

Abstract
Over 50% of multiple sclerosis (MS) patients experience cognitive deficits, and hippocampal-dependent memory impairment has been reported in >30% of these patients. While postmortem pathology studies and in vivo magnetic resonance imaging demonstrate that the hippocampus is targeted in MS, the neuropathology underlying hippocampal dysfunction remains unknown. Furthermore, there are no treatments available to date to effectively prevent neurodegeneration and associated cognitive dysfunction in MS. We have recently demonstrated that the hippocampus is also targeted in experimental autoimmune encephalomyelitis (EAE), the most widely used animal model of MS. The objective of this study was to assess whether a candidate treatment (testosterone) could prevent hippocampal synaptic dysfunction and underlying pathology when administered in either a preventative or a therapeutic (postdisease induction) manner. Electrophysiological studies revealed impairments in basal excitatory synaptic transmission that involved both AMPA receptor-mediated changes in synaptic currents, and faster decay rates of NMDA receptor-mediated currents in mice with EAE. Neuropathology revealed atrophy of the pyramidal and dendritic layers of hippocampal CA1, decreased presynaptic (Synapsin-1) and postsynaptic (postsynaptic density 95; PSD-95) staining, diffuse demyelination, and microglial activation. Testosterone treatment administered either before or after disease induction restores excitatory synaptic transmission as well as presynaptic and postsynaptic protein levels within the hippocampus. Furthermore, cross-modality correlations demonstrate that fluctuations in EPSPs are significantly correlated to changes in postsynaptic protein levels and suggest that PSD-95 is a neuropathological substrate to impaired synaptic transmission in the hippocampus during EAE. This is the first report demonstrating that testosterone is a viable therapeutic treatment option that can restore both hippocampal function and disease-associated pathology that occur during autoimmune disease.
Introduction
Cognitive impairment occurs in >50% of multiple sclerosis (MS) patients (Benedict et al., 2006; Chiaravalloti and DeLuca, 2008), and may include abnormalities in information processing speed and attention, as well as learning and memory (Amato et al., 2010). Cognitive disability greatly impacts the quality of life of MS patients, but approved disease-modifying drugs have little to no effect on cognition. Thus, there is an unmet need to identify treatments that could specifically improve cognition during MS.
To develop such treatments, we must first understand the mechanisms underlying cognitive disability. Data from in vivo magnetic resonance imaging (MRI) studies suggest that hippocampal atrophy begins early in MS (Roosendaal et al., 2008; Anderson et al., 2010), and this atrophy has been correlated with impaired performance on visuospatial memory testing (Sicotte et al., 2008). Visuospatial and verbal learning and memory are commonly affected in MS patients (Thornton and Raz, 1997; Benedict et al., 2009). However, tissue is not available from MRI studies, so the involved neuropathology remains unknown. Alternatively, postmortem studies in MS have demonstrated that demyelination and neuropathology occur in the hippocampus (Geurts et al., 2007; Papadopoulos et al., 2009), and include a variety of cellular and molecular changes involved with synaptic integrity, axonal transport, and glutamate homeostasis (Dutta et al., 2011). Unfortunately, cognitive testing was not available from patients, so functional significance of described changes remains unknown. Thus, while accumulating data indicate that both hippocampal dysfunction and pathology occur during MS, the relationship between the two is unclear.
To explore the relationship between hippocampal dysfunction and neuropathology during MS, we turned to experimental autoimmune encephalomyelitis (EAE). Recently researchers have begun to use this model to address the neurodegenerative processes that occur within the brain during disease (Rasmussen et al., 2007; Centonze et al., 2009; MacKenzie-Graham et al., 2009; Crawford et al., 2010). Nonetheless, our understanding of how pathology correlates to cognitive dysfunction remains limited. Indeed, hippocampal pathology occurs during EAE and has functional significance, since hippocampal-dependent spatial learning was impaired at a late stage of disease (Ziehn et al., 2010). In this study, we will use electrophysiology to understand the effects of EAE on excitatory synaptic transmission and furthermore identify the neuropathological substrates underlying hippocampal dysfunction.
Beyond understanding the relationship between dysfunction and neuropathology, it is also important to develop therapeutic treatments aimed at preserving hippocampal-dependent learning and memory. In this study, testosterone was chosen since it has been previously shown to be beneficial in the CNS by improving synaptogenesis and enhancing cognitive performance (Frye et al., 2004). Testosterone can also prevent neuronal and dendritic atrophy (Fargo and Sengelaub, 2007) and prevent astrogliosis and microglial activation (Barreto et al., 2007). In a pilot MS trial, testosterone treatment reduced the rate of whole brain atrophy (Sicotte et al., 2007) and improved performance in cognitive testing (Gold et al., 2008). Here, testosterone will be used as a treatment to test whether hippocampal pathology and dysfunction can be prevented during EAE.
Materials and Methods
Animals and experimental treatment.
Male C57BL/6 mice, age 12–16 weeks, were bred in-house from animals purchased from The Jackson Laboratory. All mice were castrated as described previously (Palaszynski et al., 2004), and given a 7 d recovery period before experiment initiation. Ninety day release pellets containing either testosterone propionate (T) at a 5 mg dose, dihydrotestosterone (DHT) at a 5 mg dose, or placebo pellets (PLAC) with carrier binder alone (Innovative Research of America) were implanted. All animals were maintained in accordance with guidelines of University of California, Los Angeles' Chancellor's Animal Research Committee, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
EAE induction and clinical disease scoring.
Active EAE was induced by immunizing mice with 200 μg of myelin oligodendrocyte glycoprotein (MOG) peptide, amino acids 35–55 (Chiron Mimotopes), and 300 μg of Mycobacterium tuberculosis in complete Freund's adjuvant, over two sites: the right draining inguinal and axillary lymph nodes. One week later, a booster immunization was delivered subcutaneously, over the contralateral draining lymph nodes, as described previously (Ziehn et al., 2010). For MOG immunizations mice were temporarily anesthetized with isoflurane. On days 0 and 2, EAE mice received intraperitoneal injections of Bordatella pertussis toxin (Ptx; 500 ng/mouse). Healthy control mice received saline injections (0.9% sterile saline; ThermoFisher Scientific) in place of MOG and Ptx injections in identical doses and locations to correspond with injections given to EAE mice. Mice were monitored daily throughout the experiment, and clinical disease severity was measured using the standard EAE grading scale (Pettinelli and McFarlin, 1981). Clinical scores were averaged across all animals per day, yielding a mean clinical disease index per group.
Slice electrophysiology.
Mice were randomly selected for electrophysiological recording, daily, corresponding to EAE day 21 through 45. Hippocampi were obtained from mice that were deeply anesthetized with halothane and then killed by cervical dislocation. The brain was removed and placed in cold (4°C), oxygenated (95% O2/5% CO2) artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 25 Na2HCO3, 4.4 KCl, 1 NaH2PO4, 1.2 MgSO4, 2 CaCl2, and 10 glucose. The left hemisphere was collected for subsequent immunohistochemical staining (see below) and the hippocampus from the right hemisphere of each animal was then dissected free from the rest of the brain. The hippocampus was then cut into 400-μm-thick slices parallel to its long axis and 3–4 slices from the extreme dorsal and ventral ends were discarded. The remaining slices were maintained in interface-type recording chambers perfused at a constant rate (2–3 ml/min) with a warmed (30°C), oxygenated ACSF and allowed to recover for at least 2 h before an experiment. Low resistance (5–10 MΩ) glass microelectrodes filled with ACSF were placed into stratum radiatum of the hippocampal CA1 region to record field EPSPs (fEPSPs). Presynaptic stimulation pulses were delivered once every 50 s to the Schaffer collateral/commissural fibers via a bipolar nichrome wire stimulating electrode. Basal synaptic transmission was examined by generating input/output curves where fiber volley amplitude (input) and fEPSPs slopes (output) were compared across a range of presynaptic fiber stimulation intensities (Komiyama et al., 2002; Cuthbert et al., 2007; Wiltgen et al., 2010). Whole-cell voltage-clamp recording was performed using low resistance (4–7 MΩ) microelectrodes filled with a solution containing the following (in mm): 102 cesium gluconate, 17.5 CsCl, 10 TEA-Cl, 5 QX-314, 4.0 Mg-ATP, 0.3 Tris-GTP, and 20 HEPES, pH = 7.2. Slices with the CA3 region removed were bathed in a modified ACSF containing 4.0 mm CaCl2, 2.4 mm MgSO4, 2.4 mm KCl, and 100 μm picrotoxin (to block inhibitory synaptic transmission). Evoked EPSCs were elicited by Schaffer collateral fiber stimulation and recorded at postsynaptic membrane potentials of −80 mV or +40 mV. The amplitudes of EPSCs 5 and 50 ms post-EPSC onset were used to estimate AMPA and NMDA receptor-mediated components of the compound EPSCs. Double exponential fits to the decay of synaptic currents recorded at +40 mV were used to calculate weighted mean decay time constants (Rumbaugh and Vicini, 1999). Miniature EPSCs (mEPSCs) were recorded at −70 mV in the presence of 1.0 μm tetrodotoxin (TTX) and 100 μm picrotoxin. A template-based event detection routine in pClamp 10 (Molecular Devices) and a threshold of 6 pA was used for mEPSC analysis. Paired-pulse facilitation (PPF) was measured by delivering pairs of presynaptic fiber stimulation pulses with varying interpulse intervals (25, 50, 100, 200, and 275 ms) and measured as the ratio of the slope of the fEPSP evoked by the second stimulation pulse relative to that produced by the first stimulation pulse.
Immunohistochemistry.
To determine the neuropathological correlates that might be related to changes in synaptic transmission, immunohistochemistry was performed on corresponding left hemispheres of mice from all experimental conditions. Left hemispheres were fixed in 4% paraformaldehyde overnight then transferred to a 30% sucrose/saline solution. Once tissue from all animals in experiment had been collected, all samples were gelatin embedded, frozen, and sliced thin using a cryostat at −20°C, as previously described (Ziehn et al., 2010). To determine hippocampal CA1 volume, Nissl staining was performed using standard protocols on every fifth sagittal 20-μm-thick section spanning the left hippocampus ∼640 μm (from bregma: lateral coordinates 0.12–1.08 mm; Paxinos and Franklin, 2001). For all other immunostaining conditions, three sagittal sections were used from each mouse in each condition. To detect synaptic proteins, myelin basic protein (MBP), axons, microglia, and neurons, and the following primary antibodies were used: polyclonal anti-Synapsin-1 (Syn-1) 1:500, monoclonal anti-postsynaptic density protein 95 (PSD-95) 1:500 (Millipore Bioscience Research Reagents), monoclonal anti-myelin basic protein (MBP) 1:500, polyclonal anti-neurofilament-200 (NF-200) 1:500 (Millipore Bioscience Research Reagents), polyclonal anti-Iba1 1:1000 (Wako Chemicals), and monoclonal anti-neuronal-specific nuclear protein (NeuN) 1:500 (Millipore Bioscience Research Reagents). Fluorescent-conjugated secondary antibodies (goat anti-mouse Cy5 IgG [1:750; Millipore Bioscience Research Reagents] and goat anti-rabbit Cy3 IgG [1:1000; Vector Laboratories] in 2% normal goat serum in TBS solution) were used to visualize staining. Then 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI; 1:1000; Invitrogen) staining was used in all fluorescence staining conditions to identify nuclear DNA in all cell types. To confirm neuropathological findings, all immunohistochemistry was repeated in tissue from mice killed at one time point, day 21 postimmunization. In this follow-up experiment there were five mice per condition.
Microscopy and stereological analysis.
Immunostaining was quantified using unbiased stereology, and experimenter was blind to experimental groups. Identical light intensity and exposure settings were used to acquire all images (RGB), which were split and separated by color channel, using ImageJ version 1.29, downloaded from http://rsb.info.nih.gov/ij. CA1 volume was estimated using the rigorous Cavalieri method (Gundersen and Jensen, 1987; Cruz-Orive, 1999; Ziehn et al., 2010). Iba1+ cells morphologically representative of reactive microglia in CA1 were counted and presented as number of cells per mm3 of hippocampal volume. Area of myelin and synaptic protein density within the CA1 were measured as percentage area of immunoreactivity, using ImageJ. To quantify Synapsin-1 immunoreactive (Syn-1-IR) presynaptic puncta in CA1, puncta size threshold range was determined by sampling several Syn-1-IR puncta from various animals, and averaging both upper and lower threshold limits: minimum puncta size (0.816 μm2) and maximum puncta size (3.264 μm2). This range was then used to filter for puncta particle size throughout the experiment. The number of puncta in each optical image was averaged across z-stack, brain section, and animal to attain a composite average of presynaptic puncta within the sampled hippocampal area in different group conditions.
Statistical analysis.
All quantitative measures are presented as mean ± SEM and analyzed by Student's t test or one-way ANOVA. Post hoc analyses were conducted only if ANOVA yielded p < 0.05, with either Newman–Keuls or Dunnet's test comparisons. Pearson's linear regression analysis was used to determine correlations between electrophysiology and pathology using GraphPad Software version 4.0. Cross-modality correlations have been made in other previously published work (Aubert et al., 2009; Lehmann et al., 2010; Arezzo et al., 2011).
Results
Active EAE was induced using MOG 35–55 peptide and standard EAE clinical scores were attained, primarily reflecting the level of motor disability. Experimental design is depicted to demonstrate the creation of three experimental groups: healthy controls, placebo-treated EAE mice, and testosterone-treated mice (Fig. 1A). As expected, clinical motor disability began in placebo-treated EAE mice by 14 d post immunization and persisted through experiment (Fig. 1B, red triangles). Consistent with previous findings (Palaszynski et al., 2004), testosterone-treated mice experience significantly lower clinical disability compared with placebo treatment.

Experimental design and standard clinical scores in EAE. A, Experimental design depicting the timing of gonadectomy (Day −14, denoted by x), placebo or testosterone pellet implantation (day −7, denoted by circles), EAE induction (day 0 and 7, denoted by arrows), daily clinical scoring (day 7–45, denoted by dashed line), and electrophysiology (day 21–45, denoted by gray line). Pertussis injections (intraperitoneal, i.p.) were given to both EAE groups on day 0 and 2, as part of standard EAE induction (data not shown). All mice were age-matched C57BL/6 adult males that were selected for electrophysiological recording in an intermixed fashion (one per day), so that mice from each experimental group were measured every 3 d. Brain tissue was separated depending on type of study: right hemispheres were used for electrophysiology; respective left hemispheres were prepared for pathology studies. All pathology experiments were conducted once all left hemisphere tissue from mice in all conditions had been collected. B, EAE clinical scores were recorded in placebo-treated (PLAC + EAE, red) or testosterone-treated (T + EAE, blue) mice as well as in non-EAE-induced healthy controls (NL, black). Placebo-treated mice with EAE exhibited a moderately severe clinical course, while testosterone-treated mice exhibited significantly reduced clinical severity. It is important to note that statistical variance was not altered as mice were individually selected for electrophysiological recording. Data are representative of two separate experiments. Repeated-measures ANOVA with post hoc pairwise comparisons revealed that PLAC + EAE was significantly different from other two groups, p < 0.05, n = 5 mice per group.
CA1 atrophy and disruption of synaptic integrity occurred during EAE
CA1 atrophy has been observed in MS patients using in vivo MRI (Sicotte et al., 2008), thus it was important to determine the extent of CA1 atrophy in the animal model of MS. Using the rigorous Cavalieri method of volume estimation for histopathological data, we measured CA1 atrophy during EAE. This included volume quantification of the whole CA1, as well as volume quantification of the separate CA1 layers: CA1 pyramidale, CA1 stratum oriens, and CA1 stratum radiatum. The volume of nearby CA3 was also measured. EAE caused a significant decrease in CA1 pyramidal layer volume compared with healthy controls (Fig. 2A–G), and testosterone treatment during EAE prevented this atrophy. These results were confirmed in subsequent experiments where immunohistochemical staining was performed on tissue from animals killed at one time point.

Hippocampal atrophy occurs during EAE and testosterone treatment prevents this. A–C, Representative hippocampal sections stained with Nissl from normal (NL; A), placebo-treated EAE (PLAC + EAE; B), and testosterone-treated EAE (T + EAE; C) mice depict 4× magnification of the various hippocampal subregions: CA1 pyramidal layer (pyr), CA1 stratum oriens (so), CA1 stratum radiatum (sr), CA2/3, dentate gyrus (DG), and corpus callosum (CC). D–F, The CA1 pyramidal layer shown at 40× magnification, had significantly reduced volume in placebo-treated EAE mice (E), compared with healthy controls (D), quantification in G. With testosterone treatment during EAE (F), the CA1 pyr volume was preserved to levels similar to those in controls (D and G). H, CA1 stratum radiatum (CA1 sr) area was also significantly reduced in placebo-treated EAE mice compared with healthy controls, and this atrophy was prevented in testosterone-treated EAE mice. I, Alternatively, stratum oriens (CA1 so) area was not significantly changed with EAE or testosterone treatment of EAE, compared with healthy controls. J, CA3 area was significantly reduced in placebo-treated mice with EAE, compared with healthy controls, and preserved in testosterone-treated mice with EAE. Estimated CA1 pyr volume (mm3) and CA1 sr, CA1 so, and CA3 area (mm2) depict representative means normalized to mean area and volume of healthy controls (NL). One-way ANOVA and Newman–Keuls post hoc analysis revealed significant difference between three groups. *p = 0.05, n = 3 mice per group (5 sections per mouse). Scale bars, 20 μm.
When volume of the CA1 stratum radiatum, rich in CA1 apical dendrites, was compared across groups, significant differences were again found between healthy controls and placebo-treated EAE, with EAE having decreased stratum radiatum volume (Fig. 2A–C,H). Moreover, testosterone treatment of EAE mice also prevented CA1 stratum radiatum atrophy (Fig. 2H). In contrast, stratum oriens volume analyses did not reveal significant differences (Fig. 2I). Additionally, CA3 volume was also significantly decreased in hippocampi from mice with EAE, but not in hippocampi from mice treated with testosterone, compared with healthy control mice (Fig. 2J). Thus, these results demonstrate that EAE causes atrophy within the hippocampus, specifically within the CA1 and CA3 regions containing pyramidal neurons and apical dendrites.
Since the hippocampus governs mammalian learning and memory, and synaptic integrity and transmission are critical to hippocampal function, presynaptic (Synapsin-1) and postsynaptic (PSD-95) protein markers were used to assess synaptic integrity within the CA1. Synapsin-1+ puncta were significantly decreased in CA1 (pyramidale and stratum radiatum combined) of EAE mice as compared with healthy controls (Fig. 3A,C vs D,F,J). Further, the percentage area of PSD-95 staining was also significantly decreased in EAE mice compared with healthy controls (Fig. 3B,C vs E,F,K). In contrast, both presynaptic and postsynaptic expression levels were similar to those of healthy controls when EAE mice were treated with testosterone (Fig. 3G–K). Follow-up studies confirmed these findings.

Synaptic staining in the hippocampus is disrupted in EAE and preserved with testosterone treatment. A–I, Fluorescent images depict representative hippocampal sections from healthy (A–C), placebo-treated EAE (D–F), and testosterone-treated EAE (G–I) mice, where Synapsin-1 (Syn-1; Cy3-green), PSD-95 (Cy5-red), and DAPI (blue) staining are shown at 60× magnification. During EAE, Syn-1+ puncta were significantly decreased in the CA1 region of the hippocampus (D), compared with healthy controls (A). This decrease was prevented with testosterone treatment during EAE (G), quantified in J. Similarly, PSD-95 was also significantly decreased in placebo-treated EAE mice, and preserved in testosterone-treated EAE mice (B, E, H, respectively, and quantified in K). C, F, I, Merged images of both presynaptic (Syn-1) and postsynaptic (PSD-95) staining in selected areas (dashed square) of CA1 sr demonstrate the extent of colocalization within CA1 dendrites. One-way ANOVA and Newman–Keuls post hoc analysis revealed statistical significance in presynaptic and postsynaptic integrity where *p = 0.04, n = 3 mice per group, and **p < 0.001, n = 3 mice per group. Scale bars, 10 μm.
We next wanted to assess whether decreases in synaptic protein levels corresponded with decreases in hippocampal CA1 volumes. Correlation analyses revealed that only PSD-95 levels significantly correlated with changes in both CA1 pyramidale and CA1 stratum radiatum volumes (Table 1), while Syn-1 levels did not. Together these data suggest that EAE causes significant atrophy in distinct CA1 regions, and furthermore, that this CA1 atrophy correlates significantly with decreases in PSD-95 levels.
Correlation coefficients and coefficients of determination between synaptic protein levels and CA1 subregional volume
Excitatory synaptic transmission in the hippocampus is impaired during EAE and testosterone treatment prevents this functional deficit
Previous studies have found that PSD-95 strongly regulates the trafficking of postsynaptic AMPA-type glutamate receptors at excitatory synapses (Ehrlich and Malinow, 2004) and that decreases in PSD-95 levels lead to decreases in synaptic strength (Elias et al., 2006). Thus, to examine whether the decrease in PSD-95 levels in EAE is associated with deficits in excitatory synaptic transmission in the hippocampal CA1 region we used extracellular recordings to measure basal excitatory synaptic function in the in vitro hippocampal slice preparation. In these experiments the amplitude of presynaptic fiber volleys and the slope of the EPSPs evoked by different intensities of Schaffer collateral fiber stimulation were compared with the input (fiber volley)/output (fEPSP slopes) relationships of basal synaptic transmission. Interestingly, when comparing fiber volley amplitudes ranging between 1.2 and 2.0 mV, maximal fEPSP slopes were significantly lower in hippocampal slices from placebo-treated EAE mice compared with normal. Thus, stimulating the same number of presynaptic fibers (Schaffer collaterals) in slices from placebo-treated EAE mice elicited smaller postsynaptic responses compared with healthy controls. Within this range of fiber volley amplitude (1.2–2.0 mV) testosterone treatment during EAE did not improve this decreased basal synaptic transmission (Fig. 4A). At the highest stimulation strength, however, presynaptic fiber stimulation evoked fEPSPs in slices from testosterone treatment in mice with EAE that were similar to those seen in slices from healthy controls. Thus, when maximal fEPSP slopes were compared across the three groups, there was a significant deficit in postsynaptic responses in slices from EAE mice compared with healthy controls, and this decrease in the postsynaptic response was partially prevented with testosterone treatment during EAE (Fig. 4B,C). Although EAE was associated with deficits in the input/output relationship for basal excitatory synaptic transmission, PPF, a short-term form of synaptic plasticity dependent on presynaptic release probability, was not altered in slices from EAE plus placebo or EAE plus testosterone mice compared with healthy controls (Fig. 4D).
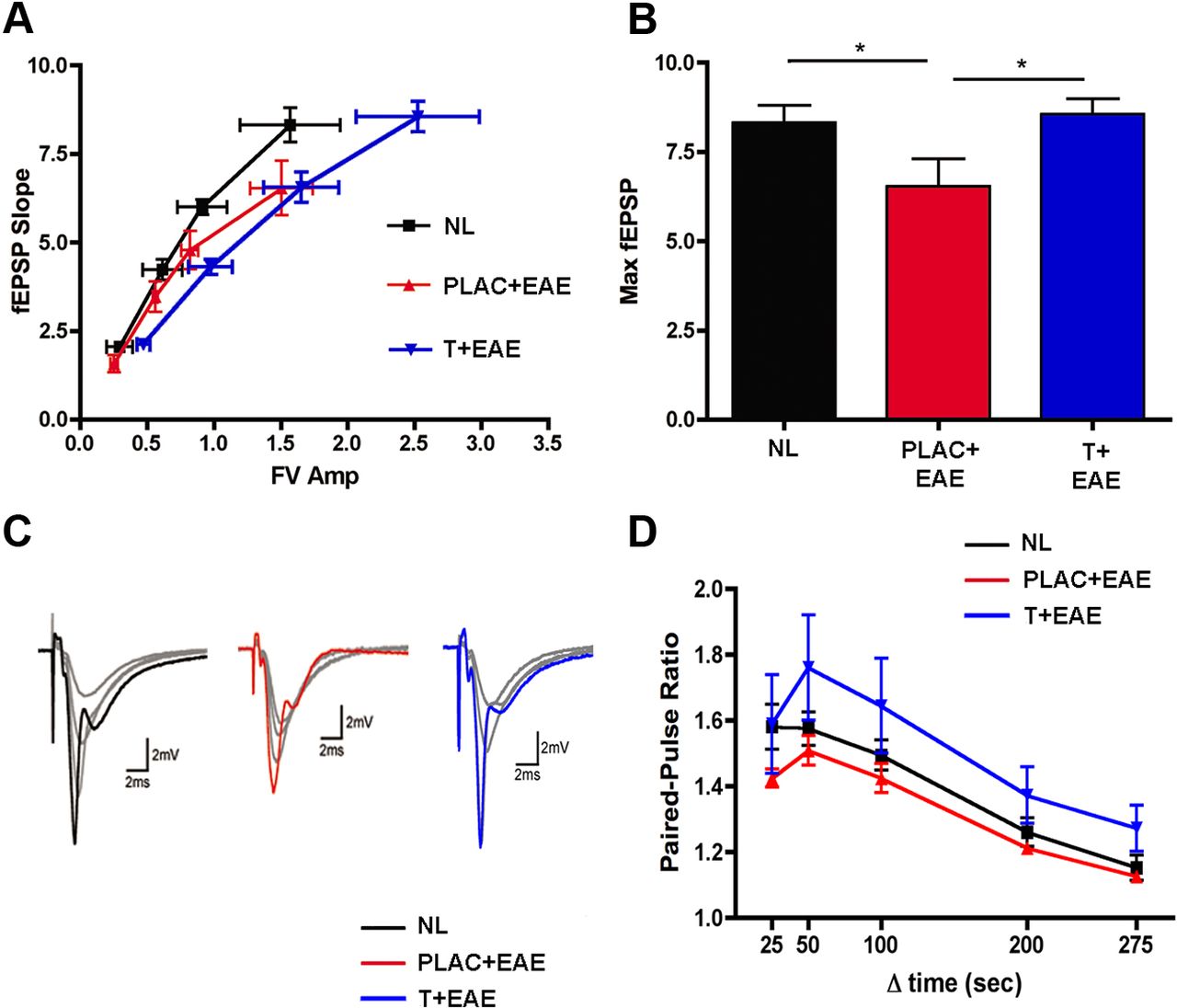
Excitatory synaptic transmission is impaired in EAE, and testosterone treatment prevents this. Input/output relationships were measured by electrophysiology. A, When comparing fiber volley amplitudes ranging between 1.2 and 2.0 mV, maximal fEPSP slopes were significantly lower in EAE (PLAC + EAE, red, n = 10 slices from 3 animals) compared with normal (NL, black, n = 10 slices from 4 animals). With testosterone treatment during EAE (T + EAE, blue, n = 12 slices from 5 animals) increased fiber volley amplitudes were attained (2.0 mV and greater), and fEPSP responses were similar to those of normal controls. B, When maximal fEPSPs were compared across all groups, placebo-treated EAE mice had significantly lower responses as compared with healthy controls, while maximal fEPSPs in testosterone-treated EAE mice were not significantly different from controls. Individual datasets were compared using Student's t test, where * indicated groups significantly different, p = 0.04. C, Sample waveforms depict representative input/output responses recorded from animals in each experimental condition. Each set of waveforms shows fEPSP responses 25, 50, and 75% of maximal fEPSP slope in gray, while each maximal fEPSP slope (100% response) is color coded to experimental condition (black, NL; red, PLAC + EAE; blue, T + EAE). D, Pairs of presynaptic fiber stimulation pulses were delivered with interpulse intervals of 25 ms, 50 ms, 100 ms, 200 ms, or 275 ms). PPF was unchanged in the hippocampal CA1 region of placebo-treated EAE mice (n = 7 slices from 3 animals) and testosterone-treated EAE mice (n = 8 slices from 5 animals), compared with healthy control mice (n = 6 slices from 4 animals).
We also examined the effects of EAE on basal synaptic transmission using whole-cell voltage-clamp techniques to record AMPA receptor-mediated, spontaneous mEPSCs in CA1 pyramidal cells. Although the mean mEPSC amplitude was not found to be significantly different between groups (Fig. 5A,B,D), there was a significant reduction in the mEPSC frequency in placebo-treated mice with EAE (Fig. 5A,C,E). A decrease in mEPSC frequency may indicate either a decrease in the probability of transmitter release or a decrease in the number of synapses. Since PPF was not significantly different in mice with EAE (Fig. 4D), our data suggest that the reduction in mEPSC frequency is most likely due to a reduction in the number of synapses. In this case, testosterone treatment during EAE was not able to prevent the reduction in mEPSC frequency (Fig. 5E).

AMPAR-mediated synaptic current frequency and NMDAR-mediated synaptic current decay rate are reduced during EAE. A, mEPSCs were recorded at −70 mV in the presence of 1.0 μm TTX and 100 μm picrotoxin. B, mEPSC amplitude cumulative probability distribution was not significantly different for cells from healthy control mice (NL, black, n = 17 cells from 5 mice) and placebo-treated EAE mice (PLAC + EAE, red, n = 9 cells from 4 mice). C, The interevent interval cumulative probability distribution in cells from placebo-treated EAE (PLAC + EAE, red) mice was shifted right, compared with healthy control (NL, black) mice, indicating longer interevent intervals between mEPSCs. D, Mean mEPSC amplitude was not considerably altered in cells from placebo- or testosterone-treated mice with EAE (T + EAE, blue, n = 13 cells from 4 mice), compared with healthy control mice. E, mEPSC frequency was strongly reduced, however, in cells from placebo-treated EAE mice and testosterone-treated EAE mice compared with mEPSC frequency in cells from healthy control mice, where *p < 0.05, one-way ANOVA followed by Dunnet's test comparisons to control. F, Examples of evoked EPSCs recorded at holding potentials of −80 mV and +40 mV in CA1 pyramidal cells from healthy control (NL, black), placebo-treated EAE (PLAC + EAE, red), and testosterone-treated EAE (T + EAE, blue) mice. G, NMDA receptor/AMPA receptor ratios of evoked EPSCs recorded at either −80 mV or +40 mV are not altered in EAE. H, The decay of NMDA receptor-mediated EPSCs recorded at +40 mV was significantly faster in cells from placebo-treated EAE mice (PLAC + EAE, red, n = 11 cells from 4 mice) compared with healthy control mice (NL, black, n = 12 cells from 5 mice). Interestingly, NMDAR-mediated synaptic current decay rate was not significantly different in testosterone-treated EAE mice (T + EAE, blue, n = 12 cells from 4 mice), compared with healthy control mice, where *p < 0.05, one-way ANOVA followed by Dunnet's test comparisons to control.
Last, the relative contribution of AMPA and NMDA receptors to evoked EPSCs in CA1 pyramidal cells was compared in mice with EAE and testosterone-treated mice with EAE versus healthy control mice. The ratio of NMDAR- to AMPAR-mediated EPSCs was not different across condition, whether recorded at holding potentials of −80 mV or + 40 mV (Fig. 5F,G). However, the decay rate of synaptic currents at holding potential +40 mV was significantly faster in cells from EAE mice, compared with cells from healthy control mice (Fig. 5H). Here, testosterone treatment during EAE prevented this increase in decay rate, and was statistically similar to EPSC decay rate in cells from healthy control mice (Fig. 5H).
Deficits in excitatory synaptic transmission during EAE significantly corresponded with disruption of PSD-95 integrity
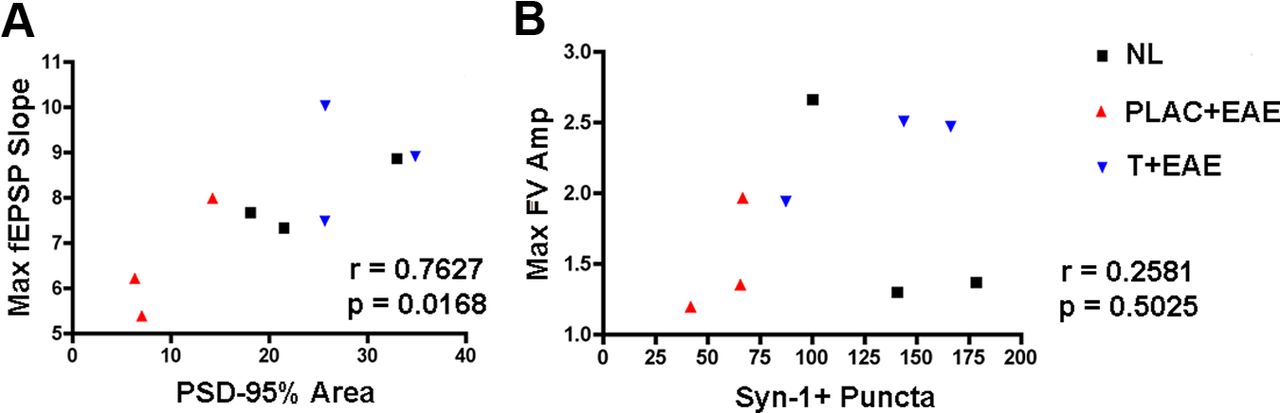
To determine which neuropathological substrate(s) might underlie the observed deficit in basal synaptic transmission in the hippocampus of mice with EAE, we used cross-modality correlations between pathological outcome measures and electrophysiological outcome measures. These comparisons were possible because electrophysiological and pathological outcomes were measured within the same hippocampal tissues. Because we had found that PSD-95 levels significantly correlated with decreases in CA1 stratum radiatum volume, we next compared PSD-95 levels with fEPSP slopes. Lower levels of PSD-95 correlated with decreased field EPSP responses (Table 2; Fig. 6A). In contrast, Syn-1 levels did not correlate with electrophysiological outcome measures (Table 2; Fig. 6B). Thus, these cross-modality correlations demonstrated that PSD-95 levels significantly correlated with excitatory postsynaptic responses.
Correlational coefficients and coefficients of determination between synaptic protein levels and electrophysiological input/output responses
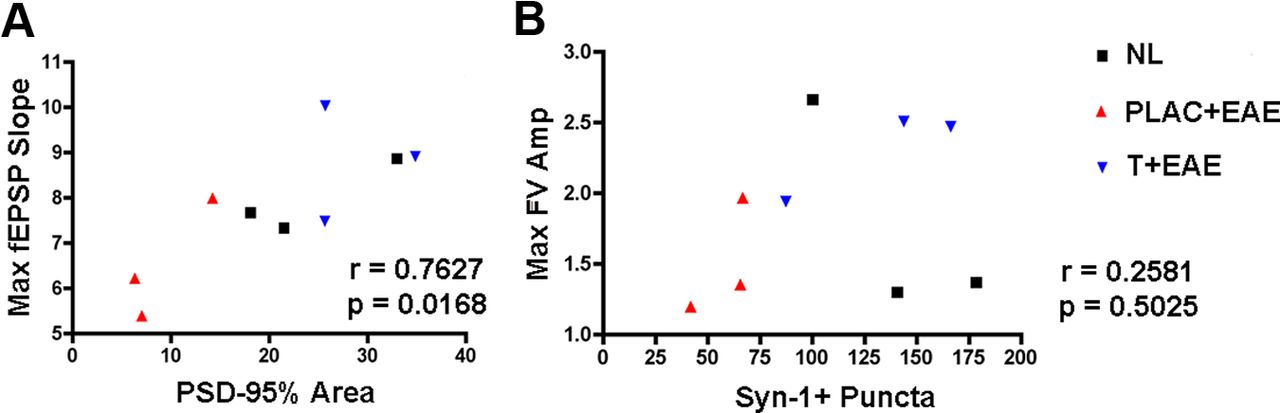
PSD-95 levels are strongly correlated to maximal postsynaptic responses. Linear regression analyses were performed to determine potential relationships between pathology and electrophysiological response. A, PSD-95% area was plotted in the x-axis, while maximal fEPSP slopes corresponding to each animal were plotted on the y-axis. When Pearson's correlation test was calculated across conditions, the correlation coefficient was large, ρ = 0.7627, indicating a strong linear relationship between PSD-95 and fEPSP slope, p = 0.0168, n = 9 mice total. B, Synapsin-1, however, was not significantly correlated to fiber volley amplitude slope, as ρ = 0. 2581, p = 0.5025, n = 9 mice, when compared across all conditions.
Demyelination and microglial activation occur in the hippocampus during EAE, and testosterone treatment prevents both
Focal demyelination is a classic neuropathological finding in spinal cords of mice with EAE. Focal demyelination has also been observed in the hippocampus of MS patients (Geurts et al., 2007; Dutta et al., 2011). Because EAE caused a deficit in hippocampal synaptic function, we next assessed the extent of demyelination within the hippocampal CA1 regions. MBP fluorescence intensity measurements indicated significant reductions in myelin staining within both the stratum oriens and the stratum radiatum of the CA1 region, of placebo-treated EAE mice, compared with healthy controls (Fig. 7A,B,D). In contrast, myelin staining levels in testosterone-treated EAE mice were not decreased as compared with healthy controls. MBP staining was further characterized by double-labeling tissue sections with an axonal protein marker, NF-200. When NF-200 (Fig. 7, green) fluorescent images were merged with MBP (Fig. 7, red) fluorescent images, we observed that in addition to significant decreases in myelin staining, NF-200 staining was also considerably disrupted in mice with EAE compared with healthy controls (Fig. 7E,F). These results demonstrate that diffuse demyelination occurs in the axon-rich regions of the hippocampus, and additional studies confirmed this decreased myelin staining.

Decreased MBP immunoreactivity occurs in the hippocampal CA1 regions of mice with EAE and is preserved with testosterone treatment. A–C, Representative fluorescent images depict MBP immunoreactivity (Cy5-red) in the stratum oriens (so) region of CA1 within healthy (A), placebo-treated EAE (B), and testosterone-treated EAE (C) mice at 40× magnification. EAE caused a significant reduction in myelin staining within the CA1 region of the hippocampus in placebo-treated mice (B), compared with myelin staining in hippocampus of healthy control mice (A, D). In testosterone-treated EAE mice, myelin levels were similar to those found in healthy controls (C). Analysis was conducted on 10× magnification images where MBP percentage area represents total MBP immunoreactivity as a percentage of total CA1 area imaged. One-way ANOVA revealed significant differences in myelin staining across conditions, *p < 0.05, n = 3 per group, and Newman–Keuls post hoc tests indicated that placebo-treated EAE mice were significantly different from both other groups. E–G, Additional magnified images are depicted to verify specificity of MBP stain within each representative hippocampal section, taken from A–C, dashed white squares and rotated clockwise 90°. Here, MBP (red) colocalizes with NF-200 (Cy3-green) to create a bright yellow fluorescence (indicated by arrowheads) which is more often observed in the hippocampal CA1 so of healthy control mice (E), and testosterone-treated EAE mice (G), but not commonly seen in the hippocampal CA1 so of placebo-treated EAE mice (F). Scale bars, 10 μm.
Reactive microglia with infiltration of T-lymphocytes and macrophages characterize the inflammation that occurs in spinal cords of EAE mice. Herein we assessed microglial activation in the hippocampus of mice with EAE. Iba1 is a protein highly expressed by resident microglia in the hippocampus (Kuzumaki et al., 2010), as well as by macrophages (Voskuhl et al., 2009). Thus, Iba1 staining was used to label cells of microglial/macrophage lineage. We previously used globoid versus ramified morphology to distinguish between macrophages and microglia, respectively, in spinal cords of mice with EAE (Voskuhl et al., 2009). Therefore, hippocampal Iba1+ cells were counted, and the morphology of those cells was analyzed. Hippocampal slices from EAE mice demonstrated significantly increased quantities of ramified microglia as compared with healthy controls (Fig. 8A,B,D), while testosterone treatment in EAE mice reduced the number of ramified microglia (Fig. 8C,D). To further characterize reactive microglia in the hippocampus, colabeling experiments were performed combining Iba1 staining with either a neuronal marker, NeuN, or myelin marker, MBP. Interestingly, Iba1+ cells with morphology of reactive microglia were found commonly associated with neuronal perikarya in the CA1 pyramidale (Fig. 8E–G) and also along regions of demyelinated axons (data not shown) in hippocampal slices from placebo-treated EAE mice. We confirmed this increased microglial activation in EAE with repeated immunohistochemical studies on tissue collected at one time point. In addition, consistent with our previous report (Ziehn et al., 2010), there was minimal infiltration of macrophages and T-lymphocytes in hippocampi from EAE mice (data not shown).

Microglial activation was significantly elevated in the hippocampus during EAE, and not during testosterone treatment of EAE. A–C, Fluorescent images depicted here are 10× magnified images representative of hippocampal sections from healthy control mice (A), placebo-treated EAE mice (B), and testosterone-treated EAE mice (C), where cells of microglial/macrophage lineage are stained with Iba1 (Cy3-green) and DAPI (blue). Iba1+ cells with morphology characteristic of reactive microglia were relatively low in healthy controls (A), significantly increased in placebo-treated EAE mice (B), and reduced to levels similar to normal in testosterone-treated EAE mice (C). The number of Iba1+ cells per mm3 were counted and presented as whole numbers (D). E–G, Representative images of microglia (Iba-1, Cy3-green) and neuronal perikarya (NeuN, Cy5-red) of CA1 pyramidale (pyr) layer within the hippocampus of animals from each condition, shown here at 40× magnification, demonstrate that some cells with morphology indicative of reactive microglia associate with pyramidal neurons of CA1, and more so in placebo-treated EAE mice (F). One-way ANOVA revealed significant differences in microglia activation, *p < 0.05, n = 3 per group, and Newman–Keuls post hoc tests showed that placebo-treated EAE mice (PLAC + EAE) were different from mice in other two groups. Scale bars, 10 μm.
We next investigated whether demyelination and microglial activation in the hippocampus were correlated with electrophysiological deficits seen in EAE. When demyelination was compared with electrophysiological input/output responses, there were no significant associations found. Interestingly, the changes in microglial activation were significantly correlated to changes in maximal fEPSP slopes (Table 3) suggesting that increased microglial activation was related to decreased excitatory postsynaptic responses in the CA1.
Correlation coefficients and coefficients of determination between myelin staining, microglial activation, and electrophysiological input/output responses
Testosterone-mediated neuroprotection is unlikely to involve androgen receptor activation
How does testosterone impart neuroprotective benefits within the hippocampus during EAE? Testosterone can either directly activate androgen receptors in the hippocampus, or it may indirectly activate estrogen receptors after being locally converted into estradiol via aromatization (Brodie, 1979). In either case, it is widely known that both estrogens and androgens can have widespread effects in the hippocampus (Cooke and Woolley, 2005; Galea et al., 2008). Other work has previously shown that androgen receptors are distributed largely throughout the CA1, CA2, and dentate gyrus of the hippocampus in adult male mice (Simerly et al., 1990; Young and Chang, 1998). Due to this previously established fact, we conducted additional experiments to see if treatment with DHT, a nonaromatizable androgen, could impart similar neuroprotective benefits as testosterone treatment. Interestingly, DHT treatment did not prevent disease as measured by clinical EAE scoring (Fig. 9A). Furthermore, when CA1 atrophy was measured in placebo-treated versus DHT-treated mice with EAE, we found that DHT was not capable of preventing EAE-mediated CA1 atrophy in the hippocampus (Fig. 9B). These results suggest that direct androgen receptor activation is not a mutually exclusive requirement of testosterone-mediated neuroprotection.

DHT is not capable of ameliorating EAE clinical severity or preventing CA1 atrophy. A, DHT-treated mice with EAE (DHT + EAE, green circles, n = 5 mice) demonstrated clinical scores similar to placebo-treated mice with EAE (PLAC +EAE, red triangles, n = 5 mice), which contrasted from testosterone-treated mice with EAE (T + EAE, blue triangles, n = 5 mice) that had clinical scores ameliorated and not different from placebo-treated healthy control mice (NL, black squares, n = 5 mice). B, When CA1 pyramidal (pyr) layer volume was compared across conditions, DHT-treated mice with EAE (n = 5 mice) had significantly reduced volumes compared with normal healthy controls (n = 35 mice), and DHT animals were not different from placebo-treated mice with EAE (n = 5 mice). This finding is in direct contrast to the effect testosterone had on CA1 pyr layer volume (T + EAE, n = 5 mice), which was to preserve CA1 pyr layer volume to levels similar to healthy control animals. One-way ANOVA demonstrated an effect of condition on EAE clinical score (p = 0.0131, n = 5 mice per group). Newman–Keuls multiple-comparisons post hoc analysis indicated that placebo-treated mice with EAE and DHT-treated mice with EAE both differed in CA1 pyr volume compared with healthy control animals and testosterone-treated mice with EAE, *p < 0.05, n = 5 mice per group.
Therapeutic testosterone administration ameliorates clinical disease severity and restores excitatory synaptic transmission
We next wanted to determine whether therapeutic treatment administration of testosterone (given after disease onset) could prevent clinical walking disability as well as hippocampal synaptic transmission and synaptic pathology. To that end, we designed and performed additional experiments where testosterone treatment was administered after disease onset, when the first clinical signs of disability presented. Here, castrated male mice were immunized using our standard protocol to induce autoimmune encephalomyelitis (Fig. 10A) and scored daily from experimental day 0 forward. When mice began to show signs of clinical disability [ranging from decreased muscle tone in the tail (score of 1) to inability to right itself (score of 2)], they were stratified into three conditions by clinical score. The following day, 10 d postimmunization, mice received a 90 d release pellet containing either testosterone (5 mg) or placebo. In contrast to pretreatment with testosterone, treatment after disease induction did not decrease standard EAE scores until ∼3 weeks after it was initiated, at a time point later in disease (day 35) when comparing testosterone-treated EAE (EAE + T, blue, n = 5 mice) with placebo-treated EAE (EAE + PLAC, red; n = 5 mice) to healthy control mice (Fig. 10B).

Testosterone treatment after EAE induction restores synaptic transmission and corresponding synaptic protein levels within the hippocampus during EAE. A, Experimental design highlighting testosterone (or placebo) treatment after disease induction (day 10) when standard EAE signs first appeared. B, In contrast to pretreatment (Fig. 1), treatment after disease induction did not decrease standard EAE scores until ∼3 weeks after it was initiated, a time point later in disease (day 35) when comparing testosterone-treated EAE (EAE + T, blue, n = 5 mice) with placebo-treated EAE (EAE + PLAC, red, n = 5 mice). C, Maximal fEPSP slopes were reduced in placebo-treated mice with EAE (EAE + PLAC, red, n = 21 slices from 7 mice), but restored in testosterone-treated EAE mice (EAE + T, blue, n = 24 slices from 8 mice) to levels similar to healthy controls (NL, black, n = 15 slices from 5 mice); *p < 0.05, one-way ANOVA with Newman–Keuls multiple comparisons. D, PSD-95% area and Syn-1+ puncta were significantly reduced in EAE mice that received placebo treatment after disease induction (iii and iv, respectively, n = 7 mice) compared with healthy control mice (i and ii, n = 5 mice), but testosterone treatment after EAE induction restored both synaptic protein levels (v and vi, graphs vii and viii, n = 8 mice). One-way ANOVA with Newman–Keuls multiple-comparisons analyses revealed significance where *p < 0.05, **p < 0.01. E, Cross-modality correlation analyses revealed that testosterone-mediated effects on synaptic transmission (maximal fEPSP slope) were significantly correlated to effects on PSD-95 synaptic protein levels, where Pearson's correlation test indicated a strong linear relationship between PSD-95 and fEPSP slope, ρ = 0.63898 p = 0.0032, n = 20 mice total. F, Syn-1+ puncta were not significantly correlated to fiber volley amplitude slope, as ρ = 0.0608, p = 0.8045, n = 20 mice total, when compared across all conditions.
To ascertain whether testosterone treatment given after disease onset could benefit excitatory synaptic transmission, we performed hippocampal slice recordings on two mice per day that were selected at random from experimental day 21 through 40 postimmunization. As expected, excitatory synaptic transmission was decreased in the hippocampus during EAE (Fig. 10C, EAE + PLAC; n = 7 mice) compared with healthy controls (NL, n = 5 mice). Interestingly, maximal fEPSPs were restored to levels similar to healthy control animals in mice that received therapeutic testosterone administration after disease onset (EAE + T, n = 8 mice). We next determined whether the therapeutic effects of testosterone affected synaptic protein levels within the hippocampus (Fig. 10D). Within the same tissue that we recorded from, testosterone also restored synaptic protein levels of Synapsin-1 (Fig. 10D, vi and viii) to levels similar to healthy controls (ii and viii), while placebo-treated EAE mice had significantly reduced Syn-1 levels (iv and viii). PSD-95 protein levels were also restored within the stratum radiatum of CA1 with therapeutic testosterone treatment (v and vii) similar to controls (i and vii), but decreased during placebo treatment of EAE (iii and vii). Cross-modality correlation analyses revealed that testosterone-mediated restoration of synaptic transmission (maximal fEPSP slope) was significantly correlated to restoration of PSD-95 synaptic protein levels (Fig. 10E). The changes in Synapsin-1 puncta numbers were not correlated to changes in maximal fEPSP, however (Fig. 10F). These results confirm our studies by suggesting that testosterone can, in fact, reverse the detrimental effects of EAE on excitatory synaptic transmission and also reverse changes in synaptic protein levels. These studies not only strengthen the impact of our findings, but also give tremendous credence to the use of testosterone in disease intervention.
Discussion
Despite the fact that cognitive deficits occur in >50% of MS patients, and hippocampal-dependent memory impairment occurs in >30%, there is currently no treatment designed to prevent this disability in MS. Recently, a postmortem study in MS revealed that hippocampal demyelination was associated with altered levels of expression of a variety of molecules potentially involved in pathogenesis (Dutta et al., 2011). However, since cognitive deficits were not established before autopsy, there were no correlations of molecular changes with functionally significant cognitive impairment. Imaging studies in MS have demonstrated hippocampal abnormalities that were indeed correlated with hippocampal-dependent memory impairment (Sicotte et al., 2008; Anderson et al., 2010), but imaging studies are inherently limited by their lack of cellular specificity with respect to ascertaining underlying neuropathogenesis. Animal models can reveal insight into cellular structure function relationships since cellular neuropathology can be assessed in a setting of established functional impairment. Thus, herein we used cross-modality correlations between hippocampal neuropathology and hippocampal function in EAE, the most widely used animal model for MS.
Assessing hippocampal-dependent learning and memory in EAE is challenging since the disease entails motor impairment. Previously we had shown that EAE mice performed worse than healthy control mice in a hippocampal-dependent spatial memory task (Ziehn et al., 2010). In the current study we used an electrophysiological approach to examine hippocampal function during EAE for two reasons. First, using electrophysiology we could assess hippocampal function without depending on animal locomotion, and second, electrophysiology can offer novel insight into the cellular and molecular mechanisms underlying deficits in hippocampal-dependent learning and memory. Herein, we demonstrate for the first time that excitatory synaptic transmission is decreased in EAE and that these changes involve both AMPA and NMDA receptor-mediated changes. Furthermore, our study demonstrates that several key neuropathological outcomes are strongly correlated with deficits in excitatory synaptic transmission in the hippocampus. Specifically, deficits in excitatory synaptic transmission were correlated with CA1 atrophy, decreased PSD-95 levels, and increased microglial activation.
Our finding of hippocampal CA1 atrophy here is in line with previous findings in both EAE (Ziehn et al., 2010) and MS (Sicotte et al., 2008). However, in the current study we were able to determine which regions of the CA1 were significantly reduced in size by using enhanced resolution microscopy and unbiased stereology. For the first time we demonstrate that CA1 atrophy includes significant decreases in volumes of dendritic layers, and furthermore, that PSD-95 levels are strongly correlated with these volumetric changes. PSD-95 is of particular interest, because it is a member of a dense network of proteins found at excitatory synapses that mediate protein–protein interactions in the synaptic membrane critical for synaptic transmission (Kim and Sheng, 2004). In addition, PSD-95 has a unique role in AMPA receptor trafficking at excitatory synapses in adult mouse hippocampi (Ehrlich and Malinow,, 2004). Acute removal of PSD-95 using short hairpin RNA targeted deletion in neurons significantly impairs excitatory synaptic transmission in cultured hippocampal neurons (Elias et al., 2006), and results from studies of PSD-95 knock out in mice indicate that deletion of PSD-95 causes an impairment of AMPA receptor-mediated synaptic transmission in hippocampal slices (Béïque et al., 2006; Carlisle et al., 2008). Thus, the deficits in basal, AMPA receptor-mediated synaptic transmission seen in EAE may, at least in part, be due to alterations in PSD-95 expression. Interestingly, we find that the decay time course of NMDA receptor-mediated EPSCs is significantly faster in CA1 pyramidal cells during EAE. This effect is unlikely to involve changes in PSD-95 expression, as PSD-95 is not thought to be importantly involved in NMDA receptor trafficking at excitatory synapses (Elias and Nicoll, 2007). Instead, the decay time course of NMDA receptor-mediated EPSCs is highly dependent on receptor subunit composition (Cull-Candy and Leszkiewicz, 2004). Thus, one possibility is that EAE is associated with a decrease in the expression of GluN2B subunit-containing NMDA receptors, which exhibit slower decay rates than GluN2A subunit-containing receptors.
Synaptic stripping has been shown in the spinal cords of mice with EAE (Trapp et al., 2007). It is known that activated microglia can release pro-inflammatory cytokines and contribute to synaptic stripping (Pickering and O'Connor, 2007). Here, we have demonstrated increased microglial activation in the hippocampus during EAE, and that these microglia associate with neuronal perikarya and axons in the CA1. Furthermore, microglial activation was directly correlated with deficits in excitatory postsynaptic transmission. Our findings are consistent with previous reports that activated microglia in the hippocampus in non-EAE conditions contribute to synaptic stripping (Schafer and Stevens, 2010) and impaired synaptic transmission (Hanak et al., 2004).
This study demonstrates that testosterone treatment either before or after EAE disease induction partially restores deficits in synaptic transmission, preserves presynaptic and postsynaptic integrity, and prevents hippocampal pathology. A growing body of evidence suggests that testosterone enhances hippocampal synaptogenesis (Frye et al., 2004), and moreover, that testosterone is important to the maintenance of normal synaptic spine density in the hippocampus, since castration leads to decreased spine density and testosterone treatment restores synaptic spine formation in the castrated animal (MacLusky et al., 2006). Testosterone can bind either to androgen receptors, which are widely distributed through the mouse hippocampus (Simerly et al., 1990; Young and Chang, 1998), or it can bind to estrogen receptors after being aromatized to estradiol (Brodie, 1979). It is widely known that both androgens and estrogens can have widespread effects in the hippocampus (Cooke and Woolley, 2005; Galea et al., 2008). Interestingly, treatment with a nonaromatizable androgen, DHT, did not prevent the EAE-mediated decrease in CA1 volume. This would suggest that testosterone might be acting principally through its conversion to estradiol and subsequent activation of the estrogen receptors α and/or β.
Indeed, there is substantial evidence suggesting that estrogens are both anti-inflammatory and neuroprotective during EAE. Estrogen treatment during EAE significantly reduces clinical severity of EAE in several strains of mice, and has been shown to reduce inflammatory responses of the immune system (Kim et al., 1999; Bebo et al., 2001; Ito et al., 2001; Liu et al., 2002, 2003; Matejuk et al., 2005). These data suggest that anti-inflammatory processes are upregulated by estrogen treatment. Cytokines, chemokines, extracellular matrix proteins (Gold et al., 2008), antigen presentation (Matejuk et al., 2005; Polanczyk et al., 2003), and dendritic cell function (Subramanian et al., 2003; Du et al., 2011) are all significantly modulated by estrogen treatment. The anti-inflammatory effects of estrogen, do not, however, exclude estrogen-mediated neuroprotective effects. Aside from ameliorating EAE disease severity and inducing favorable changes in immune system production of cytokines, estrogen treatment also decreases white matter inflammation, demyelination, and axonal loss in the spinal cord during EAE (Morales et al., 2006). Estrogen treatment is capable of decreasing neuronal pathology in spinal cord gray matter (Tiwari-Woodruff et al., 2007) and modulating neuroprotective effects via astrocytes (Spence et al., 2011). Indeed, we have recently demonstrated that treatment with a pregnancy estrogen, estriol, can prevent deficits in excitatory synaptic transmission in the hippocampus during EAE (Ziehn et al., 2012). Interestingly, estriol treatment was also capable of preserving levels of synaptic proteins that are known to orchestrate functional synaptic transmission within the hippocampus.
Estriol is a therapeutic candidate in MS because it has widespread effects on the immune system and the CNS (Gold and Voskuhl, 2009). MS patients have significantly decreased relapse rates during the third trimester of pregnancy, when estriol levels are most elevated, and relapse rates rebound during the postpartum period coinciding with an abrupt decline in serum estriol levels (Confavreux et al., 1998). In nonpregnant MS patients, estriol treatment has been shown to significantly reduce gadolinium-enhancing lesion number and volumes measured by MRI (Sicotte et al., 2002). Estriol treatment also significantly improved cognitive function as measured by the paced auditory serial addition task in relapsing-remitting MS patients, although this may have been in part due to a practice effect.
Thus surmounting evidence suggests that estrogens can have direct neuroprotective effects during autoimmune demyelinating disease. Since testosterone is thought to improve cognition at least in part through its conversion to estrogen, and subsequent binding to estrogen receptors, both sex hormones should be considered as candidate treatments to improve cognition during MS. One could potentially tailor sex hormone treatments in accord with desirable or undesirable effects in each gender, using estrogen treatment for women and testosterone treatment for men, while ending up with a common mechanism of hormone action on estrogen receptors in the brain. For these reasons, and considering the findings of this study, further research is warranted regarding testosterone treatment in MS and possibly other neurodegenerative diseases that target synapses in the hippocampus (Driscoll and Resnick, 2007).
Footnotes
This work was supported by the National Institutes of Health (NIH) Grant K24NS052117 and National Multiple Sclerosis Society Grants RG4033 and RG4364 (R.R.V.), as well as funding from the Skirball Foundation, the Conrad Hilton Foundation, and the Sherak Family Foundation. M.O.Z. was supported in part by the UCLA Laboratory of Neuroendocrinology, funded by NIH Grant T32 HD07228-26, and in part by the National Science Foundation GK-12 UCLA SEE-LA program, Grant DGE-074241. This work was also supported by the NIH Grant MH609197 (T.J.O.). We would like to acknowledge Noriko Ito and Erin Gray for their technical assistance and contribution to this work.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Rhonda R. Voskuhl, University of California, Los Angeles, 635 Charles E. Young Drive South, Neuroscience Research Building 1, Room 475D, Los Angeles, CA 90095. rvoskuhl{at}ucla.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}