

Abstract
According to the amyloid cascade hypothesis, cerebral deposition of amyloid-β peptide (Aβ) is critical for Alzheimer's disease (AD) pathogenesis. Aβ generation is initiated when β-secretase (BACE1) cleaves the amyloid precursor protein. For more than a decade, BACE1 has been a prime target for designing drugs to prevent or treat AD. However, development of such agents has turned out to be extremely challenging, with major hurdles in cell penetration, oral bioavailability/metabolic clearance, and brain access. Using a fragment-based chemistry strategy, we have generated LY2811376 [(S)-4-(2,4-difluoro-5-pyrimidin-5-yl-phenyl)-4-methyl-5,6-dihydro-4H-[1,3]thiazin-2-ylamine], the first orally available non-peptidic BACE1 inhibitor that produces profound Aβ-lowering effects in animals. The biomarker changes obtained in preclinical animal models translate into man at doses of LY2811376 that were safe and well tolerated in healthy volunteers. Prominent and long-lasting Aβ reductions in lumbar CSF were measured after oral dosing of 30 or 90 mg of LY2811376. This represents the first translation of BACE1-driven biomarker changes in CNS from preclinical animal models to man. Because of toxicology findings identified in longer-term preclinical studies, this compound is no longer progressing in clinical development. However, BACE1 remains a viable target because the adverse effects reported here were recapitulated in LY2811376-treated BACE1 KO mice and thus are unrelated to BACE1 inhibition. The magnitude and duration of central Aβ reduction obtainable with BACE1 inhibition positions this protease as a tractable small-molecule target through which to test the amyloid hypothesis in man.
Introduction
Alzheimer's disease (AD) is characterized by the generation, aggregation, and deposition of amyloid-β peptide (Aβ) in the brain. According to the amyloid cascade hypothesis, Aβ initiates a neurodegenerative cascade as either a soluble oligomer or a major constituent of cerebral amyloid plaques (Hardy and Selkoe, 2002). Aβ is generated from the membrane-spanning β-amyloid precursor protein (APP) by sequential endoproteolytic cleavages. First β-secretase cleaves APP at the NH2 terminus of Aβ to release sAPPβ and C99, a COOH-terminal fragment that remains membrane bound. Then C99 is further processed by γ-secretase to release various isoforms of Aβ, of which Aβ42 appears most pathogenic (Younkin, 1995). Therefore, β-secretase is a prime target for the development of Aβ-lowering therapeutics for the prevention and treatment of AD. Compared to γ-secretase inhibitor development, β-secretase is perceived to be a more promising target, given that its activity is conferred by a single transmembrane aspartic protease, BACE1 (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000), whose crystal structure was solved and published early on (Hong et al., 2000). Unlike targeted deletion of the PSEN-1 gene, which is embryonic lethal, BACE1 knock-outs are viable and fertile and initially were reported to be without a major phenotype (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001). Recent analyses of BACE1 KOs have revealed morphologic and functional deficits in BACE1 KOs and have introduced a level of caution but, in general, have not limited enthusiasm for developing BACE1 inhibitors. Overall, BACE1 has represented an attractive target for medicinal chemists across the pharmaceutical industry for more than a decade (Stachel, 2009), but progress has been slow. Despite impressive intrinsic potency, the compounds reported to date have struggled to produce in vivo, CNS pharmacodynamic (PD) effects under standard treatment protocols (Rajendran et al., 2008; Sankaranarayanan et al., 2009; Zhu et al., 2009; Malamas et al., 2010). Common limitations have included loss of potency in cellular systems, low oral bioavailability/high metabolic clearance, and inadequate access to the target compartment within the CNS, often driven by P-glycoprotein or other transport systems. High throughput screening (HTS) across the industry on a massive scale has produced few alternative options. Consequently, no BACE inhibitor has been reported to be in advanced clinical development to this point, and growing concerns have emerged regarding the tractability of the target as the basis for agents capable of producing a robust PD effect in the central compartment.
Here we report the generation of LY2811376, a potent drug-like BACE1 inhibitor. Robust BACE1-mediated biomarker changes in APP cleavage products translated from preclinical animal models to humans treated with LY2811376. Although the clinical development of this molecule was discontinued as a result of nonclinical non-target-related pathology findings, these data provide critical support for BACE1 as a tractable target for small-molecule intervention to test the amyloid hypothesis.
Materials and Methods
Crystallography
BACE1 crystals were prepared with minor modifications of previously described methods (Hong et al., 2000). Briefly, crystals were soaked with 2.5 mm compound and flash cooled in liquid nitrogen using glycerol as a cryoprotectant. x-ray diffraction data were collected at 100,000 at the Advanced Photon Source beamline 31-ID and were integrated and scaled using MOSFLM version 6.2.6 and SCALA version 5.0, respectively (Collaborative Computational Project, Number 4, 1994). The BACE crystal structure was solved by molecular replacement and was subject to iterative cycles of restrained refinement using REFMAC version 5.2.0019 and model building with COOT version 0.5 (Emsley et al., 2010). Figures were produced using PyMOL version 1.1r1 (http://www.pymol.org/).
Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly & Co., who operates the facility. See Table 1 for crystallographic data.
Crystallographic data
Recombinant human BACE1 assays
Human BACE1 was cloned, expressed, and purified essentially as described by Yang et al. (2004) and assayed using a synthetic peptide in a FRET format or using a MBP–C125Swe polypeptide substrate assay essentially as described by Yang et al. (2004) or Sinha et al. (1999), respectively.
Aspartyl protease selectivity enzyme assays
Human BACE2 was cloned, expressed, and purified essentially as described by Yang et al. (2004). The MBP–C125Swe assay was conducted essentially as described by Sinha et al. (1999). The 7-methoxycoumarin-4-yl-acetyl (mca) FRET assay for BACE2 was conducted essentially as described by Yang et al. (2004). Human liver cathepsin D was purchased from Calbiochem (catalog #219041) and used as a source of enzyme for cathepsin D assays following the instructions of the manufacturer. Porcine gastric mucosa pepsin was purchased from Sigma (catalog #P-6887) and used as a source of enzyme for pepsin assays following the instructions of the manufacturer. Recombinant renin from the SensoLyte Tm 520 renin assay kit (catalog #72040; AnaSpec) was used as a source of enzyme for renin assays following the instructions of the manufacturer. For all enzyme assays, the 10-point inhibition curve was plotted and fitted with the four-parameter logistic equation to obtain the IC50 values.
Cell-based assays
The human embryonic kidney cell line HEK293 (ATCC accession number CRL-1573) stably expressing a human APP751 cDNA containing the naturally occurring double mutation N670L671 (APP770 amino acid numbering), commonly called the Swedish mutation and shown to overproduce Aβ (Citron et al., 1992), was used routinely for the measurement of inhibition of BACE1 activity in whole cells (HEK293Swe assay). Media was analyzed for Aβ peptides by specific ELISAs as described by Dovey et al. (2001). Compound cytotoxicity in the HEK293Swe cell model was assessed using a CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay from Promega (catalog #G5430) according to the recommendations of the manufacturer.
Primary neuronal cultures from embryonic PDAPP mice were used as a secondary whole-cell assay for BACE1 inhibition (PDAPP 1° neuronal assay). PDAPP is a transgenic mouse animal model overexpressing a human APP mini-gene that harbors a familial mutation (APPV717F), which recapitulates much of the amyloid pathology observed in AD (Games et al., 1995). Primary cortical neurons from embryonic day 16 PDAPP embryos were incubated in the presence/absence of inhibitors at the desired concentration. Assays were conducted in triplicate. At the end of the incubation, conditioned media were analyzed for Aβ peptides by specific ELISAs as described in the section below. Compound cytotoxicity in the PDAPP primary neuronal cultures was assessed using the MTT assay well described in the literature.
Aβ peptides were measured by sandwich ELISAs, using monoclonal 2G3 as a capture antibody for Aβ1–40, monoclonal 21F12 as a capture antibody for Aβ1–42, and biotinylated 3D6 as reporting antibody (for description of Aβ monoclonal antibodies, see Johnson-Wood et al., 1997)). Culture media was diluted to allow determination of Aβs within the range of standards (0–1000 pg/ml). For both cell culture systems, the concentration of Aβ released in the conditioned media was considered reflective of BACE1 inhibition. The 10-point inhibition curve was plotted and fitted with the four-parameter logistic equation to obtain the EC50 or relative IC50 values for the compound effect.
PDAPP mouse in vivo pharmacology
In vivo pharmacology studies were conducted in young PDAPP transgenic mice essentially as described by Dovey et al. (2001). Young (2–3 months old) female hemizygous APPV717F transgenic mice (PDAPP) from Taconic (private line 6042T) were used for these studies. Typically, 32–40 mice were randomly assigned by parental lineage into groups of six to eight. Studies included vehicle-treatment groups administered in 7% Pharmasolve by oral gavage in a 10 ml/kg dose volume. LY2811376-treated mice were orally gavaged with the compound and killed at the indicated times. The use of animals in these pharmacology studies conformed to the Institutional Animal Care and Use Guidelines for Eli Lilly & Co.. For analysis of parenchymal Aβ, C99 and sAPPβ brain samples were homogenized in 5.5 m guanidine-HCl buffer, and extracts were collected, diluted, and filtered before ELISA.
Aβ ELISA determination
Parenchymal Aβ levels were determined from appropriately diluted guanidine homogenates by sandwich ELISA as described above. Aβ levels were interpolated from standard curves using XL-Fit for Excel. C99 protein levels in brain homogenates were determined by an automated sandwich ELISA using an anti-APP C-terminal antibody (catalog #8717; Sigma) to capture the analyte and biotinylated anti-APP antibody (3D6) to detect the N terminus of C99. sAPPβ protein levels in brain homogenates and CSF were determined by a manual sandwich ELISA using an anti-APP antibody (8E5) to capture the analyte, an anti-neo epitope antibody (GN2114 rabbit polyclonal) to bind the C terminus of the sAPPβ revealed by BACE enzyme cleavage, and an anti-rabbit HRP conjugate for colorometric detection. Unknowns were assayed in triplicate, and the concentration of analyte in a sample was interpolated from a four-parameter fit of the reference curve (XL-Fit for Excel). Replicate samples with a coefficient of variation of >15% or falling above or below the formal limits of quantitation were excluded from the analyses. ELISA values were normalized to protein levels (determined in duplicate by the Bradford Coomassie Plus Protein method) and expressed as picograms per milligrams (Aβ) or nanograms per milligrams (C99 and sAPPβ) protein.
Beagle dog in vivo pharmacology
Beagle dogs were used as the secondary in vivo pharmacology model to assess BACE1 inhibition in a nontransgenic animal model with physiologic levels of APP expression. A plasma pharmacokinetics (PK)/PD study was conducted in beagle dogs. On day 0 of this study, three plasma samples were collected from four dogs (two male and two female) at −1, −0.5, and 0 h before receiving a vehicle (PBS) dose. The mean value of plasma concentrations of Aβ1-x from these three samples were used as a baseline for each animal. The average predose baseline plasma Aβ1-x for the four dogs was 340 ± 14.2 pg/ml (mean ± SEM, n = 4). After dosing, plasma was collected at multiple time points out to 48 h and frozen for later analysis. Dogs were allowed 1 week to recover and then on day 7 were administered a 5 mg/kg oral dose of LY2811376·2HCl, and plasma samples were collected under the same schedule as the week before. After another week for recovery, on day 14, the dogs received another 5 mg/kg dose of LY2811376·2HCl, and plasma samples were collected as before.
As a companion to the plasma PK/PD study described above, efficacy in a central compartment was evaluated in the same study animals by collecting cisternal CSF at selected time points after vehicle or 5 mg/kg doses of LY2811376·2HCl and measuring a variety of Aβ species. For these studies, dogs were anesthetized, and 1 ml samples of CSF were collected by a cisternal tap. CSF samples that were grossly bloody were excluded from the analyses. For the day 0 vehicle-treatment group, CSF taps were collected at 3 and 24 h after dosing. These two values were averaged and used as the baseline for each dog. The average baseline CSF Aβ1-x for the four dogs was 12,409 ± 1014 pg/ml (mean ± SEM, n = 4). After a 7 d recovery period, the same dogs were administered a 5 mg/kg dose of LY2811376·2HCl, and cisternal CSF samples again were collected at 3 and 24 h after dose. Finally on day 14, the same dogs were administered another 5 mg/kg dose, and cisternal CSF samples were collected at 9 h as well as 24 and 48 h after dose. The use of animals in these pharmacology studies conformed to the Institutional Animal Care and Use Guidelines for Eli Lilly & Co.
Cisternal CSF and plasma concentrations of Aβ1-x (also known as Aβ total) were determined by standard sandwich ELISA protocols using m266.2 and biotinylated 3D6 (anti-Aβ1–5) as the capture and reporter antibodies, respectively. Unknowns were assayed in duplicate, and picograms per milliliters was determined by interpolating (Soft Max Pro version 5.0.1; Molecular Dynamics) from eight-point standard curves and then adjusted for dilution. Replicate samples with a coefficient of variation of >15% or falling above or below the formal limits of quantitation were excluded from the analyses. Grossly bloody cisternal CSF taps were excluded from the analysis.
Statistical methods
Statistical significance was determined by ANOVA using GraphPad Prism software. If significant, treatment groups were compared with controls using Dunnett's method. Significance was set as a p value of ≤0.05.
Preclinical toxicology studies
Sprague Dawley [Crl:CD(SD)] rats from Charles River Laboratories (10 per sex per group), ∼7 weeks of age, were given 0 (vehicle only), 10, 30, or 100 mg/kg LY2811376 by daily oral gavage. Vehicle consisted of 1% (w/v) hydroxyethylcellulose, 0.25% (v/v) polysorbate 80, and 0.05% (v/v) Dow Corning Antifoam 1510-US in reverse osmosis water. At necropsy after 3 months of treatment, tissues were immersion fixed in 10% neutral-buffered formalin (brain) or modified Davidson's solution (eyes) and then processed by routine methods to paraffin block and hematoxylin–eosin (H&E)-stained histologic slides. From selected animals given 100 mg/kg on a subsequent investigative study, eyes were fixed in modified Karnovsky's solution, processed routinely into epoxy resin, and then ultrathin sections stained with uranyl acetate and Sato's lead citrate were examined in a transmission electron microscope. The use of animals in these toxicology studies conformed to the Institutional Animal Care and Use Guidelines for the contract laboratory and sponsor. In a separate study, BACE1 knock-out mice (B6.129-Bace1tm1Pcw/J) were given 0 or 100 mg/kg LY2811376 by daily oral gavage for 9 weeks, and then necropsied tissues were collected and examined by light microscopy as described above for the rat toxicology study.
Methods for the clinical study
Study design.
The CSF sampling portion of the study was a subject- and investigator-blind, placebo-controlled, randomized, single-dose design in healthy subjects. The study assessed the safety and tolerability as well as plasma and CSF PK/PD. The study was conducted at PAREXEL International Early Phase Los Angeles, from February to June 2009. The California Institutional Review Board approved the study. All subjects provided written informed consent before the beginning of the study. The trial was conducted in compliance with the Declaration of Helsinki and International Conference on Harmonisation/Good Clinical Practice guidelines.
Subjects.
Thirty healthy subjects (21–49 years old, 27 males) participated in the CSF sampling portion of the study.
Procedures.
Subjects were randomly assigned to receive a single dose of 30 mg of LY2811376, 90 mg of LY2811376, or placebo in the fasted state. Safety was assessed before and after dosing by recording adverse events, physical and neurological examinations, vital signs, electrocardiograms, and results of clinical safety laboratory tests. The indwelling lumbar catheter was placed by anesthesiologists ∼4 h before administration of study drug, and subjects remained supine for the duration of the CSF sample collection period. Up to 22 CSF samples were collected at regular intervals, from 4 h before study drug administration up to 36 h after study drug administration (approximately −4, −2, 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 18, 20, 24, 28, 32, and 36 h after dose). All CSF samples were collected in polypropylene tubes and stored at −20°C or below. In addition, up to 13 plasma samples (before dose and ∼0.5, 1, 2, 4, 6, 8, 12, 24, 36, 48, 60, and 96 h after dose) were collected on each subject; the plasma time points were selected based on the preclinical plasma Aβ data.
Bioanalytical methods.
Plasma Aβ1–40 and Aβ1-x and CSF Aβ1–40, Aβ1–42, sAPPα, and sAPPβ were measured by PPD Inc. using validated ELISAs. Aβ1–40, Aβ1–42, and Aβ1-x captured using monoclonal 2G3, 21F12, and 266.2, respectively, were colorimetrically quantified using biotinylated monoclonal 3D6, a streptavidin–HRP conjugate, and TMB substrate. Plasma Aβ1–40 and Aβ1-x assays were validated over concentration ranges of 25–800 and 28–600 pg/ml, respectively. The CSF Aβ1–40 and Aβ1–42 assays were validated over a concentration range of 25–400 pg/ml for both analytes. CSF sAPPα and sAPPβ were measured using a commercially available assay kit (Meso Scale Discovery) validated over a concentration range of 200–100,000 pg/ml for both analytes.
Plasma and CSF samples were analyzed for LY2811376 using validated liquid chromatography with tandem mass spectrometric detection methods by Covance Laboratories. Deuterated LY2811376 was added to each sample as an internal standard. Separation was achieved using a gradient HPLC system with a Prodigy ODS(3) column (5 μm, 50 × 2.0 mm; Phenomenex) using mobile phase A (0.2% formic acid in water) and mobile phase B (0.2% formic acid in acetonitrile). After separation, LY2811376 was detected using a Sciex API 4000 in positive ion mode monitoring the transition mass/charge 321 → 217. The validated concentration range for plasma was 1–5000 ng/ml and for CSF was 0.5–500 ng/ml. CSF red blood cell counts were also measured on two predose samples at the local laboratory (Glendale Adventist Medical Center Laboratory).
Clinical statistical analyses.
Statistical analysis of percentage change from baseline of CSF Aβ1–40 concentrations obtained over the fixed-schedule sampling period were analyzed using a repeated-measures analysis with the primary statistical inference of pairwise comparison of the overall mean difference of low-dose LY2811376 (30 mg) with placebo and high-dose LY2811376 (90 mg) with placebo using an 80% confidence interval (this is equivalent to a one-sided hypothesis test with α = 0.1). The statistical model included the baseline CSF Aβ1–40 concentrations obtained during predose CSF collection periods and fixed effects of dose groups (placebo, low-dose LY2811376, and high-dose LY2811376), scheduled CSF sampling time, and the interaction between dose groups and sampling times. A compound symmetric covariance structure was used. Additional exploratory analyses included the analysis of percentage change from baseline of CSF Aβ1–42, sAPPα, and sAPPβ. The statistical model included fixed effects of dose, sampling time, and the statistical interaction term with a compound symmetric covariance structure.
Results
Fragment-based discovery and optimization of LY2811376
Informed by the community experience with peptidomimetic and HTS-derived inhibitors of BACE1, we chose to explore a fragment-based screening approach (Erlanson et al., 2004), keenly focused on identifying a ligand-efficient starting point suitable for producing a lead compound capable of facile target access within the CNS. Ligand efficiency can be variably defined (Hopkins et al., 2004) but in this context is a measurement of the contribution of each individual atom to the overall binding energy of a ligand to its protein target.
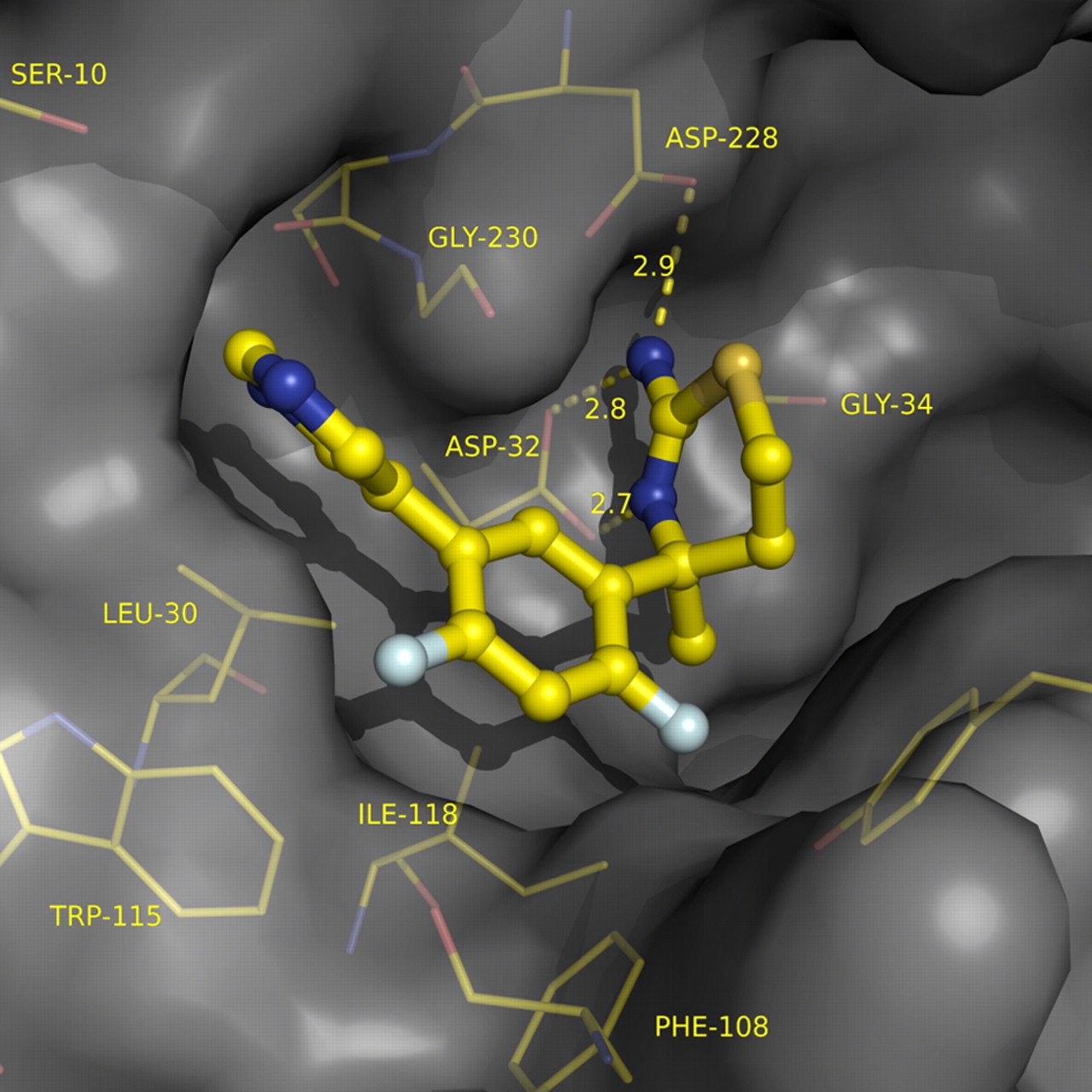
Screening a collection of ∼8000 fragments at high concentration (4.76 mm) against BACE1 with a maltose binding protein–fusion protein substrate (Sinha et al., 1999) produced a number of low-affinity fragment hits with high LEAN values [LEAN is an internal metric for ligand efficiency and is defined as −log(IC50)/number of non-hydrogen atoms]. Of particular interest were amino-benzothiazine (Fig. 1, 1) and amino-thiadiazine (Fig. 1, 2) (Sharma et al., 1963), which met our initial criteria for ligand efficiency and possessed attractive physicochemical properties for subsequent optimization. An important advance for our effort was the successful crystal soaking experiment of 1 in Figure 1 with a soluble construct for BACE1, revealing two copies of the inhibitor with high active-site occupancy and an open-flap protein conformation. One copy engaged in an efficient H-bonding interaction with the catalytic diad of BACE1, whereas a second copy extended across the S1–S3 cavity. Recognizing that the rigid bicyclic scaffold of the aspartate-binding benzothiazine failed to afford optimal vectors for fragment growth into S1–S3, we excised the fused aryl ring, effectively deplanarizing the aminothiazine, and introduced a quaternary methyl group to both exacerbate this topology and enhance chemical stability, yielding 3 in Figure 1. BACE1 recognized only a single enantiomer of this now chiral inhibitor, achieving a substantial increase in potency while maintaining excellent efficiency of the ligand–protein interactions. Surprisingly, however, inhibitor 3 in Figure 1 also revealed two copies bound to the BACE1 active site during crystal-soaking experiments, again with one copy productively engaged with the catalytic aspartates and the second in an S1–S3 role. Extension of the inhibitor with a suitable S3 motif such as a pyrimidine now produced an inhibitor competent to play both roles. A series of tricyclic aminothiazines similar to 4 in Figure 1 soaked into BACE1 crystals with a single bound copy, now achieving micromolar potency with only 20 heavy atoms. Deactivation of the metabolically labile central aryl ring with fluorines reduced in vivo clearance and also provided a welcome enhancement in potency, breaking the micromolar barrier with high ligand efficiency. As evidenced by x-ray co-crystal structure (Fig. 2), LY2811376 (Fig. 1, 5) retained an optimal H-bonding network with the catalytic aspartates, efficiently traversed S1 and projected the pyrimidine into the opening of S3.

Optimization of BACE1 inhibitor from initial fragment hits entailed deplanarization of bicyclic fragment to enhance occupancy of S1 pocket, introduction of an S3 binding element to increase affinity, and optimization of final inhibitor for in vivo performance. * hBACE1 MBP–C125 ELISA assay; ** hBACE1 mcaFRET assay.

Co-crystal structure of LY2811376 in BACE1 active site (flap not shown for clarity). Structure factors and protein coordinates to be deposited into Protein Data Bank.
In vitro characterization of LY2811376
In recombinant enzyme assays, LY2811376 demonstrated concentration-dependent inhibition of hBACE1 with an IC50 of 239 and 249 nm against a small synthetic peptide or a larger chimeric protein substrate, respectively (Table 2). Because of limitations of substrate solubility (MBP–C125 assay) or inner-filter effects (mcaFRET assay), bona fide Km values for each substrate were not determined. However, rather than being used for detailed kinetic analyses, these enzyme assays were optimized to be highly reproducible and detect differences in potency from compound to compound to drive the structure–activity relationship. LY2811376 demonstrated ∼10-fold selectivity toward BACE1 over BACE2, a related aspartyl protease, and 50-fold or greater separation between inhibition of BACE1 and the more distantly related aspartyl proteases cathepsin D, pepsin, or renin (Table 2). In an APP-overexpressing human embryonic kidney cell line, LY2811376 treatment yielded a concentration-dependent decrease in Aβ secretion with a half-maximal effective concentration (EC50) of ∼300 nm (Table 3). LY2811376 treatment of primary neuronal cultures of PDAPP transgenic mouse produced a concentration-dependent decrease in Aβ secretion with an EC50 of ∼100 nm (Table 3). These reductions in Aβ were achieved in the absence of any detectable cytotoxic effects on cells, as shown in Table 3. Thus, although LY2811376 may not display the exquisite intrinsic potency of other reported BACE1 inhibitors, it is highly cell penetrant and as a result efficiently inhibits native murine and human BACE1 in a cellular environment.
Potency of LY2811376 in BACE1 and related aspartyl protease assays
Potency of LY2811376 cellular APP processing assays
In vivo characterization of LY2811376 in preclinical animal models
To assess BACE1 inhibition in vivo, we used the APPV717F mouse, a transgenic animal model of Aβ pathology (Games et al., 1995). Young mice were administered LY2811376 by oral gavage and killed at 3 h after dosing to assess brain levels of relevant PD markers of BACE target engagement. Systemic exposures averaged 667, 1830, and 7200 ng/ml at the 10, 30, and 100 mg/kg doses, respectively. The optimized physiochemical properties of the molecule, e.g., molecular weight of 320, logP of 2.38, and polar surface area of 64, resulted in a molecule that was highly brain penetrant with brain exposures of 1460, 3352, and 12,120 ng/g achieved for the 10, 30, and 100 mg/kg doses, respectively. Consistent with brain exposures exceeding the cellular IC50, administration of LY2811376 resulted in dose-dependent, significant reductions in Aβ (Fig. 3A), as well as sAPPβ and C99, the proximal cleavage products of APP proteolysis by BACE1 (Fig. 3B,C). The concordant inhibition of all three relevant biomarkers supports an in vivo mechanism of action for LY2811376 consistent with BACE1 inhibition in brain after acute administration of LY2811376. However, LY2811376 is rapidly cleared in mice, resulting in overall low sustained exposure profiles at orally administered pharmacological doses, which dissuaded us from pursuing chronic administration studies in transgenic mice to investigate effects on Aβ amyloid plaque lowering.

Pharmacologic effects in vivo of oral administration of LY2811376. A–C, PDAPP mice (n = 6 per group) were treated with increasing doses of LY2811376 or vehicle, and Aβ (A), sAPPβ (B), or C99 (C) levels were determined from cortical extracts obtained 3 h after dosing. LY2811376 produced dose-dependent decreases in all APP-related PD markers of BACE1 inhibition in PDAPP mice, p < 0.01 versus vehicle control, ANOVA/Dunnett's post hoc analysis. D, Beagle dogs (n = 4) were treated sequentially with vehicle or 5 mg/kg LY2811376, and plasma and CSF samples were collected at various times after dosing. The average predose baseline plasma Aβ1-x was 340 ± 14.2 pg/ml (mean ± SEM, n = 4). The average vehicle baseline CSF Aβ1-x was 12409 ± 1014 pg/ml (mean ± SEM, n = 4). LY2811376 produced robust and time-dependent decreases in Aβ1-x in both plasma and CSF of dog compared with baseline control values. *p < 0.05, **p < 0.01 versus baseline controls, ANOVA/Dunnett's post hoc analysis.
Beagle dogs permitted confirmation of the in vivo pharmacology in a nontransgenic animal model. Effects on both plasma and CSF Aβ were assessed after oral gavage of a 5 mg/kg dose of LY2811376. LY2811376 was rapidly absorbed with Cmax values of 1915 ng/ml after ∼1 h after dosing (Tmax = 0.81 h). Overall plasma exposure averaged 12,300 ng · ml−1 · h−1 area under the concentration time curve (AUC), with a T1/2 of 6.8 h. After treatment with LY2811376, reductions in Aβ1-x were observed in plasma, with a maximal 85% reduction observed from 4 to 12 h after dosing (Fig. 3D). Plasma Aβ1-x levels remained below baseline 24 h after dosing. These significant changes in plasma Aβ were mirrored in the CSF compartment. Relative to baseline, CSF Aβ1-x levels were reduced 43% by 3 h after oral administration of 5 mg/kg LY2811376 (Fig. 3D). By 9 h, the effect size increased to ∼70% reduction in CSF Aβ1-x. Comparable reductions in CSF Aβ1–40 and Aβ1–42 as well as the potentially N-terminal truncated species Aβx-40 and Aβx-42 were observed at these same time points after LY2811376 treatment (results not shown). The concordant inhibition of various Aβ isoforms in CSF supports an in vivo mechanism of action for LY2811376 consistent with BACE1 inhibition in brain.
PK/PD characterization of LY2811376 in healthy volunteers
A single ascending dose (SAD) study and a follow-up CSF sampling study were conducted in healthy human subjects to characterize peripheral and central LY2811376 PK/PD. Plasma LY2811376, Aβ1–40, and Aβ1-x concentrations were measured in the SAD study over 96 h with oral administration of placebo or LY2811376 at doses ranging from 5 to 90 mg. Low (30 mg) and high (90 mg) doses of LY2811376 investigated in the CSF sampling study were based on PK and plasma Aβ1–40 PD observed in the SAD study (Fig. 4). Individuals participating in the CSF sampling study were randomly assigned to oral administration of placebo, 30 mg of LY2811376, or 90 mg of LY2811376 (n = 10 in each group). Concentrations of LY2811376, Aβ1–40, Aβ1–42, sAPPα, and sAPPβ were measured in CSF collected for 36 h via an indwelling lumbar sac catheter. Plasma concentrations of LY2811376, Aβ1–40, and Aβ1-x also were measured in the CSF sampling study.

SAD study in healthy volunteers. Mean plasma Aβ1–40 (A) and Aβ1-x (B) change from baseline after single doses of LY2811376. After single doses of LY2811376 between 5 and 90 mg, plasma concentrations of both Aβ1–40 and Aβ1-x decreased, reached a nadir, and then slowly returned to their predose baseline values. The time at which the nadir occurred ranged from a mean of 6–12 h and appeared to be independent of dose. The magnitude of the decrease in plasma Aβ1–40 and Aβ1-x, as measured by either the nadir or the average reduction over the first 24 h tended to increase with increasing doses of LY2811376. Plasma concentrations of Aβ1–40 and Aβ1-x after the highest dose of 90 mg did not fully return to their predose baseline values within the 120 h sampling period of the study. Plasma Aβ1–40 and Aβ1-x PD response provided guidance for dose selection to the second part of the trial looking at PD effect in CSF.
Maximum plasma concentrations of LY2811376 were reached 2 h after dosing, and the mean terminal half-life was ∼40 h. At 90 mg, the mean observed maximum concentration and AUC (time, 0 − ∞) were 242 ng/ml and 4560 ng · ml−1 · h−1, respectively. Maximum concentrations of LY2811376 in CSF were reached at ∼5 h, which is delayed relative to plasma concentrations. This delay is consistent with expectations (Bateman et al., 2009) based on the CSF samples being collected from lumbar region rather than the cisterna magna. The relationship between plasma and CSF LY2811376 AUC appeared to be linear, with the AUC in CSF approximately threefold lower than in plasma.
Single doses of LY2811376 resulted in significant and dose-dependent reductions in plasma Aβ1–40 (Fig. 5A), with a mean reduction over the first 24 h of 64% and reductions at the observed nadir (∼7 h after dosing) of 80% after the 90 mg dose. After the 90 mg dose, plasma concentrations of Aβ1–40 did not fully return to their predose baseline values within the 120 h sampling period of the dose-escalation part of the study. The reduction of plasma Aβ1-x was similar to plasma Aβ1–40.

SAD CSF sampling study in healthy volunteers. Single doses of 30 or 90 mg of BACE inhibitor LY2811376 administered to healthy human subjects resulted in significant biomarker changes in plasma and CSF. A, Plasma concentrations of Aβ1–40 decreased in a dose-dependent manner, with the 90 mg dose producing an observed mean nadir of −80.0%. B, C, CSF concentrations of Aβ1–40 and Aβ1–42 significantly decreased relative to placebo, with a 90 mg dose producing observed mean nadirs of −58.0 and −58.1%, respectively. In placebo-treated subjects, CSF concentrations of Aβ1–40 and Aβ1–42 increased over time without a return to baseline, suggesting a procedural phenomenon rather than diurnal effect. The average predose baseline CSF concentrations of Aβ1–40 and Aβ1–42 were ∼5300 and 650 pg/ml, respectively. D, E, Administration of LY2811376 resulted in decreases in CSF concentration of sAPPβ and increases in CSF concentration of sAPPα, providing direct evidence of the mechanism of action of LY2811376 in the CNS of man. Data are presented as mean ± SD and n = 10 for all graphs.
Single doses of LY2811376 also resulted in significant and dose-dependent reductions in CSF Aβ1–40 and Aβ1–42 (Fig. 5B,C). A repeated-measures mixed-model analysis was used to compare differences among the treatment groups during the 36 h sampling period. The maximum mean reduction from baseline concentration for the 30 and 90 mg doses was ∼20 ± 17 and 54 ± 17%, respectively. The 30 and 90 mg doses showed a significant mean reduction compared with placebo by 7 h after dose and reached a maximum reduction at 12–14 h after dose. The average reduction in CSF Aβ1–40 over the first 24 h was 33%, and the reduction at the observed nadir (∼18 h after dosing) was 54% after the 90 mg dose. In the placebo-treated subjects, concentrations of CSF Aβ1–40 and Aβ1–42 increased over time without a return to baseline after 24 h. This phenomenon appears to be related to the CSF collection procedure and has been reported by other researchers (Bateman et al., 2007). Accounting for the increase in the placebo group, the average reduction over the first 24 h in the CSF concentration of Aβ1–40 for the 90 mg dose relative to placebo was ∼56%. The magnitude of the reduction in CSF Aβ1–42 was very similar to that in CSF Aβ1–40.
Single doses of LY2811376 resulted in significant and dose-dependent reductions in CSF sAPPβ (Fig. 5E), providing direct evidence of β-secretase inhibition in the human CNS. The 90 mg LY2811376 dose showed a maximum reduction of sAPPβ concentration of 42%, which occurred after 20 h. LY2811376 also resulted in increased concentrations of sAPPα (Fig. 5D) with 76% maximum increase for the 90 mg dose of LY2811376, which suggests that inhibition of β-secretase in humans resulted in a compensatory increase in APP cleavage at the α site. Concentrations of sAPPα appeared to continue to increase during the 36 h sampling period.
Overall, LY2811376 was well tolerated in the CSF study. There were no serious adverse events, and all 27 treatment-emergent adverse events were mild or moderate in severity. The most frequent treatment-emergent adverse events were procedural headache (n = 15) and catheter site pain (n = 7). There were no clinically significant alterations in vital signs, laboratory analytes, or electrocardiograms associated with LY2811376 treatment during the dosing period.
Preclinical toxicologic findings of retinal pathology
In parallel to the phase 1 studies in healthy volunteers, a 3 month rat toxicology study was performed to prepare for longer exposures in phase 2 clinical trials. LY2811376 caused cytoplasmic accumulations of finely granular autofluorescent material dispersed within the retinal epithelium (Fig. 6) and less prominently within neurons and glial cells in the brain at doses ≥30 mg/kg. The autofluorescence was observed when H&E-stained slides were observed with epifluorescent illumination (Leica cube I3: excitation, 450–490 nm bandpass; emission barrier filter, 515 nm long pass). The retinal epithelial cells were enlarged because of the burden of accumulated material, there was photoreceptor degeneration within the sensitive retina, and neuronal degeneration was present in brain. In a subsequent repeat-dosing investigative study, the retinal epithelial change was characterized ultrastructurally by notably increased numbers of secondary lysosomes containing partially degraded photoreceptor outer segment material, which caused the cellular enlargement.

LY2811376-related changes in the retinal epithelium of Sprague Dawley [Crl:CD(SD)] rats (top) and BACE1−/− knock-out (BACE1tm1Pcw) mice (bottom). Retinas from the vehicle-treated rats (A) and mice (C) were normal. Retinal epithelial cells from LY2811376-treated rats (B) and mice (D) were enlarged and distended with autofluorescent granular material (insets). The retinal epithelial layer is labeled with arrow and/or bracket. Note the brown pigment granules within the retinal epithelial layer and underlying choroid, which are a normal feature of the eye from this strain of pigmented mouse (C, D) but absent in the eye from the albino Sprague Dawley rat (A, B). Additionally, the bright yellow–green zone in the bottom right of the inset for C is attributable to autofluorescence of the scleral collagen, a normal structure that is not included in the insets for A, B, or D. H&E stain. Insets are H&E-stained sections viewed with epifluorescence.
A subsequent study using LY2811376 in BACE1−/− mice demonstrated that the findings observed in the retinal epithelium and brain were unrelated to the BACE1 pharmacological target. Treatment of BACE1−/− mice from The Jackson Laboratory (strain BACE1tm1Pcw; Johns Hopkins University, Baltimore, MD) with 100 mg/kg LY2811376 for 9 weeks resulted in accumulated autofluorescent material and degenerative changes in the retinal epithelium comparable with the changes observed in rats given similar doses, whereas the eyes of vehicle-treated BACE1−/− mice were histologically unremarkable. Intracellular autofluorescent granular material was present in the brains of treated BACE1−/− mice, although at considerably lower levels than occurred in treated rats; the material was not present in the untreated mice.
Importantly, as soon as these preclinical pathology data became available, clinical dosing of LY2811376 was discontinued, the Food and Drug Administration was notified, and the studies were terminated in agreement with the Food and Drug Administration. As a safety follow-up, all study participants were contacted for follow-up eye examinations. The examinations were conducted ∼6–10 months after completion of the trial. They revealed no clinically significant observations in the 45 of the 61 enrolled subjects, who agreed to participate.
Discussion
Here, we report on the first small-molecule BACE1 inhibitor demonstrating robust translation from preclinical animal models to early-stage clinical testing in humans. A fragment-based, LEAN-driven drug discovery effort was used to circumvent many of the poor physiochemical properties that plagued the early BACE1 inhibitors. LY2811376 efficiently inhibits BACE1 activity in vivo as demonstrated by the coordinate reduction of the two primary APP cleavage products generated by BACE1, e.g., C99 and sAPPβ, coupled with a reduction of various Aβ peptides beginning with the canonical BACE1 cleavage site as well as N-terminal truncated Aβ species. Thus, small oral doses of LY2811376 effectively attenuate the amyloidogenic processing pathway of APP.
The significant reduction in brain and CSF Aβ levels observed in preclinical animal models after oral administration of 5–10 mg/kg doses of LY2811376 contrasts sharply with in vivo pharmacology studies reported for earlier BACE1 inhibitors. Although extremely potent BACE1 inhibitors have been optimized in vitro, their in vivo performance has suffered as a result of P-glycoprotein efflux issues, poor oral bioavailability and/or rapid clearance. To obtain comparable central effect sizes observed with LY2811376, other BACE1 inhibitors have had to be dosed in MDR1a KO mice (Meredith et al., 2008) or in conjunction with a P-glycoprotein inhibitor (Hussain et al., 2007) to permit sufficient brain exposure to be achieved. Similarly, coadministration of a Cyp3A4 inhibitor to block first-pass metabolism was required to permit sufficient systemic exposure after oral gavage of a BACE1 inhibitor to achieve central BACE1 inhibition in a nonhuman primate model (Sankaranarayanan et al., 2009).
Although some progress with BACE1 inhibitors in preclinical models has been reported at recent meetings, comparable clinical PD responses in plasma and CSF in humans have not been reported previously. The group at CoMentis has reported in a preliminary communication robust changes in plasma Aβ after intravenous infusion of CTS 21166, a potent BACE1 inhibitor to healthy volunteers (Strobel, 2008), but no central pharmacology has been reported to date. Thus, to our knowledge, our data with LY2811376 represent the first demonstration of robust and sustained reduction of lumbar CSF Aβ in healthy volunteers dosed with a BACE1 inhibitor. Importantly, the reduction in Aβ levels is accompanied by sustained reduction of lumbar CSF sAPPβ, the primary cleavage product of BACE1 cleavage of APP.
Our study of LY2811376 revealed retinal pathology in longer-term nonclinical toxicology studies. Although the mechanism driving this pathology is not clearly understood, it appears to be off-target because a similar adverse effect was obtained with long-term dosing of BACE1 KO mice with LY2811376. It cannot presently be ruled out that, at toxicologic doses, activity against other aspartyl proteases, such as BACE2 or even cathepsin D, might play a role in the retinal pathology observed with LY2811376. More research in this area is necessary. Importantly, however, safety follow-up exams in the clinical trial participants revealed no clinically significant observations from administration of LY2811376.
In addition to the retinal pathology observed in preclinical toxicology studies, there are theoretical concerns with BACE1 inhibition that have not been specifically addressed with LY2811376. These concerns stem from theoretical risks inferred from the study of BACE1 KO animals as well as an expanding complement of additional substrates posited for BACE1. Perhaps the best characterized substrate for BACE1 in addition to APP is Neuregulin-1 (Hu et al., 2006; Willem et al., 2006). Although initially characterized as having a minimal phenotype (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001), more careful study of BACE1 KOs revealed hypomyelination of the peripheral nervous system in neonates (Hu et al., 2006; Willem et al., 2006). Given the role that Type 3 Neuregulin-1 is known to play in myelination (Michailov et al., 2004), inappropriate processing of Neuregulin-1 would be consistent with the hypomyelination observed in neonates harboring a BACE1 deficiency. The hypomyelination phenotype was not noted in BACE1 hemizygous mice (Hu et al., 2006), and the phenotype noted in homozygous KOs appeared to diminish with age, suggesting that BACE1 KO delayed but did not block myelination. Consistent with this interpretation, genetic deficiency of BACE1 also delayed remyelination of the sciatic nerves in mice following an experimental neuronal crush model (Hu et al., 2008).
The voltage-gated sodium channel (VGSC; Nav1) β-subunits are another recently described substrate for BACE1 (Wong et al., 2005; Kim et al., 2007). These β-subunits are auxiliary proteins to the pore-forming α subunits and play a role in trafficking and regulation of VGSC (Brackenbury and Isom, 2008). Enhanced excitability and frank behavioral seizures have been noted in BACE1 KOs, a behavioral phenotype that may result from hyperexcitability of hippocampal circuitry attributable to altered VGSCs (Evin et al., 2003). However, a similar behavioral phenotype was also noted by Hitt et al. (2010) in a subset of BACE1 KO mice without altered VGSCs expression, so other mechanisms may be at play, and clearly additional investigation is needed.
There is currently no evidence that pharmacologic inhibition of BACE1 in adults would result in a similar phenotype as that observed with genetic knock-out. As noted above, the hypomyelination phenotype was not noted in BACE1 hemizygous mice expressing 50% of BACE1 in brain. In addition, pharmacologic inhibition achieved by direct central administration of a BACE1 inhibitor did not appear to alter Neuregulin-1 cleavage patterns in adult mouse brain (Sankaranarayanan et al., 2008), but functional deficits were not evaluated. Similarly, the deficits in behavioral cognitive tests, e.g., Morris water maze, Y maze, etc., observed in BACE knock-outs were not present in BACE1 hemizygous mice (Laird et al., 2005), suggesting that partial reduction in BACE1 activity may be better tolerated even when it already occurs during development. Finally, as a cautionary note about over-interpreting theoretical safety concerns from KO data, it should be noted that genetic KO of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase results in embryonic lethality (Ohashi et al., 2003), but statins that target HMG-CoA reductase remain one of the most widely prescribed classes of drugs in the world (Simmons, 2003).
In conclusion, this first description of an orally available CNS-active β-secretase inhibitor after more than a decade of research clearly demonstrates that BACE1 is a tractable target and that profound central Aβ lowering can be achieved by BACE1 inhibition in humans. The magnitude and duration of central Aβ reduction obtainable with BACE1 inhibition needs to be balanced against the possible on-target effects against other known substrates of BACE, unknown substrates yet to be discovered, as well as off-target idiosyncratic toxicity profiles to establish an adequate safety margin. Once established, adequately powered long-term clinical trials of similar compounds with comparable PD responses in appropriate populations will be required.
Footnotes
This research was funded by Eli Lilly & Co.
S.S.J., M.Y., and L.E. are employees of PAREXEL. All other authors are or were employees of Eli Lilly & Co..
- Correspondence should be addressed to Dr. Martin Citron, Lilly Research Laboratories, Eli Lilly and Co., Lilly Corporate Center, Indianapolis, IN 46285. citronma{at}lilly.com





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}