

Abstract
TrkA high-affinity receptors are essential for the normal development of sympathetic paravertebral neurons and subpopulations of sensory neurons. Paravertebral sympathetic neurons and chromaffin cells of the adrenal medulla share an ontogenetic origin, responsiveness to NGF, and expression of TrkA. Which aspects of development of the adrenal medulla might be regulated via TrkA are unknown. In the present study we demonstrate that mice deficient for TrkA, but not the neurotrophin receptor TrkB, show an early postnatal progressive reduction of acetylcholinesterase (AChE) enzymatic activity in the adrenal medulla and in preganglionic sympathetic neurons within the thoracic spinal cord, which are also significantly reduced in number. Quantitative determinations of specific AChE activity revealed a massive decrease (−62%) in the adrenal gland and a lesser, but still pronounced, reduction in the thoracic spinal cord (−40%). Other markers of the adrenal medulla and its innervation, including various neuropeptides, chromogranin B, secretogranin II, amine transporters, the catecholamine-synthesizing enzymes tyrosine hydroxylase and PNMT, synaptophysin, and L1, essentially were unchanged. Interestingly, AChE immunoreactivity appeared unaltered, too. Preganglionic sympathetic neurons, in contrast to adrenal medullary cells, do not express TrkA. They must, therefore, be affected indirectly by the TrkA knock-out, possibly via a retrograde signal from chromaffin cells. Our results suggest that signaling via TrkA, but not TrkB, may be involved in the postnatal regulation of AChE activity in the adrenal medulla and its preganglionic nerves.
- adrenal gland
- spinal cord neurons
- acetylcholinesterase
- chromaffin cells
- neurotrophin receptors
- knock-out mice
TrkA is a high-affinity neurotrophin receptor that mediates essential actions of nerve growth factor (NGF) on subsets of sympathetic and sensory neurons as well as CNS forebrain cholinergic neurons (Thoenen and Barde, 1980; Levi-Montalcini, 1987; Barde, 1989;Snider, 1994). In rodents, targeted mutations of the genes coding for TrkA (Smeyne et al., 1994), NGF (Crowley et al., 1994) or blockade of endogenous NGF with neutralizing antibodies (Angeletti et al., 1972;Thoenen, 1972) cause the loss of paravertebral sympathetic neurons, thereby demonstrating the essential roles of TrkA and NGF in their development.
Paravertebral sympathetic neurons and the neuroendocrine chromaffin cells of the adrenal medulla and paraganglia share an origin from the neural crest (cf. Unsicker, 1993), responsiveness to exogenous NGFin vitro and in vivo (Unsicker et al., 1978; Aloe and Levi-Montalcini, 1979; Müller and Unsicker, 1986), and expression of TrkA (Snider, 1994; this study). It is not clear, however, which aspects of development of the adrenal medulla may be regulated via TrkA. Transgenic mice deficient for each of the known members of the Trk receptor family have been generated recently (Klein et al., 1993, 1994; Smeyne et al., 1994) (for review, see Klein, 1994;Snider, 1994). Whether the adrenal medulla is affected in any of these knock-outs has not been reported.
Beyond cell survival, transmitter synthesis, and neurite growth, TrkA-mediated mechanisms have long been known to regulate expression of cell surface molecules as, e.g., neural cell adhesion molecule (N-CAM) (Prentice et al., 1987), NGF-inducible large external protein (NILE) (McGuire et al., 1978), and various proteases (Machida et al., 1989,1991). Acetylcholinesterase (AChE; E.C. 3.1.1.8) is a prominent intra- and extracellularly located enzyme in cholinergic and many noncholinergic neurons, including chromaffin cells (Lewis and Shute, 1969; Massoulié et al., 1993). Its principal function in cholinergic tissues is the hydrolysis of acetylcholine (Massouliéet al., 1993), but other functions also have been discussed (Small, 1989; Layer and Willbold, 1995). NGF has long been known to regulatein vitro acetylcholinesterase activity in PC12 pheochromocytoma cells and adrenal chromaffin cells (for references, see Discussion).
Adrenal medullary chromaffin cells receive a prominent cholinergic innervation from preganglionic sympathetic neurons, which, in their vast majority, are located in the intermediolateral (IML) column of the thoracic spinal cord (Kesse et al., 1988; Strack et al., 1988; Pyner and Coote, 1994a,b) (for review, see Parker, 1996). Nerve fibers, presumably of preganglionic origin, can be found in the rat adrenal gland as early as embryonic day (E) 15 (Millar and Unsicker, 1981). These nerve fibers become AChE-positive during the first postnatal week (Millar and Unsicker, 1981), and a functional innervation of chromaffin cells, i.e., discharge of catecholamines by neurogenic stimuli, commences in rat at the end of the first week (Slotkin, 1986; Parker et al., 1988).
The present study shows that mice deficient in the TrkA, but not the TrkB receptor, display a significant reduction in AChE activity in the adrenal medulla and its preganglionic innervation. In addition, numbers of preganglionic sympathetic neurons in the IML column of the spinal cord are reduced significantly in TrkA-deficient, but not TrkB-deficient, animals.
MATERIALS AND METHODS
TrkA- and TrkB-deficient (−/−) and wild-type (+/+) mice aged 0–12 d were used. Numbers of animals per phenotype were postnatal (P) P0, n = 15; P3, n = 15; P7,n = 25; and P12, n = 25. The TrkA and TrkB knock-out mice were generated by breeding heterozygous mutant mice kept on a mixed 129/sv × C57BL/6 background. Standard procedures (Laird et al., 1991) were used for the genomic DNA extraction from tail biopsies of mice. We determined TrkA and TrkB genotypes by PCR amplification using, respectively, a common 5′ primer (5′-GACCCTGCACTGTCGAGTTTGC-3′) and either a 3′ primer for wild-type allele (5′-CGGACCTCAGTGTTGGAGAGCTGG-3′) or a primer from the pgk-1 promoter of the neo cassette (5′-GCTCCCGATTCGCAGCGCATCG-3′) and a common 5′ primer (5′-TCGCGTAAAGACGGAACATGATCC-3′) and either a 3′ primer for the wild-type allele (5′-AGACCTGATGAGTGGGTCGCC-3′) or a 3′ primer from the pgk-1 promoter of the neo cassette (5′-GATGTGGAATGTGTGCGAGGCC-3′). The PCR product was analyzed on a 1.5% agarose gel.
In situ hybridization of TrkA mRNA. Wild-type mice (P6,n = 2; P12, n = 2; adult,n = 2) were anesthetized and perfused with 4% paraformaldehyde (PFA). Adrenal glands were dissected, post-fixed (12 hr), and processed for paraffin embedding. Deparaffinized sections (7 μm) were rehydrated and washed in 0.83% NaCl and in PBS. Then sections were post-fixed for 10 min in 4% PFA, washed two times in PBS, and incubated for 30 min with proteinase K (20 μg/ml in 50 mm Tris/0.5 m EDTA), followed by washes in PBS/0.2% glycine and PBS. Sections again were post-fixed for 10 min in 4% PFA, rinsed in PBS and distilled water, and thereafter incubated for 10 min in 1.3% triethanolamine/0.31% acetic anhydrate in 0.05N HCl. Finally, slides were washed in PBS and 0.83% NaCl, dehydrated, and air-dried.
Hybridization was performed in 50% formamid, 0.3 m NaCl, 20 mm Tris, pH 7.5, 5 mm EDTA, 10% dextran sulfate, 1× Denhardt’s solution, 0.5 mg/ml total yeast tRNA, and 10 mm dithiothreitol (DTT) with 1 × 107 cpm of 35S-UTP-labeled cRNA probe, which was transcribed with T7 polymerase from pRB6, a pBS KS− carrying a 398 bp insert from the TrkA receptor. Sections were hybridized overnight at 60°C in a humidified chamber. On the next day slides were washed for 1 hr at 55°C in 2× SSC, 50% formamid, and 10 mm DTT and then for 1 hr at 55°C in 2× SSC, 50% formamid, and 10 mm DTT. Subsequently, slides were rinsed three times for 10 min at 37°C in NTE buffer (0.5 m NaCl, 10 mmTris, and 5 mm EDTA) and then incubated for 30 min at 37°C with 20 μg/ml RNase A in NTE buffer. After a 15 min processing in NTE buffer, slides were washed for 1 hr at 55°C in 2× SSC, 50% formamid, and 10 mm DTT and then for 15 min at room temperature (RT) in 0.2× SSC. After dehydration, sections were air-dried, dipped in Kodak NTB-2 emulsion (diluted 1:1 in water), exposed for 4 weeks at 4°C, developed, fixed, and counterstained with hematoxylin.
AChE histochemistry. Staining was performed according to a modification of the direct coloring thiocholine method of Karnovsky and Roots (1964) for histochemical detection of AChE activity (Andräand Lojda, 1986). Adrenal glands and the full length of the thoracic spinal cord were removed from perfused TrkA (−/−), TrkB (−/−), and wild-type (+/+) animals (4% PFA in phosphate buffer, pH 7.4), cryoprotected (30% sucrose), frozen on dry ice, and cut into 20-μm-thick sections. Sections were mounted on slides and stained for 1 hr at 37°C in the following solution (60 ml): 30.0 mg of acetylthiocholine iodide (Serva Feinbiochemica, Heidelberg, Germany), 44.4 ml of 0.1 m Tris-maleate buffer, pH 5.0 (containing 0.1% Triton X-100), 6.0 ml of 0.4 m sodium citrate, 6.0 ml of 0.12 m copper sulfate, 3.0 ml of 0.16 mpotassium ferricyanide, and 0.6 ml of 10−3mtetraisopropylpyrophosphoramide (iso-OMPA; Sigma, St. Louis, MO). Sections from knock-out and wild-type animals were always stained in parallel.
Immunocytochemistry. Perfusion, fixation, and preparation of sections were performed as described above. Cryostat sections (14, 20 μm) were mounted on gelatin-coated slides, dried at RT (for 30 min), and placed in 0.1 m phosphate buffer (PB), pH 7.4. Nonspecific bindings were blocked by preincubation with 5% normal goat serum and 0.1% Triton X-100 in PB for 1 hr at RT. Sections were immunostained as follows: (1) incubation with primary antiserum (for details, see Table 1) and diluted in PB (containing 2% normal goat serum, 1% bovine serum albumin, and 0.1% Triton X-100) for 12 hr at RT; (2) incubation with Cy3-conjugated goat anti-rabbit IgG (Dianova, Hamburg, Germany) and diluted 1:2000 in PB for 1 hr at RT. Controls were performed by using rabbit normal serum or by omitting the respective antiserum. Finally, all sections were rinsed three times in PB, dried, and embedded in Kaiser’s glycerol gelatin.
Polyclonal primary antibodies used for immunocytochemistry
Western blot analysis. Fresh adrenal glands from wild-type (+/+), TrkA (+/−;−/−), and TrkB (−/−) animals (P6; pooled from two animals per genotype) were homogenized in the presence of detergent, and proteins were separated on a 7.5% acrylamide-containing SDS-gel (2 μg/lane) in a mini chamber (Bio-Rad, Richmond, CA). After transfer to nitrocellulose, the proteins were incubated with a monoclonal AChE antibody (diluted 1:2500; Transduction Laboratories, Lexington, KY) overnight. The primary antibody was visualized via a mouse Vectastain kit (Vector Laboratories, Burlingame, CA). Finally, the bands were analyzed by computer densitometry (ImageQuant).
Determination of AChE activity. AChE activity was determined in homogenates of fresh adrenal glands (pooled from three animals) and the thoracic spinal cord by a microtiter plate-adapted modification of the Ellman method (Ellman et al., 1961) at 405 nm (20 min). The substrate concentration for acetylthiocholine iodide was 1.5 mm. To inhibit any butyrylcholine esterase activity, we determined the AChE activities in the presence of 0.1 mmiso-OMPA. The protein content in each of the homogenates was quantified according to the Bradford method (1976), using bovine serum albumin as a standard. Results were given as mean values ± SEM and tested for significance by Student’s t test.
Determinations of catecholamines. Catecholamines of single adrenal glands (P6) were quantified by high performance liquid chromatography (HPLC) and electrochemical detection essentially as described by Müller and Unsicker (1981). The amounts of catecholamines were averaged and the SEM was calculated. Statistical significance was determined by Student’s t test.
Cell counts. Nissl-stained thoracic spinal cord sections (TrkA −/−, n = 4; TrkB −/−, n = 3; wild-type +/+, n = 3) were used for determining numbers of sympathetic preganglionic neurons (100 adjacent cross sections per animal, 20 μm, segments T8–T10). To identify sympathetic preganglionic neurons reliably, we counterstained Nissl-stained sections weakly by AChE histochemistry as described above. Only neurons with a clearly visible nucleus were counted, and the total number of labeled neurons was estimated according to Abercrombie’s formula (Konigsmark, 1970). Results were given as mean values SD and tested for statistical significance by Student’s t test.
RESULTS
TrkA is expressed in the developing and adult mouse adrenal medulla
To establish that NGF responsiveness and high-affinity binding of NGF to chromaffin cells (for references, see Discussion) reflect an expression of TrkA, we performed in situhybridization studies. Figure 1 shows the localization of TrkA mRNA and corresponding sense controls in mouse adrenal glands at postnatal ages P6 (Fig. 1a,b), P12 (Fig.1c,d), and in the adult (Fig. 1e,f). During all postnatal ages, labeling was confined to the center portion of the gland, consistent with the expression of TrkA within the adrenal medulla and its chromaffin cells. No signal was detectable at P0 (data not shown). Adrenal chromaffin cells did not express TrkB at these ages in mice (data not shown).

In situ hybridization of TrkA mRNA using an antisense probe (a, c, e) and a sense control (b, d, f) in the P6, P12, and adult mouse adrenal glands shows specific labeling of the adrenal medulla (am), which is particularly prominent over clusters of chromaffin cells. The surrounding cortex (ac) shows background labeling. Scale bars, 200 μm.
AChE histochemical staining of the adrenal medulla is reduced in TrkA (−/−) mice
Figure 2 illustrates the temporal development of histochemically demonstrable AChE activity in the early postnatal adrenal gland of wild-type mice (+/+; Fig. 2a,c,e) and animals deficient for TrkA (−/−; Fig. 2b,d,f). At postnatal days P0 (data not shown) and P3 (Fig. 2a,b), there were no detectable differences in AChE staining between wild-type and TrkA knock-out mice. At these early ages, AChE activity was very weak but clearly confined to the adrenal medulla and nerve fibers traversing the cortex. (Fig. 2a,b). In wild-type animals (Fig.2c,e) AChE activity clearly was increased at P7 and P12, as compared with P3, and associated with cells, fiber bundles, and delicate strands of axons innervating the adrenal medulla. In TrkA (−/−) mice a dramatic decrease in AChE histochemical staining became apparent at P7 (Fig. 2d), resulting in an almost complete loss of adrenal AChE staining in P12 animals (Fig.2f).

Localization of AChE activity by histochemistry in the adrenal gland. At P3 (a, b), there are no differences in AChE activity and localization between wild-type (+/+) and TrkA knock-out (−/−) animals. The gross morphology of the adrenal gland apparently is unchanged in the knock-outs. At P7 (c, d) and P12 (e, f), AChE activity is reduced in TrkA-deficient mice (d, f), as compared with wild-type (c, e) animals. Scale bars, 200 μm.
AChE histochemical staining of preganglionic sympathetic neurons, but not motoneurons, in the spinal cord is reduced in TrkA (−/−) mice
To investigate whether the reduction in AChE activity of adrenal medullary nerve fibers in TrkA (−/−) mice was accompanied by a decrease of AChE activity within the perikarya of these neurons and specific for this neuron population, we studied the thoracic spinal cord, where these neurons are located in the IML column (Strack et al., 1988). Figure 3a–d shows that within the thoracic spinal cord of TrkA (−/−) mice (P12) AChE staining intensity clearly was reduced, as compared with wild-type specimens. This reduction was very pronounced in cell bodies of preganglionic sympathetic neurons located in the IML column and in nerve fibers within the dorsal horn. AChE staining in motoneurons appeared to be unaffected. As in the adrenal medulla, alterations in spinal cord AChE staining became apparent at P7 and progressed toward P12 (data not shown).

Localization of AChE activity by histochemistry in thoracic spinal cord. In TrkA-deficient mice (b), there is a strong reduction of AChE activity in autonomic nuclei as well as in the superficial layers of the dorsal horn (DH), as compared with the wild-type (a). IML, Intermediolateral column;NC, nucleus centralis; VH, ventral horn.c, Longitudinal section of thoracic spinal cord (T7–T10) showing AChE-positive preganglionic sympathetic neurons located in IML and NC of a wild-type animal. d, Both the number of reactive neurons and their AChE activity are decreased dramatically in TrkA knock-out mice. Scale bars, 200 μm.
AChE histochemical staining is unaltered in adrenal gland and spinal cord IML neurons of TrkB (−/−) mice
To exclude that alterations in AChE activity seen in adrenal gland and spinal cord of TrkA-deficient mice were unspecific, resulting from severe illness of these animals, we investigated TrkB (−/−) mice that also died during the early postnatal period. Patterns and intensities of AChE staining in adrenal medullary nerve fibers and thoracic spinal cord IML neurons were undistinguishable in TrkB (−/−) and wild-type (+/+) mice (Fig. 4a–d). This result suggests that TrkA, but not TrkB, is involved in the postnatal regulation of AChE activity in preganglionic sympathetic neurons of the spinal cord.

AChE histochemical staining in adrenal medulla (a, b) and thoracic spinal cord (T7–T10; c, d) of TrkB-deficient mice, as compared with wild-type littermates. At P12 there is no difference in AChE activity and localization detectable between TrkB knock-outs (b, d) and wild-type controls (a, c). IML, Intermediolateral column; NC, nucleus centralis. Scale bars, 200 μm.
Quantitative determination of specific AChE activity reveals a pronounced reduction in adrenal gland and thoracic spinal cord homogenates of TrkA (−/−) mice, but not in TrkB-deficient mice
Quantitative analysis of AChE activity in homogenates from whole adrenal glands provided evidence that the decrease in AChE staining intensity seen in TrkA (−/−) mice was, in fact, attributable to a substantial reduction in AChE activity (P7; −62% relative to wild-type littermates, Fig. 5a). In the thoracic spinal cord of the same animals, a 40% decrease in specific AChE activity could be demonstrated (Fig. 5b). In adrenal glands and thoracic spinal cords of TrkB knock-outs, the specific AChE activity was not altered significantly (Fig. 5a,b).

Biochemical determination of AChE activity in homogenates. At P7, specific AChE activity is decreased significantly (−62%) in the adrenal gland (a) of TrkA knock-out animals (+/+, n = 3; −/−, n = 3; *p > 0.05). In thoracic spinal cord (b), there is a 40% reduction of specific AChE activity (+/+, n = 12; −/−, n = 9) relative to the wild-type. In contrast, levels of AChE activity of adrenal glands and spinal cords in TrkB knock-out animals are not altered significantly. Error bars, ± SEM.
Chromaffin cell-associated markers detected by immunocytochemistry are not changed overtly in TrkA (−/−) and TrkB (−/−) mice
To monitor other putative deficits of adrenal medullary development in TrkA knock-outs, we used immunocytochemistry with antibodies to a number of markers associated with chromaffin cells and their preganglionic nerve fibers (see Table 2). Neuropeptide Y (NPY) immunoreactivity was present in most, if not all, adrenal chromaffin cells of wild-type mice as well TrkA- and TrkB-deficient animals at P0, P7, and P12. There was a minor, yet inconsistent, increase in the intensity of immunofluorescence in the knock-out animals (Fig. 6c,d). Subpopulations of chromaffin cells displayed immunoreactivities for somatostatin, met-enkephalin, and galanin, which were weak or nonexistent at P0 and P6 but clearly apparent at P12, with no notable differences between wild-type and knock-out animals. Immunoreactivities for synaptophysin and the adhesion molecule L1 were associated with fibers and varicosities surrounding chromaffin cells and cell clusters. Both the distributional patterns and fluorescence intensities were unaltered in the knock-out animals. Likewise, patterns and intensities of the immunoreactivities for tyrosine hydroxylase (TH) and phenylethanolamineN-methyltransferase (PNMT), chromogranin B, secretogranin II, and the vesicular monoamine transporters I and II (VMAT-1, VMAT-2) were not affected by the TrkA and TrkB deficits.
Summary of semiquantitative data revealed by immunocytochemistry

AChE-immunoreactive nerve fibers (a, b) and NPY immunoreactivity (c, d) in the adrenal medulla. At P7 (a, b), density and distribution pattern of AChE-immunostained fibers are not distinguishable in wild-type (a) and TrkA knock-out mice (b). NPY immunoreactivity in chromaffin cells is increased slightly in knock-out, as compared with wild-type, mice at P12. Scale bars, 100 μm.
Immunocytochemical staining of AChE is unaltered in adrenal medullae of TrkA (−/−) mice
Given the lack of alterations in any of the above axonal and neuroendocrine markers, we investigated whether changes in AChE staining using enzyme activity as an indicator were reflected in alterations of immunocytochemically demonstrable AChE protein. As shown in Figure 6a,b, the polyclonal antibody to AChE clearly revealed the localization of AChE in nerve fibers supplying adrenal medullary cells without showing any differences in the intensities of the immunocytochemical staining. These results suggest that the TrkA receptor knock-out clearly affects the activity of the enzyme, without affecting the presence of preganglionic nerve fibers within the adrenal gland. Moreover, expression of the protein and enzyme activity seems to be differentially regulated.
Western blot analysis of AChE protein
To support further the notion that AChE immunoreactivity in adrenal glands of TrkA (−/−) animals was primarily unchanged, we performed Western blot analysis. As shown in Figure 7, the authentic 68 kDa band of AChE is equally prominent in homogenates from wild-type, TrkA hetero- and homozygotes, and TrkB homozygote mice. Densitometric analysis revealed a 3.2% decrease of the band from TrkA (−/−) mice, as compared with wild-type littermates.

Western blot analysis showing immunoreactive 68 kDa AChE protein in adrenal glands of P6 wild-type, TrkA hetero- and homozygote, and TrkB-deficient animals.
Cell counts reveal a reduction in IML neuron numbers in TrkA-deficient, but not in TrkB-deficient, mice
Cell counts performed on double-stained (Nissl/AChE) serial cryosections (100 adjacent cross sections/animal) through thoracic spinal cord levels T8–T10 of TrkA-deficient mice revealed a reduction in IML neuron numbers by 41.5%, as compared with wild-type controls (Figs. 8, 9). In contrast, the number of IML neurons in TrkB-deficient mice was not affected (Figs. 8, 9).

Cell counts of serial transverse sections through spinal cord segments T8–T10 reveal a 41.5% reduction in IML neuron numbers in TrkA (−/−), as compared with wild-type mice. In TrkB-deficient mice numbers of IML neurons are not affected,p > 0.01.
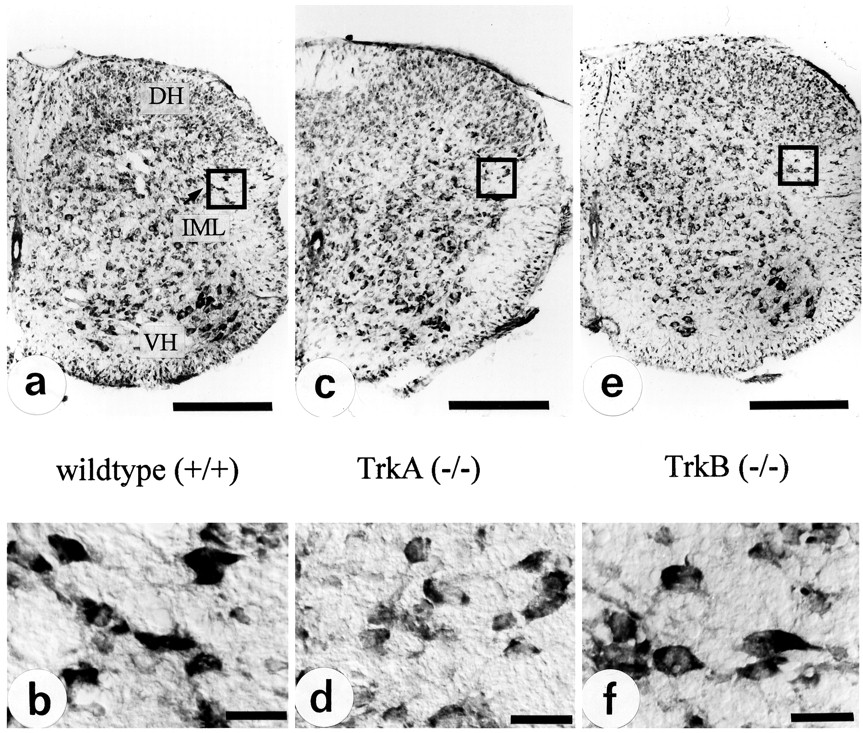
Illustration of the quantitative data presented in Figure 7. Nissl-stained sections from spinal cord segments T8–T10 (a, c, e) and IML (b, d, f).DH, Dorsal horn; IML, intermediolateral column; NC, nucleus centralis; VH, ventral horn. Scale bars: a c, e, 200 μm; b, d, f, 30 μm.
Catecholamines are reduced in adrenal glands of TrkA (−/−) mice
To investigate whether the chronic loss of AChE activity in TrkA-deficient animals had an impact on the catecholamine storage and secretion of chromaffin cells, we determined the catecholamines adrenaline and noradrenaline by HPLC–amperometric detection. As shown in Figure 10, there was a significant reduction in the medullary levels of both amines (noradrenaline, −63.5%; adrenaline, −70.7%), probably because of the prolonged activation of catecholamine secretion by acetylcholine at reduced activity of the hydrolyzing enzyme.

Quantitative determination of catecholamines in P6 adrenal glands reveals significant (*p > 0.01) reductions in the noradrenaline (−65.5%) and adrenaline (−70.7%) content in TrkA (−/−) mice, as compared with wild-type littermates.
DISCUSSION
The present results add a novel feature to the previously established TrkA (−/−) phenotype, a severe deficit within adrenal gland and preganglionic sympathetic spinal cord neurons regarding the activity of AChE, an enzyme with a well documented crucial role in terminating transmitter actions at cholinergic synapses (for review, see Massoulié et al., 1993). The evidence that the TrkA knock-out affects AChE activity is based on a specific histochemical staining technique (Andrä and Lojda, 1986) and a quantitative photometric method for determining specific AChE activity (Ellman et al., 1961) in homogenates. AChE staining pattern and intensity reflect the quantitative distribution of AChE activity, because there is a linear correlation between enzyme activity quantified photometrically and staining intensity (Andrä and van Duijn, 1985). The deficit in AChE activity seen in adrenal gland and spinal cord of TrkA (−/−) mice fails to correlate with an overt change in the immunocytochemically demonstrable AChE and AChE protein detectable in Western blots, suggesting that AChE activity is compromised more severely than AChE protein expression. This adds to the growing evidence that a pool of inactive AChE protein can be activated post-translationally in an environment-dependent manner (for review, see Massoulié et al., 1993; Layer and Willbold, 1995). Uncompromised AChE protein expression in the adrenal gland of TrkA (−/−) mice is also in accord with an unimpaired localization of synaptophysin within the adrenal gland.
The neuroendocrine chromaffin cells of the adrenal medulla and sympathetic neurons share an ontogenetic origin from the neural crest (cf. Unsicker, 1993) and many structural and functional features, including a prominent cholinergic innervation (Coupland and Holmes, 1958; Lewis and Shute, 1969; Millar and Unsicker, 1981; Ahonen, 1991). Both pre- and postganglionic noradrenergic sympathetic neurons, as well as chromaffin cells, synthesize and secrete AChE (Lewis and Shute, 1969; Mizobe and Livett, 1980, 1984; Millar and Unsicker, 1981; Hefti et al., 1982; Ahonen, 1991; Parker et al., 1993; Small et al., 1993). The functional implications of TrkA and NGF for the development and maintenance for each of these neural crest derivatives seem to diverge considerably. Both the recent targeted mutations of the TrkA and NGF genes (Crowley et al., 1994; Smeyne et al., 1994) and the early immunosympathectomy experiments (Angeletti et al., 1972) support the essential physiological role of TrkA and retrogradely acting NGF to prevent ontogenetic death of the paravertebral sympathetic neurons [for review, cf. Snider (1994) and Rush et al. (1995)]. In contrast, chromaffin cells of the adrenal medulla do not die on NGF withdrawal by treatment with NGF antibodies (Bode et al., 1986). They do respond, however, to NGF in vitro with neurite outgrowth (Unsicker et al., 1978; Doupe et al., 1985), a moderate increase in survival (Unsicker et al., 1985a,b), induction of TH, and AChE activity (Acheson et al., 1984; Müller and Unsicker, 1986). High-affinity binding sites for NGF on chromaffin cells (Hofmann et al., 1987), TrkA mRNA in newborn rat adrenal medulla (Suter-Crazzolara et al., 1997), and localization of TrkA mRNA in the adult rat (Michael et al., 1995) and mouse adrenal medulla (this study) strongly argue in favor of TrkA being expressed by chromaffin cells. The 62% reduction in adrenal AChE of TrkA −/− mutants shown in the present study probably can be attributed to reduced AChE activity in both the preganglionic nerve cells and fibers as well as chromaffin cells (compare Figs. 2, 3). AChE has been reported to appear in adrenal nerve terminals and very few chromaffin cells of the rat around birth, gradually increasing toward adulthood (Millar and Unsicker, 1981). This suggests that AChE activity in nerve fibers and chromaffin cells may not have reached adult levels at P7 and P12.
AChE-positive neurons within the IML column sending axons to sympathetic ganglia and adrenal medulla have not been reported to express TrkA. In fact, small interneurons, but not autonomic preganglionic neurons within the rat spinal cord, are immunoreactive for TrkA and express TrkA mRNA (Michael et al., 1995; K. Huber, unpublished observation). Consistent with this observation, NGF, in contrast to fibroblast growth factor-2, ciliary neurotrophic factor, and transforming growth factor-β, does not rescue IML neurons after ablation of one of their prominent targets, the adrenal medulla (Blottner et al., 1989a,b, 1996). Nonetheless, cell counts of Nissl-stained neurons in the IML of the thoracic spinal cord between segments T8 and T10 in TrkA-deficient and wild-type mice show a >40% reduction, reflecting neuron death or shrinkage. From these segments 25% of the IML neurons are known to project to the adrenal medulla, while the remaining 75% of the neurons project to prevertebral (coeliac, aorticorenal, superior mesenteric, other) and paravertebral sympathetic ganglia (Strack et al., 1988; Blottner et al., 1996). Our calculations, based on published cell counts and retrograde tracings (Strack et al., 1988), indicate that at least 65% of the IML neurons projecting to paravertebral ganglia could have disappeared in the TrkA knock-outs (compare Fig. 11). Consistent with this notion, it has been shown in chick embryos that NGF apparently indirectly regulates the survival of these preganglionic sympathetic (the so-called Terni column) neurons within the spinal cord by affecting the survival of their target cells, the postganglionic sympathetic neurons (Oppenheim et al., 1982). Thus, the reduction of AChE activity in autonomic neurons within the spinal cord of TrkA mutants is most likely attributable to a reduction of enzyme activity in spared IML neurons that project to the adrenal medulla and prevertebral ganglia.

Schematic drawing summarizing the basic findings of this report. Two populations of IML neurons are shown. First, a population of neurons (a, right) innervates sympathetic neurons within sympathetic paravertebral ganglia. These neurons express AChE activity (black dots). Their numbers are reduced in TrkA (−/−) mice (b, right) because of the loss of paravertebral ganglia. IML neurons innervating chromaffin cells of the adrenal medulla (a, left) also have AChE activity. In TrkA (−/−) mice, these neurons do not die but lose AChE activity (b, left).
In contrast to postganglionic paravertebral sympathetic neurons, which virtually all disappear in TrkA knock-outs (Smeyne et al., 1994), many aspects of the structure and chemistry of the adrenal medulla appear unchanged. Established markers for chromaffin cells, including various neuropeptides (NPY, met-ENK, somatostatin, galanin), markers of chromaffin granules (chromogranin B, secretogranin II), and vesicular amine transporters (VMAT-1/VMAT-2), are normally expressed and appear not to be changed overtly. As in the wild-type mouse, all chromaffin cells have TH immunoreactivity, and ∼75% of them express PNMT. In addition to the reduction of AChE activity, a significant decrease in the catecholamine content is the only other hallmark of the TrkA −/− adrenal medullary phenotype. Both phenomena probably are causally linked in that loss of the acetylcholine-hydrolyzing enzyme probably accounts for a prolonged and chronic activation of secretion of catecholamines, leading to a partial depletion of catecholamines from the adrenal medulla.
Although a reduction of AChE activity in TrkA-expressing chromaffin cells may be accepted readily as a feature of the TrkA knock-out, the reduction in IML neurons lacking TrkA is more difficult to explain. We assume that the reduction in AChE activity of IML neurons is likely to be a second-order effect. One possible explanation might be that chromaffin cells provide IML neurons with a signal, the expression of which in chromaffin cells can be affected by a TrkA-mediated mechanism. The molecular nature of such a retrograde AChE-regulating messenger remains to be elucidated.
Several lines of evidence indicate that NGF is synthesized within the adrenal gland. NGF mRNA levels shown by Northern blotting in adult rabbit adrenals amount to ∼25% of mRNA levels in the spleen and ∼20% of heart atrium and ventricle, all of which are densely innervated by sympathetic nerves (Shelton and Reichardt, 1984). Furthermore, mouse adrenal explants secrete a neurotrophic activity into their culture medium, which can be blocked by NGF antibodies (Harper, 1976). Taken together, these data suggest that the adrenal gland in vivo is a source of NGF and that chromaffin cells may be the target for the adrenal NGF.
In the spinal cord of TrkA mutants, AChE activity was not only affected in the IML neurons but also with regard to AChE-positive nerve fibers in the superficial and deeper layers of the dorsal horn. These fibers are in their majority axons of dorsal root ganglionic (DRG) neurons, which are virtually all AChE-positive (Gruber et al., 1971). The decrease in AChE staining in the dorsal horn of TrkA mutants is, therefore, likely to reflect the 70–90% loss of DRG neurons and loss of small sensory afferents in the TrkA knock-out (Smeyne et al., 1994).
AChE activity in spinal cord somatic motoneurons apparently was unaltered in TrkA (−/−), as well as in TrkB (−/−) mice, as compared with wild-type littermates. Somatic motoneurons express TrkB, TrkC, and p75 neurotrophin receptors (Koliatsos et al., 1991; Ernfors et al., 1993; Henderson et al., 1993; Yan et al., 1993). Interestingly, they retrogradely transport not only BDNF (and NT-3) (DiStefano et al., 1992) but, during a limited period of their development, also NGF (Yan et al., 1988), implying the transient presence of p75 and possibly also TrkA. Apparently, none of these properties of motoneurons affects the regulation of AChE activity by NGF or BDNF. The fact that AChE activity of motoneurons was affected neither in TrkA or TrkB mutants underscores the specificity in the TrkA-mediated regulation of AChE within a discrete neuron population, the preganglionic sympathetic neurons.
In conclusion, our data suggest a novel phenotypic feature of TrkA mutants: a loss of AChE activity in the adrenal medulla, in preganglionic nerves to the adrenal medulla, and in autonomic spinal cord neurons (compare Fig. 11). This alteration is specific in that it is not seen in TrkB mutants nor in motoneurons of TrkA- and TrkB-deficient mice. It remains to be investigated whether other AChE-expressing neuron populations within the CNS that are coupled to TrkA-expressing systems also are affected in TrkA-deficient animals.
Footnotes
This study was supported by Deutsche Forschungsgemeinschaft (SFB317/C8/D4, SFB269/A2). L.M. is supported by a long-term European Molecular Biology fellowship. J.E.G.A. and J.L.R.L. were funded by the Minority International Research Training program of National Institutes of Health–Fogarty International Center and National Institutes of Health Grant RR-8102-18. We thank Richard Hertel and Martin Barth for their expert technical assistance. We thank Drs. N. Wolf, K. Krieglstein, N. Kahane, and C. Kalcheim for sharing unpublished results on adrenal TrkB expression. Antibodies to AChE, L1, chromogranin B, secretogranin II, VMAT-1, and VMAT-2 were generously provided by Drs. J. Massoulié, A. Faissner, W. Huttner, and M. Hannah.
Correspondence should be addressed to Dr. Klaus Unsicker, Department of Anatomy and Cell Biology III, University of Heidelberg, Im Neuenheimer Feld 307, D-69120 Heidelberg, Germany.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}