stimuli in WT and Kir4.1 cKO mice. Note the larger undershoot induced in the recording from the cKO mouse (stimulation artifacts were removed). B, Description of the parameters considered for the comparison of the [K+]o responses in WT and cKO mice (B1). The time constant was evaluated with an exponential curve fit between the end of the stimulation and the maximum amplitude of the undershoot. Maximum amplitudes of the [K+]o during stimulations and during undershoot were evaluated with regard to the basal [K+]o level. The duration of the undershoot (“time of return”) corresponds to the time needed for [K+]o to recover to the basal level after the end of the stimulation. The total surface area of the undershoot was also estimated (gray area). No difference was found between WT and Kir4.1 cKO recordings for [K+]o clearance (as reflected by the time constant) and amplitude of [K+]o responses (B2,B3). However, the [K+]o undershoot is statistically more pronounced in the cKO. Area and amplitude of [K+]o undershoot are significantly different between the cKO and WT (B4,B5). The time for the [K+]o return to basal level is also longer in the cKO (B6, cKO mice: n = 5, WT mice: n = 5; Mann–Whitney U test, *p < 0.05, **p < 0.02).
Glial cells from Kir4.1 cKO mice show decreased potassium permeability
One would have expected that the depletion of Kir4.1 channels would result in larger differences in the [K+]o clearance parameters between cKO and WT mice. We therefore investigated the behavior of glial cells from cKO mice in regard to their K+ permeability. It has been well documented that, due to the overwhelming abundance of K+ channels in glia of normal brains, of which inward rectifiers such as Kir4.1 are predominantly active at resting membrane potentials (for review, see Duffy et al., 1995; Olsen and Sontheimer, 2008), glial cell membrane behaves as an almost perfect K+-electrode, whereby glial membrane potential tightly follows the [K+]o variations (Orkand et al., 1966; Somjen, 1975; Amzica and Steriade, 2000; Amzica et al., 2002). In other terms, and as a reflection of the high permeability of glia to K+, the glial membrane potential evolves along the K+ equilibrium potential predicted by the Nernst equation. Figure 5A illustrates such an example of the tight link between the WT glial cell membrane potential and [K+]o during an intense stimulation. Glial cells depolarized in parallel with the [K+]o increase, reaching a steady-state when [K+]o was reaching the ceiling level. In addition, their membrane repolarization was closely associated with the [K+]o recovery. The average correlation peak was 94% (n = 6, WT glia) (Fig. 5A), suggesting a good agreement with the Nernst equation.

Kir4.1 cKO glial cells display a decrease in potassium permeability. A, Representative traces from a WT glial cell showing parallel evolution of the cell's membrane potential and [K+]o variations induced by a train of 300 stimuli. Bottom left panel shows a detail of the period during stimulation with the stimulation artifact removed, clearly demonstrating that WT glial cells behave as “[K+]o sensitive-electrodes.” Bottom right panel shows cross-correlation ± SD between membrane potential of WT glial cells and [K+]o dynamic (n = 6 recordings). The positive peak of the cross-correlation suggests in-phase relationship between the two waves, whereas its high amplitude (93.9%; n = 6 recordings) indicates the high degree of global resemblance of the two waves. B, Representative traces depicting decorrelation between the Kir4.1 cKO glial cell membrane potential and [K+]o induced by a train of 300 stimuli, suggesting a decrease in the cell's K+ permeability. C, The magnitude, but not the trend, of the cKO membrane potential response is dependent on the number of stimulations.
In contrast to WT glia, the membrane potential of Kir4.1 cKO glial cells displayed an uncoupled behavior with respect to the [K+]o. First, no membrane potential variations were observed in any of the recorded cells during moderated [K+]o increases induced by stimulations with 5, 10, or 20 pulses at 10 Hz (data not shown; n = 8 cKO glial cells). Membrane potential variations were evident and systematic only with intense stimulation (>50 electrical pulses) (Fig. 5B,C) when cKO glia started to depolarize very slowly at 16.67 ± 2.61 s after the onset of the stimulation (n = 21 trials, n = 9 cells). The maximum depolarization was on average 2.48 ± 0.35 mV and was reached during the [K+]o undershoot, 72.83 ± 2.61 s after the end of the stimulation (n = 21 trials, n = 9 cells). The amount of intraglial depolarization was proportional to the number of pulses in the stimulation train but was always slow and uncoupled from the [K+]o variations (Fig. 5C). This behavior was common to all cKO glial cells. The fact that the membrane potential of Kir4.1 cKO glial cells does not follow [K+]o variations strongly suggests a decrease in their K+ permeability and implicates the Kir4.1 channel as the main glial K+ conductance.
Discussion
Absence of Kir4.1 channels leads to depolarization of the hippocampal glia
In vivo impalements of hippocampal glial cells in Kir4.1 cKO mice revealed that the Kir4.1 depletion induces severe electrophysiological changes in these cells; cKO glial cells were depolarized by 20 mV and displayed a loss of K+ permeability. Previous in vitro reports based on studies done in Kir4.1 KO mice have already described the contribution of Kir4.1 channels in keeping a hyperpolarized membrane potential of brainstem astrocytes (Neusch et al., 2006), Müller cells in the retina (Kofuji et al., 2000), spinal astrocytes (Olsen et al., 2006), hippocampal astrocytes (Seifert et al., 2009), and spinal oligodendrocytes (Neusch et al., 2001). A more depolarized membrane of astrocytes and oligodendrocytes in the hippocampal slices of Kir4.1 cKO mice was also reported (Djukic et al., 2007). However, the use of knock-out mice does not allow to directly link Kir4.1 depletion and depolarized membrane potential. First, Kir4.1 expression could subserve alternate mechanisms involved in the development of the hyperpolarized membrane potential of mature cells (Djukic et al., 2007). Second, a developmental adaptation to the genetic deletion could occur. These shortcomings are avoided using Kir4.1 RNA interference technique. With this approach, similar depolarized membrane potentials have been observed in spinal and cortical astrocyte cultures (Olsen et al., 2006; Kucheryavykh et al., 2007), where no compensatory upregulation of other Kir genes was found (Olsen et al., 2006). Thus, all of these approaches converge toward a predominant role of the Kir4.1 channel in maintaining a hyperpolarized glial membrane potential near Ek.
The degree of membrane depolarization of Kir4.1 cKO glia observed in our in vivo study is in agreement with some studies (Neusch et al., 2006; Olsen et al., 2006), whereas others reported more depolarized KO glial cells (Kofuji et al., 2000; Djukic et al., 2007, Seifert et al., 2009). Beyond divergences attributable to age and tissue-specific characteristics, two main technical differences with in vitro and in situ patch-clamp studies could be proposed: (1) in contrast to in vitro/situ studies where the cells undergo visual identification before being recorded, our sharp electrode technique relies on blind impalement, possibly leaving out some cells that, if taken into account, might bias the final average of the membrane potential; and (2) it is possible that extra- and/or intracellular potassium concentrations are different in cKO mice with respect to WT, and reliable measures cannot be obtained in slices with whole-cell patch-clamp techniques because of controlled bath and pipette solutions. This information would be crucial to a better understanding of the impact of the Kir4.1 channels in setting the glial membrane potential.
In our study, we report no significant difference in the apparent RIN between WT and Kir4.1 cKO. Patch-clamp studies show higher RIN in (c)KO mice (Kofuji et al., 2000; Neusch et al., 2006; Olsen et al., 2006; Djukic et al., 2007). This discrepancy between in vivo and in vitro observations may result from the increase in the leak conductance of cells due to the more invasive impalement with sharp electrodes, which might shunt to some extent other currents, rendering the procedure less sensitive to small changes in input resistance. Finally, several nonexclusive, other phenomena could underlie the apparent contradiction in regard to the absence of RIN changes in depolarized cKO glia.
The absence of the Kir4.1 channels in cKO mice would produce a higher input resistance. But the more depolarized resting membrane potential (−58 mV) might open at various degrees other conductances such as K+ delayed rectifiers, Ca2+ or Cl− voltage-gated channels, yielding to an overall unchanged input resistance. Testing such a hypothesis would have been extremely difficult under in vivo conditions.
Kir4.1 channels are generally localized on the astrocytic processes, thus at some distance from the soma, where the testing of the input resistance occurs. Therefore, the low input resistance obtained in this study might reflect only the somatic domain, with possible contributions from the currents through gap junctions. Indeed, the closure of gap junctions with high concentrations of isoflurane has produced higher input resistance in a subpopulation of cat glial cells (Ferron et al., 2009).
Finally, the remote localization of Kir4.1 channels, combined with the contribution of two-pore domain K+ channels to passive somatic conductances (Zhou et al., 2009), could also explain the lack of correlation between the (depolarized) membrane potential and the input resistance. Alternatively, the steady depolarized membranes could also result from a higher resting concentration of the extracellular K+ acting upon the membrane potential in a Nernstian fashion.
Removal of Kir4.1 influences K+ permeability of glial cells
Under current-clamp conditions, normal glial cells in vivo display a membrane potential between −110 and −60 mV and respond to [K+]o variations with a change in membrane potential closely following the Nernst equation, similar to the K+-selective electrodes. This close relationship has been repeatedly reported in the literature since the works of Kuffler et al. (1966) and Orkand et al. (1966), who first discovered the electrophysiological responses of glial cells to stimulations and their relation to potassium concentrations. Such K+-dependent glial responses could be recorded during normal spontaneous activity, seizures, or induced by stimulations (Somjen, 1975; Amzica and Steriade, 2000; Amzica et al., 2002) and are attributable to the high potassium permeability of glial cells.
In vitro experiments gave rise to quantifiable measurements of potassium permeability and potassium uptake impairment in astrocytes with underexpressed or absent Kir4.1 channels. Interestingly, Kir4.1 channel depletion leads to a nearly complete loss of inward current in glial cells (Neusch et al., 2006; Kucheryavykh et al., 2007). Moreover, bath application of barium, a nonspecific Kir channel blocker, does not alter the electrophysiological pattern of Kir4.1 KO glial cells (Neusch et al., 2006; Djukic et al., 2007). This supports the idea that, at least at the resting membrane potential, the Kir4.1 channel is the main glial inward conductance, and it is responsible for the high potassium permeability of glia. Our in vivo glial cell recordings combined with simultaneous [K+]o recordings are in agreement with these results. The membrane potential of Kir4.1 cKO glial cells lacks the previously reported close relationship with [K+]o. Membrane potential variations were only observed with intense stimulation leading to weak and delayed depolarizations, without any apparent relationship with the behavior of [K+]o, suggesting a loss of high potassium permeability in Kir4.1 cKO glia. Surprisingly, the maximum depolarization of cKO glial cells occurred in conjunction with the maximum [K+]o undershoot. It was proposed that the development of the [K+]o undershoot depends on the activity of the Na+/K+-ATPase, because it is abolished by the ATPase blocker ouabain (Galvan et al., 1979; Förstl et al., 1982; D'Ambrosio et al., 2002). We might then speculate that stimulation-induced depolarization of cKO glia arises from the Na+/K+-ATPase activity. Under normal conditions, the potassium permeability of WT glial cells is high enough that the electrogenic and/or other ionic currents contributions to their membrane potential fluctuations may be negligible. Conversely, in Kir4.1 cKO glia, where the influence of [K+]o on membrane potential is decreased due to reduced potassium permeability, other mechanisms (such as the Na+/K+-ATPase activity) could significantly affect their membrane potential.
Involvement of Kir4.1 channels in K+ buffering
Alterations of potassium uptake are linked to electrophysiological changes in glia during epilepsy (for review, see Binder and Steinhäuser, 2006). Decreased conductance of Kir channels in reactive glial cells in the sclerotic tissue from epileptic patients has been observed (Bordey and Sontheimer, 1998; Hinterkeuser et al., 2000; Jauch et al., 2002). It was suggested that the loss of glial potassium conductance would favor extracellular K+ accumulation, contributing to neuronal hyperexcitability. Our initial prediction was that the Kir4.1 cKO mice would display early signs of extracellular K+ misregulation, such as spontaneous paroxysmal events, in contrast to WT mice; however, this was not the case. Moreover, in responses to stimulation, although the [K+]o recovery profile was slightly slower in cKO than in WT mice, the former did not show any evidence of sustained extracellular accumulation of K+ and precipitation of epileptic fits.
This raises the question whether Kir channels are essential for potassium clearance. Several in situ studies argue that Na+/K+ ATPase-mediated removal plays a major role in and furthermore could be responsible for the rate of K+ recovery (Xiong and Stringer, 1999; D'Ambrosio et al., 2002). It could be argued that K+ homeostasis is maintained at equilibrium by various mechanisms and that genetic depletion or pharmacological blockade of one of them could be compensated by the other mechanisms. The more pronounced [K+]o undershoot recorded in Kir4.1 cKO mice may support this hypothesis by suggesting a higher Na+/K+-ATPase activity in the cKO. A similar phenomenon was also observed in the cerebellum of conventional Kir4.1 KO mice (Neusch et al., 2006) and in vitro with application of the nonspecific Kir channel blocker barium (Ransom et al., 2000; D'Ambrosio et al., 2002). D'Ambrosio et al. (2002) speculated that Kir channels would be important as a return pathway for replenishing the extracellular K+ during the ATPase-mediated undershoot, which could explain why bath application of barium in their in vitro preparation increases the undershoot. This could also explain both the larger amplitude and the longer recovery time of this phenomenon in the Kir4.1 cKO mice. One could also speculate that a more intense ATPase activity would lead to a longer recovery to baseline and that the compensatory mechanisms for Kir depletion may rely on the ATPase. In epileptic tissue, Kir conductance, as well as ATPase activity, are decreased (Brines et al., 1995; Vaillend et al., 2002). This combination of factors could contribute to the observed impaired [K+]o homeostasis. Other glial and neuronal mechanisms, such as Na+/K+/2Cl− cotransporters or alternate K+ and Cl− channels have also been implicated in K+ uptake. A recent study emphasizes the role of the leak potassium channel P2Y in potassium buffering (Päsler et al., 2007). Whatever the underlying compensatory mechanism, further insight could be achieved by establishing the functional adaptation of such components in knock-out or pharmacological models in use.
In summary, our study does not promote the idea of a predominant role of the Kir4.1 channel in potassium clearance but rather the idea that this channel is essential for efficient potassium buffering. Kir4.1 channels are facing synapses (Higashi et al., 2001; Hibino et al., 2004) and could play a role in synaptic transmission by buffering the excess extracellular K+ in synaptic microdomains. The use of K+ electrodes in vivo during stimulations does not allow a fair enough estimation of this synaptic fine tuning. A couple of teams have reported an alteration in synaptic transmission after Kir4.1 depletion (Djukic et al., 2007; Kucheryavykh et al., 2007); however, more experiments are needed to clarify the role Kir4.1 plays in synaptic function.
Footnotes
This work was supported by the Candian Institutes for Health Research. We thank P. Giguère and S. Ftomov for technical assistance.
- Correspondence should be addressed to Florin Amzica, C.P. 6128, succursale Centre-ville, Montréal, QC H3C 3J7, Canada. florin.amzica{at}umontreal.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}