

Abstract
Cyclic nucleotide-gated (CNG) channels are important mediators in the transduction pathways of rod and cone photoreceptors. Native CNG channels are heterotetramers composed of homologous A and B subunits. In heterologous expression systems, B subunits alone cannot form functional CNG channels, but they confer a number of channel properties when coexpressed with A subunits. To investigate the importance of the CNGB subunits in vivo, we deleted the CNGB1 gene in mice. In the absence of CNGB1, only trace amounts of the CNGA1 subunit were found on the rod outer segment. As a consequence, the vast majority of isolated rod photoreceptors in mice lacking CNGB1 (CNGB1-/-) failed to respond to light. In electroretinograms (ERGs), CNGB1-/- mice showed no rod-mediated responses. The rods also showed a slow-progressing degeneration caused by apoptotic death and concurred by retinal gliosis. Cones were primarily unaffected and showed normal ERG responses up to 6 months, but they started to degenerate in later stages. At the age of ∼1 year, CNGB1-/- animals were devoid of both rods and cones. Our results show that CNGB1 is a crucial determinant of native CNG channel targeting. As a result of the lack of rod CNG channels, CNGB1-/- mice develop a retinal degeneration that resembles human retinitis pigmentosa.
- cyclic nucleotide-gated channel
- CNGB1
- channel trafficking
- rod photoreceptor
- retinitis pigmentosa
- apoptosis
Introduction
Cyclic nucleotide-gated (CNG) channels play a key role in visual and olfactory transduction (Finn et al., 1996; Biel et al., 1999a; Kaupp and Seifert, 2002; Matulef and Zagotta, 2003). In the vertebrate retina, different CNG channels are expressed in the outer segments (OSs) of rod and cone photoreceptors. In darkness, these channels are opened by cGMP, maintaining an inward current. Light induces a hydrolysis of cGMP, thus resulting in closure of the channels and hyperpolarization of the cell as the response. CNG channels are tetramers and, in their native forms, are composed of A and B subunits (Hofmann et al., 2003). Both types of subunits share a common topology characterized by six transmembrane segments (S1-S6), a pore-forming loop between S5 and S6, and a C-terminal cyclic nucleotide-binding domain (CNBD). The rod channel consists of three CNGA1 subunits and one copy of a long isoform of the CNGB1 subunit (CNGB1a) (Weitz et al., 2002; Zheng et al., 2002; Zhong et al., 2002), whereas the cone channel exhibits a 2A (CNGA3): 2B (CNGB3) stoichiometry (Peng et al., 2004). In heterologous expression systems, such as Xenopus oocytes or human embryonic kidney 293 (HEK293) cells, most A subunits (CNGA1-CNGA3) form functional homomeric channels by themselves. In contrast, the two B subunits (CNGB1 and CNGB3; CNGB2 does not exist) do not, but they confer a number of channel properties typical of native channels (e.g., flickery behavior, increased sensitivity to cAMP and l-cis- diltiazem, and weaker block by extracellular Ca2+) when coexpressed with A subunits (Finn et al., 1996; Biel et al., 1999a; Kaupp and Seifert, 2002; Matulef and Zagotta, 2003). In addition, the B subunits may have other significance in vivo. Recently, it has been proposed, based on expression experiments in Xenopus oocytes, that a specific interaction between CNGA1 and CNGB1 is required for surface expression of the CNGA1/CNGB1 complex (Trudeau and Zagotta, 2002) (but see Dryja et al., 1995; Mallouk et al., 2002).
To study the role of a CNGB subunit in a physiological context, we generated a mouse line lacking the CNGB1 subunit. The CNGB1 gene locus consists of at least 33 exons encoding several isoforms (Ardell et al., 2000). Rods express a 240 kDa isoform (CNGB1a) containing a long cytosolic N terminus that is also translated as a separate cytosolic protein [glutamic acid-rich protein (GARP) (Chen et al., 1993; Körschen et al., 1995)]. Shorter variants of the subunit lacking the GARP part are present in the native olfactory CNG channel (CNGB1b) (Sautter et al., 1998; Bönigk et al., 1999) and have been identified in sperm cells and other tissues (Biel et al., 1996; Wiesner et al., 1998). To inactivate all CNGB1 isoforms, we deleted by gene targeting exon 26 of CNGB1, which encodes the pore-forming region and the S6 segment. In this study, we report on the retinal phenotype of CNGB1-deficient (CNGB1-/-) mice.
Materials and Methods
Generation of CNGB1-deficient mice. Using a BAC clone (Genome Systems, St. Louis, MO) isolated from a genomic 129SvJ library, we constructed a targeting vector (see Fig. 1 A) such that exon 26 of the CNGB1 gene was flanked in upstream direction by a loxP-neo/tk-loxP cassette and in downstream direction by a single loxP site. The construct was electroporated into R1 embryonic stem cells, and G418-resistant clones were screened by Southern blot analysis. Two positive clones were transfected with a Cre-expressing plasmid. Cre-mediated recombination resulted in the deletion of exon 26, causing a frame shift and introducing a stop codon in the first triplet of exon 27. Correctly targeted clones were injected into C57BL/6N blastocysts, and the resulting chimeric mice were mated with C57BL/6N mice for germ-line transmission. Heterozygous (CNGB1+/-) mice were intercrossed to produce homozygous CNGB1-deficient (CNGB1-/-) mice. Transgene determination was done by multiplex PCR; for primer sequences and PCR-conditions, see supplemental Table 1 (available at www.jneurosci.org as supplemental material).

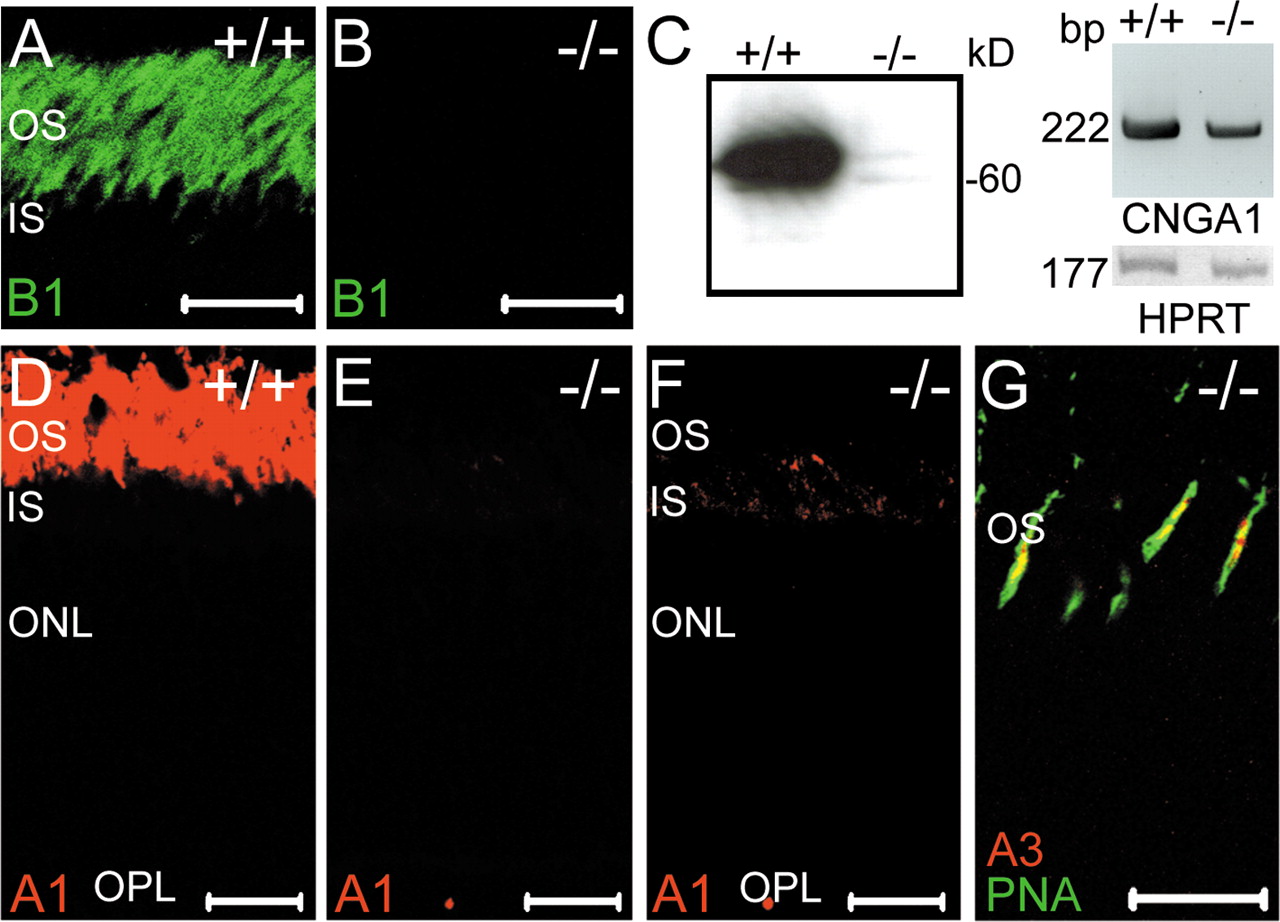
Disruption of the CNGB1 gene. A, Top, Wild-type CNGB1 locus and targeting vector. Exons 22-29 are represented by gray boxes. The targeting vector contains a neo/tk cassette flanked by two loxP sites (black triangles) inserted between exons 25 and 26 and a third loxP site inserted between exons 26 and 27. E, EcoRI; N, NheI; S, SacI; X, XmaI. Bottom, Homologously recombined allele and knock-out (ko) locus after Cre-mediated deletion of exon 26 and the neo/tk cassette in embryonic stem cells. B, Southern blot analysis of EcoRI-digested genomic DNA from CNGB1+/+, CNGB1+/-, and CNGB1-/- mice hybridized with probe (P) shown in A (black box). The 5.6 and 3.5 kb bands represent the wild-type and mutant CNGB1 allele, respectively. C, Multiplex PCR analysis with genomic DNA using primers displayed as arrows in A yielding a 480 bp fragment for wild-type and a 415 bp fragment for the knock-out allele. D, Reverse transcription (RT)-PCR analysis of CNGB1 expression in retina using primers detecting exon 26 (indicated as arrowheads in A). A 257 bp band is amplified from retinal mRNA of wild-type mice but is missing in knock-out mice. A 177 bp band specific for HPRT is amplified in both genotypes. E, Western blot analysis of membrane protein preparations of wild-type and CNGB1-deficient retinas using an antibody against the C terminus of CNGB1 demonstrates the absence of the CNGB1 protein in CNGB1-/- mice.
Dark current from single rods
Animals. For all experiments, age-matched CNGB1+/+, CNGB1+/-, and CNGB1-/- mice on a hybrid 129SvJ and C57BL/6N background were used. Mice were housed in 12 hr light/dark cycles, except for single-cell recordings (14/10 hr light/dark cycles). The animals were treated in accordance with both German legislation and National Institutes of Health guidelines on the protection of animals.
Reverse transcription-PCR. Retinas were dissected, and total RNA was isolated using the RNeasy kit (Qiagen, Hilden, Germany) and subsequently treated with DNAseI (Roche Diagnostics, Mannheim, Germany). First-strand cDNA was synthesized from equal amounts of RNA with the Superscript II H- kit (Invitrogen, Karlsruhe, Germany) using oligo-dT primers. cDNAs of CNGA1, CNGB1, and hypoxanthine phosphoribosyl-transferase (HPRT) were amplified using the primers and conditions listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Amplicons were separated on 5% polyacrylamide gels and visualized with ethidium bromide. All primer pairs used were intron spanning to avoid amplification of genomic DNA.
Generation of anti-CNGB1 antibody. A rabbit polyclonal antibody (C-AbmCNGB1) directed against a sequence in the C terminus of the murine CNGB1 protein (EAAGPPEPSVRIRVSPGP) was generated by standard techniques. C-AbmCNGB1 specifically detected a protein of the predicted size (240 kDa) in membrane extracts of HEK293 cells transfected with an expression vector carrying the CNGB1a cDNA (data not shown).
Histology. Posterior eyecups were immersion fixed in 4% paraformaldehyde or a mixture of 4% paraformaldehyde and 2% glutaraldehyde in 0.1 m phosphate buffer. For light microscopy, semithin sections of Epon-embedded retinas (Epon 812; Fluka, Buchs, Switzerland) were stained with toluidine blue (Fluka). For transmission electron microscopy, small tissue blocks were postfixed in 1% osmium tetroxide-0.1 m cacodylate buffer, stained en bloc with 2% uranyl acetate, dehydrated in ethanol and acetone, and embedded in Epon. Ultrathin sections were viewed with an electron microscope (EM 10; Zeiss, Oberkochen, Germany). For scanning electron microscopy, specimens were fixed, cryoprotected in graded DMSO, and freeze fractured in liquid nitrogen. Resulting fragments were postfixed in 1% osmium tetroxide-0.1 m cacodylate buffer, dehydrated in ethanol, air dried, and gold covered by standard methods. Specimens were analyzed with a XL 40 Philips (Aachen, Germany) scanning electron microscope.
Immunohistochemistry was performed on retinal cryosections as described previously (Claes et al., 2004). The sources and working dilutions of primary antibodies are listed in supplemental Table 2 (available at www.jneurosci.org as supplemental material). For secondary detection, we used FITC donkey anti-rabbit IgG (Dianova, Hamburg, Germany), and Alexa 594 goat anti-mouse IgG (Molecular Probes, Eugene, OR). Direct fluorescence microscopy was performed using primary antibodies labeled with DY-547-NHS ester (Dyomics, Jena, Germany). To visualize cell nuclei, specimens were treated with 5 μg/ml Hoechst 33342 (Molecular Probes).
Peanut agglutinin (PNA), a lectin that specifically labels carbohydrate moieties on the extracellular surface of cone photoreceptors (Blanks and Johnson, 1984), was used as cone marker. Either FITC-labeled PNA (1:100; Sigma, Deisenhofen, Germany) or biotinylated PNA (1: 100; Sigma), followed by detection with DY-547 streptavidin (Dyomics), was applied.
Terminal deoxynucleotide transferase (TdT)-mediated dUTP nick end labeling (TUNEL) was performed on retinal cryosections using standard methods based on Gavrieli et al. (1992). TdT was obtained from New England Biolabs (Frankfurt am Main, Germany).
All specimens were examined on a conventional light microscope (Axioskop 2; Zeiss) equipped with a CCD camera (HMRc; Zeiss) or a confocal laser scanning microscope (LSM510 Meta; Zeiss).
Western blotting. Membrane proteins were isolated from either murine retinas or transfected HEK293 cells as described previously (Much et al., 2003). Equal amounts of proteins were separated using 7-10% SDS-PAGE, followed by Western blot analysis according to standard procedures. The antibodies were used in the following dilutions: C-AbmCNGB1 (1: 1000), PMc1D1 (1:50), and PPc6N (1:2000).
Electroretinography. Ganzfeld electroretinograms (ERGs) were obtained from anesthetized mice as described previously (Seeliger et al., 2001). The ERG equipment consisted of a Ganzfeld bowl, a DC amplifier, and a personal computer-based control and recording unit (Toennies Multiliner Vision; Viasys Healthcare, Hoechberg, Germany). Bandpass filter cutoff frequencies were 0.1 and 3000 Hz. Single white-flash recordings were obtained under both dark-adapted (scotopic) and light-adapted (photopic) conditions. Light adaptation before the photopic session was performed with a background illumination of 30 cd/m2 for 10 min. Single white-flash stimulus intensities were increased from 10-4 cd/m2 to 25 cd/m2, divided into 10 steps of 0.5 and 1 log cd/m2. Ten responses were averaged with an interstimulus interval of either 5 or 17 sec (for 1, 3, 10, 25 cd/m2).
Single-cell recordings. The recording procedure was broadly as described previously (Yang et al., 1999). In brief, mice were dark adapted overnight and killed by CO2 asphyxiation under dim red light. All subsequent procedures were performed under infrared light. The eyes were removed and hemisected, and the retina was removed and stored in L-15 medium (Invitrogen, San Diego, CA) supplemented with 10 mm glucose and 0.1 mg/ml bovine serum albumin (Sigma) on ice for up to 7 hr. When used, a small piece of retina was mechanically chopped in chilled L-15 medium in a 35 mm Sylgard-coated Petri dish. The tissue was transferred to the recording chamber and perfused with bicarbonate-buffered Locke's solution containing the following: 149 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 10 mm HEPES, pH 7.4, 0.02 mm EDTA, 3 mm NaHCO3, 3 mm disodium succinate, 0.5 mm sodium glutamate, 0.1% vitamins and amino-acid supplement (Invitrogen, San Diego, CA), and 10 mm glucose, bubbled with 95% O2-5% CO2. The perfusion solution was heated to 37-38°C (Reisert and Matthews, 2001), and the temperature of the solution in the chamber was monitored continuously with a telethermometer. An individual rod outer segment (ROS) projecting from a piece of retina was drawn into a suction electrode containing the following (in mm): 140 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2, 3 HEPES, pH 7.4, 0.02 EDTA, and 10 glucose. Membrane current was monitored with a current-to-voltage amplifier (Axopatch 200B; Axon Instruments, Union City, CA). All signals were low-pass filtered at 20 Hz (eight-pole Bessel) and sampled at 500 Hz. The light stimulus consisted of brief flashes (10 msec) of unpolarized, 500 nm (10 nm band-width) light at intervals of 8 sec. Because of progressive ROS degeneration in the CNGB1-/- mouse retina, we only recorded from 17-d-old [postnatal day 17 (P17)] mouse rods.
Results
Disruption of the CNGB1 gene
We disrupted the CNGB1 gene through homologous recombination using a Cre/loxP-based strategy (Fig. 1A). To achieve a global knock-out covering all CNGB1 isoforms, we deleted the common exon 26 encoding the pore and the S6 segment of the CNGB1 subunit. The knock-out was verified at the level of genomic DNA by Southern blot analysis (Fig. 1B) and PCR (Fig. 1C). CNGB1-deficient mice (CNGB1-/-) did not express mRNA containing exon 26 (Fig. 1D). An antibody directed against the C terminus of CNGB1a detected the 240 kDa CNGB1a protein in retinal membrane fractions from wild-type mice but not from CNGB1-/- mice (Fig. 1E). An antibody directed against the cytosolic N terminus of CNGB1a (the GARP part) also detected a 240 kDa protein in CNGB1+/+ and CNGB1+/- mice but not in knock-out mice (supplemental Fig. S1G, available at www.jneurosci.org as supplemental material). Importantly, no truncated version of the CNGB1a subunit, which would have resulted from translation of exons 1 through 25, was observed with this antibody (supplemental Fig. S1G, available at www.jneurosci.org as supplemental material). The immunolabeling obtained with the N-terminal antibody in retinal slices of CNGB1-/- mice (see Fig. 5K,L) was very likely attributable to the presence of soluble GARP isoforms, which are generated by alternative splicing of the first 12-16 exons of the CNGB1 gene (Ardell et al., 2000). Heterozygous matings produced CNGB1+/+, CNGB1+/-, and CNGB1-/- mice at the expected Mendelian ratio. However, CNGB1-/- mice revealed an increased postnatal mortality compared with their littermates. Moreover, the knock-outs were smaller and had a reduced body weight (weight at P30: CNGB1+/+, 16.2 ± 0.2 gm, n = 17; CNGB1-/-, 10.4 ± 0.2 gm, n = 16; p < 0.0001). Both phenotypes were probably caused by an impairment of olfaction (our unpublished observations).

Retinal morphology of CNGB1-/- mice. A, Toluidine blue-stained vertical semithin section of a PM4 CNGB1+/+ mouse retina compared with retinal sections of CNGB1-/- mice at PM4, PM6, and PM11. All sections were from the region midway between the optic nerve head and periphery. B, C, Scanning electron micrographs of freeze-fractured retinas from PM4 CNGB1+/+ (B) and CNGB1-/- (C) mice. In the mutant retina, the outer segment layer is collapsed. D-F, Transmission electron micrographs of rod outer segments from PW6 CNGB1+/+ (D) and CNGB1-/- (E, F) mice. The arrows in E label outer segments that have started to disintegrate. G-I, Immunostaining with anti-rhodopsin antibody (Rhod, red) in P15 wild-type (G) and P15 (H) and PW8 (I) CNGB1-/- mice. J-L, Immunostaining with an antibody recognizing the N terminus common to GARP and CNGB1a (green) in P15 wild-type (J) and P15 (K) and PW8 (L) mutant mice. Please note that the wild-type immunosignal (J) represents CNGB1a and soluble GARP expression, whereas in the CNGB1-/- retina (K, L), only soluble GARP is detected. M-O, Retinal sections of knock-out mice costained with PNA (red) and anti-mid-wavelength-sensitive opsin (MWS) antibody (green) reveal secondary cone degeneration. The age of the mice was PW8 (M), PM6 (N), and PM11 (O). At PM11, cones almost completely vanished. In G-I and M-O, cell nuclei have been stained with Hoechst dye (blue). IS, Inner segments; INL, inner nuclear layer; IPL, inner plexiform layer; RPE, retinal pigment epithelium. Scale bars: A, 50 μm; B, C, 5 μm; D, E, 2 μm; F, 1 μm; G-O, 20 μm.
Requirement of CNGB1 for the presence of CNGA1 subunit in the ROS
As expected, the CNGB1 protein was abundantly expressed in the OS layer of wild-type mice (Fig. 2A) but absent in the mutants (Fig. 2B). Surprisingly, Western blots of retinal membrane fractions probed with an antibody specific for the rod photoreceptor A subunit (CNGA1) yielded only a very faint signal in CNGB1-/- mice (Fig. 2C, left). In contrast, the mRNA of CNGA1 was clearly present in both genotypes, indicating that the strong reduction in the amount of CNGA1 was not caused by a shutdown of gene transcription (Fig. 2C, right). Immunostainings of retinal sections from P15 knock-out mice confirmed the Western blot results by showing that the CNGA1 subunit was profoundly down-regulated in the outer segment layer of CNGB1-/- mice (Fig. 2D,E). Only during stimulation with strong laser intensities was some residual CNGA1 immunolabeling detected in rod outer segments (Fig. 2F). There was no evidence for mislocalization or accumulation of CNGA1 in other retinal compartments (Fig. 2 F). The CNGA3 subunit (Fig. 2G) and the CNGB3 subunit (data not shown), which together form the native cone CNG channel, were normally expressed in cone outer segments of CNGB1-/- mice. Importantly, expression of both subunits was restricted to cones, indicating that rod photoreceptors cannot compensate for the loss of CNGB1 and CNGA1 by upregulation of cone channel subunits. Finally, the olfactory CNGA2 subunit was not expressed in wild-type and knock-out retinas (data not shown).

Analysis of CNG channel expression in wild-type and mutant retina. Immunolabeling with C-AbmCNGB1 demonstrates the expression of CNGB1 inrodouter segments of wild-type retina (A) and its complete absence in the knock-out retina (B). The age of the mice in A and B was 8 weeks. C, Left, Western blot of membrane protein preparations (24 μg of protein per lane) of retinas from 8-week-old CNGB1+/+ and CNGB1-/- mice using a CNGA1-specific antibody. Right, RT-PCR with CNGA1-specific primers detects CNGA1 mRNA in wild-type and knock-out retina. HPRT was amplified as control. D-F, Confocal images of retinal sections from 15-d-old CNGB1+/+ and CNGB1-/- mice labeled with anti-CNGA1 antibody. To detect low-level expression of CNGA1 in mutant mice, three times higher laser intensity was used in F than in D and E. G, Colabeling of a retinal section from a PW8 CNGB1-/- mice with anti-CNGA3 (red) and PNA (green), showing the specific expression of the CNGA3 subunit in cone photoreceptors. Scale bars: A, B, D-G, 20 μm. IS, Photoreceptor inner segments.
Residual photocurrent in CNGB1-/- rod photoreceptors
To evaluate the effect of the deletion of CNGB1 on rod phototransduction, we recorded membrane current from single rods of P17 mice (Fig. 3). Isolated rods from wild-type and heterozygous mice responded to light, with no difference between their intensity-response relationships (Fig. 3A,B, top and middle; Table 1). In contrast, only a small percentage of CNGB1-/- rods (3 of 35 recorded cells) responded to light (Fig. 3A, bottom). The amplitudes of responses for these cells were too small (10-15% of wild-type response) for the intensity-response relationship to be characterized.

The effects of CNGB1 deletion on P17 mouse rod flash responses. A, Responses of CNGB1+/+ control (top), CNGB1+/- (middle), and CNGB1-/- (bottom) rods to 10 msec, 500 nm flashes of increasing intensities. Each trace was averaged from 7-60 flash trials. Flash was delivered at time 0. Flash strengths were as follows: 16.8, 36.8, 72.6, 136.6, 266.2, 493.9, 1010.8, and 1956. 6 photons μm-2 for the CNGB1+/+ and CNGB1+/- rods; and 1956.6 photons μm-2 for the CNGB1-/- rods. The saturating response amplitudes were 9.3 pA for CNGB1+/+ rod, 8.5 pA for CNGB1+/- rod, and 0.9 pA for CNGB1-/- rod. B, Response-intensity relationships (at the transient peak of the response) for the corresponding cells in A. The continuous curves are the saturating exponential function, r/rmax = 1 - exp (-ki), where i is flash strength, and k is a constant inversely proportional to the sensitivity of the cell. The half-saturating flash intensity, σ, is derived from σ = ln 2/k. Half-saturating responses occurred at 74.7 photons μm-2 for the CNGB1+/+ rod (top) and 61.4 photons μm-2 for the CNGB1+/- rod (bottom).
Characterization of retinal function
To investigate the effect of CNGB1 deletion on retinal function, ERGs were recorded from 1- and 6-month-old CNGB1-/- and CNGB1+/+ mice under dark-adapted (Fig. 4A-C) and light-adapted (Fig. 4D-F) conditions. In dark-adapted conditions, wild-type mice showed a response starting at a flash intensity of ∼1 mcd/m2. In contrast, first responses were discernible in both postnatal week 4 (PW4) and postnatal month 6 (PM6) mutant mice only at ∼10-30 mcd/m2, a threshold similar to those found in mouse models of pure cone function, such as the rhodopsin knock-out mouse (Jaissle et al., 2001). At higher intensities, the dark-adapted mixed response of mutant mice lacked a substantial a-wave (the initial negative deflection) and the first two major oscillations on the positive-going b-wave (Fig. 4 B), both typical indicators of rod system activity (Biel et al., 1999b). Because of the lack of the a-wave, the b-wave amplitude measured from the baseline to the b-wave peak was smaller in CNGB1-/- mice than in control animals (Fig. 4C). These findings indicate that the rod system is not functional in the CNGB1-/- mouse. In contrast, there were no response differences between knock-outs and controls in the light-adapted ERG up to PM6, indicating that the cone system is functionally intact (Fig. 4 D-F).

Electroretinographic data from wild-type and knock-out mice. Dark-adapted (A) and light-adapted (D) single-flash ERG intensity series of a control mouse (PW4) and CNGB1-/- mice (PW4 and PM6). Stimulus intensity (cd/m2) is indicated in the left panel. Vertical line crossing each trace shows the timing of the light flash. Representative dark-adapted (B) and light-adapted (E) responses of a PW4 CNGB1+/+ and CNGB1-/- mouse. The location of the a- and b-wave of the ERG response is indicated in B. Dark-adapted (C) and light-adapted (F) b-wave amplitudes from four PW4 CNGB1-/- mice are plotted as a function of the logarithm of the flash intensity. The dotted lines delimit the normal range given by the 5 and 95% quantile of the CNGB1+/+ mice (n = 4).
Loss of rod and cone photoreceptors in CNGB1-/- mice
We next examined the effect of the deletion of CNGB1 on the retinal structure. Figure 5A shows a series of representative semithin retinal sections from knock-out mice of different ages compared with wild-type animals. In CNGB1-/- mice, a slow-progressing decline in photoreceptor number with age was evident; this decline was somewhat more severe in the peripheral than in the central portion of the retina. Up to 2 months of age, wild-type and knock-out mice had the same number of nuclei in the outer nuclear layer (ONL) (data not shown). At 4 months (PM4), photoreceptor nuclei in the ONL of midcentral retina in knock-outs decreased to 8-10 rows instead of the normal 10-12 rows. At PM6, the number was further reduced to 6 rows. Finally, at time points later than PM10, only one discontinuous layer remained. We also observed that ROSs of CNGB1-/- mice were shorter than those of wild-type mice (Fig. 5D,E). Moreover, at 6 weeks, the number of ROSs was reduced in CNGB1-/- mice, and mutant ROSs frequently showed signs of structural disintegration (Fig. 5E, arrows). Interestingly, at this age, mutant ROSs revealed a by-and-large normal morphological organization of discs and rim regions, indicating that CNGB1 is not required for principal ROS formation (Fig. 5F). ROS shortening started much earlier than loss of rod nuclei, being evident at P15 (Fig. 5H,K). Scanning electron microscopy revealed a significantly thinner outer segment layer in 4-month-old knock-out retina compared with wild type, a time point when most rod nuclei were still present in the knock-outs (Fig. 5B,C). Interestingly, shortened ROSs contained rhodopsin and GARP, though at reduced amounts (Fig. 5G-L). Immunosignals for these specific proteins were detectable as long as ROSs were present in the retina. Primarily, cones were unaffected by the deletion of CNGB1. Although cone outer segments collapsed when the surrounding ROS vanished, they were functionally intact (Fig. 4) and were found at normal numbers even at PM6 (Fig. 5N). However, both mid-wavelength-sensitive (Fig. 5M-O) and short-wavelength-sensitive (data not shown) cones started to degenerate after PM6. At PM11, a few cone nuclei were still present, but only trace amounts of cone opsins were detectable (Fig. 5O). At the age of ∼1 year, CNGB1-/- mice were essentially devoid of both rods and cones. Unlike photoreceptors, the principal structure of the inner retina seemed to be unaffected by the deletion of CNGB1 (Fig. 6). However, the processes of most rod bipolar cells (Fig. 6A-F) and horizontal cells (Fig. 6G-I) were retracted in the knock-outs. Moreover, both bipolar and horizontal cells extended single sprouting processes into the ONL (Fig. 6 B, H, arrows). Occasionally, cell bodies of rod bipolar or horizontal cells were found to be misplaced into the outer plexiform layer (OPL) (Fig. 6C,H, arrowheads). There was no obvious difference in the morphology of the outer and inner retina between CNGB1+/- and wild-type mice. Moreover, cone and rod photoreceptors did not show any signs of degeneration in aged heterozygotes (supplemental Fig. S1 A-F, available at www.jneurosci.org as supplemental material).

Morphological alterations of rod bipolar and horizontal cells in aged CNGB-/- mice. A-F, Confocal images of vertical retinal sections immunostained for the rod bipolar cell marker PKCα. Rod bipolar cells of knock-out mice retracted their dendrites and developed single sprouting extensions into the ONL (arrow in B). D-F represent higher-magnification pictures from the OPL shown in A-C. G-I, The same sections stained with the horizontal cell marker calbindin. Like the rod bipolar cells, horizontal cells of knock-out mice reveal a reduced number of processes in the OPL and show sprouting extensions (arrow) into the ONL. Note that singled is placed rod bipolar and horizontal cell bodies (arrow heads in C and H) were found in the OPL of CNGB-/- mice. INL, Inner nuclear layer; IPL, inner plexiform layer. Scale bars, 20 μm.
Gliosis and apoptotic cell death in the CNGB1-/- retina
Müller glial cells respond early in degenerating retina by induction of intermediate fibers, such as glial fibrillary acid protein (GFAP) (de Raad et al., 1996; Bringmann and Reichenbach, 2001). In CNGB1-/- mice, GFAP upregulation started at P21 (Fig. 7A) and was very pronounced at later time points (Fig. 7C). In contrast, in wild-type mice, GFAP immunoreactivity was always restricted to the neurofilament layer (Fig. 7 B, D). We detected in the photoreceptors of the knock-out mice two hallmark features of apoptosis: increased nuclear DNA fragmentation (Fig. 7E) and activation of caspase 3 (Fig. 7F). Apoptosis started after P15, reaching peak between P21 and P28 and declining afterward (data not shown). We did not observe apoptotic cells in wild-type mice at all time points tested (P15 to PM3; data not shown).

Gliosis and apoptosis in the CNGB1-/- retina. A-D, Retinal sections of wild-type and mutant retinas stained with anti-GFAP (red) and the nuclear dye Hoechst (blue). GFAP is specifically expressed in Müller glia end feet and astrocytes (B, D). At P21, glia cells of knock-out mice begin to upregulate GFAP (arrows in A). At PM10, massive gliosis is observed (C). TUNEL (E, black dots) and detection of activated caspase 3 (F, yellow dots) in retinal slices of P21 knock-out mice. TUNEL-positive cells and activated caspase 3 are not detected in age-matched wild-type sections (data not shown). Scale bars: A-D, 100 μm; E, 50 μm; F, 20 μm. INL, Inner nuclear layer; IS, inner segments; act-Casp 3, activated caspase 3.
Discussion
A major finding of our study is that CNGB1 is required for the formation and/or outer segment targeting of the native rod CNG channel. Because functional homomeric CNGA1 channels can be formed in heterologous expression systems, it would appear that improper targeting is the underlying pathology in native rods, as a result of which the CNGA1 protein is presumably rapidly degraded. This rapid degradation is suggested by the lack of CNGA1 in rod inner segments and cell bodies of CNGB1-/- retinas. The detection of small light responses in a low percentage of rods suggests that homomeric CNGA1 channels, once formed, are nonetheless functional in the ROS. The mechanistic role of CNGB1 in properly targeting CNGA1 to the outer segment remains unclear. The primary sequence of CNGA1 in the cytosolic N and C termini contains several KKXX motifs known as endoplasmatic reticulum retention/retrieval motifs that mediate membrane trafficking for a host of proteins (Teasdale and Jackson, 1996). It is possible that, in cell lines that successfully target heterologously expressed CNGA1 to the plasma membrane, there happen to be factors such as chaperones or interacting proteins that can mask these retention signals. Perhaps such a factor is absent in rods, and CNGB1 is required for this purpose. In a more general sense, our results indicate that the formation and cell surface expression of ion channels critically depend on the cellular environment. Thus, studies in native cells in which the respective channel proteins are expressed in vivo will be required to confirm data obtained in heterologous systems such as Xenopus oocytes or HEK293 cells. In any case, it is tempting to speculate that the requirement for CNGB1 in targeting CNGA1 to the outer segment may have evolved to ensure that only heteromeric channels get there. The CNGB3 subunit in cones may have an equivalent function and can explain why human patients lacking CNGB3 protein attributable to genomic mutations suffer from achromatopsia, a disease resulting from the complete loss of cone function (Sundin et al., 2000). It would thus be interesting to know whether indeed all CNG channels in native tissues have a B subunit.
Another important finding of our study is that mice deficient in CNGB1 develop a retinal degeneration that is similar to human retinitis pigmentosa (RP). RP comprises a group of genetically diverse diseases that are characterized by a progressive degeneration of rods and subsequently cones (Pierce, 2001). Typically, the earliest clinical symptom of RP is an initial night blindness attributable to the dysfunctional rod system. The correlate of the subsequent degeneration of cones is a progressive loss of the visual field from the periphery toward the macula, leading to so-called “tunnel vision” and eventually to legal blindness. In CNGB1 mutant mice, initial signs of rod degeneration, including apoptosis and gliosis, were observed at the age of 2 to 3 weeks after birth. At 6 months after birth, the number of the photoreceptors was reduced by ∼50%, and it took ∼1 year for complete loss of rods. Cone photoreceptors degenerated secondary to the rods and with significantly slower kinetics. The stratification and structural organization of the inner retina of CNGB1-/- mice was by-and-large normal. Remarkably, however, rod bipolar cells and horizontal cells revealed an atrophy of dendrites and thin processes in the OPL. In addition, in both cell types, irregular processes extending to the ONL were observed. Similar morphological alterations have been found in other mouse models lacking functional cones and/or rods (Strettoi et al., 2003; Claes et al., 2004). Together, these findings strongly suggest that rod photoreceptor inputs are required for the functional maintenance of synaptic contacts.
ERG measurements were in complete agreement with the histological data, by showing that CNGB1-/- mice lack rod responses but retain normal cone-mediated responses at least up to 6 months. It appears that the light responses observed in a minor fraction of rods from young knock-out animals were incapable of producing a detectable signal in the dark-adapted ERG. The degeneration observed in the retina of CNGB1-/- mice proceeded much more slowly than in other well characterized RP mouse models, such as the rd mouse (Bowes et al., 1990) or the rhodopsin-deficient mouse (Humphries et al., 1997). At the moment, the exact sequence of events that lead to rod cell death in CNGB1-/- mice is unclear. One explanation would be that the structural scaffolds of the ROS were affected. Biochemical studies indicate that the CNGB1 channel through the GARP part binds to peripherin-2 oligomers in the rim region of outer segment disc membranes (Poetsch et al., 2001). Thus, one might have speculated that, in the absence of CNGB1, a tight coupling between the disc rim region and the plasma membrane is sufficiently impaired to result in a structural defect of the outer segment. However, our electron microscopy data do not support this hypothesis because they demonstrate the presence of normally shaped disks and rim structures in mutant ROSs, at least at the age of 6 weeks. Thus, it seems more likely that rod cell death is simply attributable to a lack of functional CNG channels on the ROS, analogous to the human RP caused by defective CNGA1 (Dryja et al., 1995; Travis, 1998). At present, the molecular pathways coupling the absence of CNG channels to the initiation of cell death are unknown.
Recently, RP patients have been identified who carry a point mutation in the CNBD of CNGB1 (Bareil et al., 2001). So far, the functional consequences of this mutation, G993V, which involves a highly conserved residue in the CNG channel family, have not been determined in expression systems. It is not known whether the mutant protein correctly assembles with the CNGA1 subunit and is targeted to the plasma membrane of ROSs. If the targeting is disturbed, the disease mechanism leading to human CNGB1-related RP would be equivalent to the one in CNGB1-deficient mice. Alternatively, if the mutant CNGB1 was produced and assembled with CNGA1, it might impair the cGMP-dependent activation of the channel. In any case, the observation that RP patients carrying the G993V mutation show no olfactory impairment (Bareil et al., 2001) would tend to suggest that the CNGB1 mutation impact photoreceptors and olfactory receptor neurons differently.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (M.B., A.G., and M.S.) and National Institutes of Health Grant EY 06837 (K.-W.Y.). We thank R. S. Molday for the gift of antibodies PMc1D1 and PPc6N, S. Mintova for help with scanning electron microscopy, and B. Wessinger for excellent technical support.
Correspondence should be addressed to Martin Biel, Department Pharmazie, Pharmakologie für Naturwissenschaften, Ludwig-Maximilians-Universität München, Butenandtstrasse 7, 81377 München, Germany. E-mail: mbiel{at}cup.uni-muenchen.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250130-09$15.00/0
↵* M.S. and D.-G.L. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}