

Abstract
Connexin45 (Cx45) is known to be expressed in the retina, but its functional analysis was problematic because general deletion of Cx45 coding DNA resulted in cardiovascular defects and embryonic lethality at embryonic day 10.5. We generated mice with neuron-directed deletion of Cx45 and concomitant activation of the enhanced green fluorescent protein (EGFP). EGFP labeling was observed in bipolar, amacrine, and ganglion cell populations. Intracellular microinjection of fluorescent dyes in EGFP-labeled somata combined with immunohistological markers revealed Cx45 expression in both ON and OFF cone bipolar cells. The scotopic electroretinogram of mutant mice revealed a normal a-wave but a 40% reduction in the b-wave amplitude, similar to that found in Cx36-deficient animals, suggesting a possible defect in the rod pathway of visual transmission. Indeed, neurotransmitter coupling between AII amacrine cells and Cx45-expressing cone bipolar cells was disrupted in Cx45-deficient mice. These data suggest that both Cx45 and Cx36 participate in the formation of functional heterotypic electrical synapses between these two types of retinal neurons that make up the major rod pathway.
Introduction
Gap junctions are intercellular channels that allow direct communication between adjacent cells. Each gap junctional channel is made up of two hemichannels (connexons), one contributed by each cell. One connexon consists of six membrane-spanning protein subunits called connexins (for review, see Willecke et al., 2002).
To date, 20 connexin genes have been identified in the murine genome and 21 in the human genome (Söhl and Willecke, 2003).
Three decades after the first description of gap junctions between photoreceptor cells in the vertebrate retina (Raviola and Gilula, 1973), specific connexin proteins could be assigned to distinct cell types in the mouse retina. Initially, connexin36 (Cx36) was discovered between AII amacrine cells as well as between AII amacrine cells and ON cone bipolar cells in the mouse and rabbit retinas (Feigenspan et al., 2001; Mills et al., 2001). Its disruption in Cx36-deficient mice impairs proper signal transmission from the rod to the cone signaling pathway (Güldenagel et al., 2001; Deans et al., 2002). Whereas Deans et al. (2002) reported additional Cx36 expression in mouse rod photoreceptors, Feigenspan et al. (2004) demonstrated its localization in cone photoreceptors and OFF cone bipolar cells. Most recently, connexin57-deficient reporter mice revealed that tracer coupling is disrupted between horizontal cells and that connexin57 (Cx57) is expressed exclusively in horizontal cells (Hombach et al., 2004). The variety of coupled neuronal networks in the retina (Vaney, 1991), together with molecular data (Güldenagel et al., 2000), suggests that additional connexins are expressed in the retina. Biochemical, electrophysiological, and immunohistological data indicate that the heterologous coupling between AII amacrine cells and ON cone bipolar cells involves a connexin isoform other than Cx36 on the bipolar cell side (Strettoi et al., 1992; Vaney et al., 1998; Massey and Mills, 1999; Feigenspan et al., 2001; Veruki and Hartveit, 2002).
In the mouse retina connexin45 (Cx45) protein has been detected in the outer and inner plexiform layers, and Cx45(lacZ) reporter mice revealed a blue punctate staining in the inner nuclear and ganglion cell layers (Güldenagel et al., 2000), making Cx45 a likely candidate for a bipolar cell connexin.
Here we demonstrate Cx45 expression in subpopulations of cone bipolar cells by using a transgenic mouse mutant that expresses enhanced green fluorescent protein (EGFP) instead of the Cx45 coding sequence under the control of the endogenous Cx45 promoter. Neuronal deletion of Cx45 was achieved by Cre-recombinase-mediated deletion of the Cx45 coding sequence with the use of Nestin-Cre (Nes-Cre) mice (Tronche et al., 1999), circumventing early embryonic lethality. In addition, these data were complemented by the analysis of transgenic mouse mutants expressing the lacZ gene under the control of the Cx45 promoter. We show that ON and OFF cone bipolar cells, but not rod bipolar cells, express Cx45. In ON cone bipolar cells Cx45 constitutes the counterpart to Cx36 in heterotypic gap junctions, which are likely to allow the passage of functional rod-derived signals between AII amacrine cells and ON cone bipolar cells.
Materials and Methods
Construction of the conditional Cx45 deletion vector. The targeting vector (pHB1) was cloned by using an ∼8 kb DNA fragment spanning all three exons of the Cx45 gene locus (Krüger et al., 2000). A frt-site-flanked selection cassette (PGK-neo, containing a neomycin-resistance gene under the control of the phosphoglycerate kinase promoter) followed by a single loxP site was cloned by EagI/ApaI digestion of the vector pM30 (Meyers et al., 1998) into an ApaI restriction site upstream of Cx45 exon3. A single loxP site was introduced by insertion of a HindIII/PvuI fragment derived from the vector ploxPvu (B. Teubner, unpublished data) into a SwaI restriction site of the 3′ homology region leading to a loxP-site-flanked (“floxed”) exon3 of the Cx45 gene locus. A HindIII/NcoI fragment containing parts of intron2 as well as the splice acceptor site of Cx45 exon3 was cloned into a NcoI site at the start codon of the EGFP coding sequence of pEGFP-N1 (Clontech, Palo Alto, CA). Finally, this HindIII/AflI fragment containing the reporter gene was cloned into a BbvCl restriction site in the 3′ homology region. Proper functionality of the frt and loxP sites of the 14 kb pHB1 vector was verified by subsequent transformation in Escherichia coli strains expressing Flp- or Cre-recombinase protein (Buchholz et al., 1998).
Transfection and screening of ES cell clones. For homologous recombination 100 μg of pHB1 plasmid DNA was linearized by Asp718I digestion and transfected by electroporation (800 V, 3 μF) into feeder-independent embryonic stem cells (HM1 ES cells) (cf. Magin et al., 1992). Selection of recombination events was performed by using 350 μg/ml G418 (Invitrogen, Karlsruhe, Germany). A total of 224 ES cell clones was tested for homologous recombination events by Southern blot hybridization, using genomic DNA restricted by XbaI digestion, and probed with a radioactively labeled XbaI/HindIII fragment from the 5′ homology region in front of exon1. Hybridization was performed by using QuikHyb hybridization solution (Stratagene, La Jolla, CA). Homologous recombination was demonstrated by the appearance of a 4.7 kb fragment, as opposed to a 5.7 kb fragment representing the wild-type allele. Recombination of the 3′ region and multiple integration were analyzed by using an EGFP probe that yielded an 8.0 kb band for the mutant allele. Finally, 6 of 224 clones showed correct recombination by Southern blot hybridization.
Generation and genotyping of transgenic mice. ES cells that showed proper recombination events and correct karyotypes were injected into C57BL/6 blastocysts as previously described (Theis et al., 2000). Male mice showing a high degree of coat-color chimerism were mated to C57BL/6 females, and agouti offspring were tested for germline transmission of the mutated allele (Cx45fl,neo) by PCR analysis of isolated tail DNA. The first generation of mice still harboring the frt-site-flanked PGK-neo cassette was tested for appearance of the 3′ loxP site (717 bp fragment) or wild-type 3′ untranslated region (UTR; 579 bp fragment), using the following primer combinations: IIpX3rev (5′-AAG AAC GGC CAC AAC TCT GGT AAC AGG AAG-3′) and IIpX3for (5′-TGG CTC TGG TTA TCT TCA GGG ATG CTG CTG TTG-3′); this was performed in the presence of 2 mm MgCl2 under the following conditions: 94°C for 5 min; 94°C for 45 sec, 70°C for 45 sec, and 72°C for 45 sec, 35 cycles; 72°C for 5 min. Heterozygous animals (Cx45fl,neo/+) were mated to transgenic mice expressing the Flp-recombinase gene (hACTB:FLPe mice) (Rodríguez et al., 2000), leading to removal of the frt-site-flanked PGK-neo selection cassette and generation of the Cx45fl allele. Subsequently, mice were backcrossed to C57BL/6 mice to increase C57BL/6 background, to remove genomic presence of the hACTB:FLPe transgene, and to generate homozygously floxed animals (i.e., Cx45fl/fl). Additionally, Cx45fl/+ mice were mated with PGK-Cre mice (Lallemand et al., 1998), resulting in mice lacking both the selection cassette and the loxP-site-flanked exon3 (Cx45del[EGFP] allele). To test for allelic recombination, we established a three primer PCR, detecting a 389 bp signal representing the wild-type allele, a 473 bp signal for the Cx45fl allele, and a 620 bp signal for the Cx45del[EGFP] allele. Because all experiments were performed by using mice with either a Cx45fl and/or a Cx45del[EGFP] allele, the PCR strategy of screening for germline transmission (see above) was replaced, and the new primer combination and PCR protocol were as follows: I5FCfor (5′-GGA TTA AAG GCA TAT GTC ACC ACT CTT GGC-3′), I3Frev (5′-CTC TAG GAA CAC TGT AAC CTG AGA TGT CCC-3′), and IIpX3rev (see above); this was performed in the presence of 2 mm MgCl2 under the following conditions: 94°C for 15 min and hot start at 80°C; 94°C for 1 min, 70°C for 1 min, and 72°C for 2 min, 40 cycles; 72°C for 10 min. Presence of the Cx45 knock-out allele, i.e., Cx45lacZ (Krüger et al., 2000), was tested as described previously. Genomic presence of the Cre-recombinase transgene, which is controlled by the nestin promoter (Tronche et al., 1999), was identified by PCR with the use of Nes-up (5′-TCC CTT CTC TAG TGC TCC ACG TCC-3′) as a forward primer annealing within the nestin promoter and Int-rev (5′-TCC ATG AGT GAA CGA ACC TGG TCG-3′) as a reverse primer annealing within the Cre-recombinase coding region. A 650 bp amplicon was observed by using the following parameters in the presence of 2 mm MgCl2: 95°C for 5 min; 95°C for 30 sec, 68°C for 50 sec, and 72°C for 1.5 min, 36 cycles; 72°C for 10 min. To prove correct genomic recombination events after Flp- and/or Cre-recombinase activity, we performed Southern blot hybridization, using as radioactive probes either a 1.2 kb PCR fragment representing the entire coding sequence for the Cx45 wild-type gene or a 0.73 kb BamHI/NotI fragment of the vector pMJ Green containing the coding sequence of the EGFP gene (J. Degen, unpublished results). Briefly, an AsnI/StuI fragment of pEGFP-N1 (Clontech), containing a CMV promoter and the coding region of the EGFP gene, was ligated into a NotI/ClaI fragment representing the vector backbone of pBEHpac18 (Horst et al., 1991). The vector pMJ Green allows for selection with puromycin and shows EGFP expression under the control of the CMV promoter. Genomic DNA from mouse liver with different allelic combinations was isolated, restricted by NcoI or XbaI digestion, electrophoretically separated, and processed for Southern blot hybridization. All mice were kept in strict accordance to governmental and institutional care regulations, maintained under standard housing conditions with a 12 hr dark/light cycle, and provided with food and water ad libitum.
Tissue preparation. Adult mice were anesthetized deeply with CO2 and killed by cervical dislocation. The eyes were enucleated and transferred to a Petri dish with mouse Ringer's solution containing the following (in mm): 137 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 d-glucose, and 5 HEPES, pH 7.4 (NaOH). The cornea and lens were removed, and the retina was dissected from the eyecup and cut in two halves with a scalpel. Each half was embedded in agar-agar (Roth, Karlsruhe, Germany), and vertical sections (200 μm) were cut with a vibratome (Leica, Nussloch, Germany). Slices were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer (PB) for 10 min and rinsed in 0.1 m PB several times before injection.
Injection of EGFP-positive bipolar cells and immunostaining. Slices were transferred to an injection chamber and injected under visual control with the use of sharp microelectrodes made of borosilicate glass (Hilgenberg, Malsfeld, Germany). EGFP-positive cells were identified by their green fluorescence and subsequently injected with Alexa-594 potassium hydrazide (Molecular Probes, Eugene, OR). The dye was diluted 1:4 with 0.1 m Tris, pH 7.4, and injected with a negative current of -1 nA for 3-10 min, using the current-clamp circuit of the EPC-9 amplifier (HEKA, Lambrecht, Germany). After injection the slices were rinsed in 0.1 m PB several times and then blocked with 5% normal goat serum (NGS) in 0.1 m PB containing 0.3% Triton X-100 for 1 hr at room temperature. Then the slices were incubated in either Cx36 polyclonal antibodies (51-6300, diluted 1:500; Zymed, San Francisco, CA) or a calretinin antibody (diluted 1:250; Swant, Bellinzona, Switzerland) at 4°C overnight. Antibodies were diluted in 0.1 m PB containing 3% NGS and 0.3 Triton X-100. On the next day the slices were rinsed for at least 30 min with 0.1 m PB and afterwards were incubated for 2 hr in 0.1 m PB containing 1% NGS, 0.3% Triton X-100, and the secondary antibody goat anti-rabbit Cy5 (1:200; Dianova, Hamburg, Germany). After three final washes the slices were mounted with Vectashield (Vector Laboratories, Burlingame, CA). Confocal micrographs of fluorescent specimen were taken with a Leica TCS SL confocal microscope. Scanning was performed with a 40×/1.25 Plan-Apochromat and a 63×/1.32 Plan-Apochromat objective at a resolution of 1024 × 1024 pixels. Scans of different wavelengths were performed sequentially to rule out overlap among red, green, and blue channels. Images were superimposed and adjusted in brightness and contrast with Photoshop 6.0 (Adobe, San Jose, CA).
Vertical cryosections (18 μm) were used for Nissl staining and detection of protein kinase Cα (PKCα) and glycine immunoreactivity by following the protocols of Feigenspan et al. (2001). Monoclonal antibodies against PKC were obtained commercially (Amersham Biosciences, Braunschweig, Germany), and polyclonal antibodies against glycine were a generous gift from Dr. David Pow (Vision, Touch, and Hearing Research Centre, University of Queensland, Brisbane, Australia). Nonspecific binding in retina sections was blocked with 10% NGS in 0.1 m PB containing 0.3% Triton X-100 for 1 hr at room temperature. Both primary antibodies were diluted 1:1000 in 0.1 m PB containing 0.3% Triton X-100, and retina sections were incubated overnight at 4°C. After several washes in 0.1 m PB, PKC and glycine immunoreactivity were visualized with goat anti-mouse Alexa 488 (Molecular Probes) (diluted 1:250 in 0.1 m PB/0.3% Triton X-100/2% NGS) and goat anti-rat Cy3 (Dianova) (diluted 1:400 in 0.1 m PB/0.3% Triton X-100/2% NGS). Sections were coverslipped, and confocal micrographs were taken and analyzed as described above.
Detection of Cx45 and mGluR6 transcripts in mouse retina and ON bipolar cells with the use of RT-PCR. RNA was extracted from wild-type mouse retina by means of an RNA preparation kit (NucleoSpin RNAII, Macherey-Nagel, Düren, Germany) according to the manufacturer's protocol. Bipolar cell RNA was obtained by patch clamping dissociated single bipolar cells, which were identified by their characteristic morphology and axon length.
The dissociation of mouse retina was performed as described previously (Feigenspan et al., 2000). In brief, retinas of 2- to 5-month-old wild-type mice were dissected in oxygenated Ca2+/Mg2+-free HBSS and transferred into 2 ml of digestion buffer containing Ca2+/Mg2+-free HBSS, 0.1 mm EDTA (Sigma, Seelze, Germany), 10 mm cysteine (Sigma), 20 U/ml papain (Worthington Biochemical, Freehold, NJ), and 200 U/ml DNase I (Sigma); then the retinas were subjected to enzymatic digestion (37°C for 45 min). To stop papain activity, we transferred retinas to trituration buffer (MEM; Sigma) containing 10% fetal calf serum (Sigma) and 100 U/ml DNase I (Sigma) and incubated them for 5 min at 37°C. The digested tissue was centrifuged at 1000 rpm (5 min, 22°C), and the pellet was resuspended in 2 ml of MEM. Trituration was performed in three steps with fire-polished Pasteur pipettes. The cell suspension containing bipolar cells was allowed to sediment on Concanavalin A-coated glass coverslips. These were kept in a 12-multiwell chamber, and sedimentation was performed in an incubator in 5% CO2/55% O2 at 37°C.
Cytoplasm of bipolar cells was aspirated with a patch pipette filled with an intracellular solution containing the following (in mm): 140 KCl, 3 MgCl2, 5 EGTA, 5 HEPES, pH 7.3 (NaOH). The tip of the pipette was broken into a PCR tube containing 20 U RNase inhibitor (RNasin; Promega, Madison, WI), and the sample (∼8 μl) was subjected to a brief centrifugation.
Amplification of contaminating genomic DNA was prevented by digestion with DNase I (amplification grade; Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. cDNA synthesis from 2-4 μg of retinal RNA and from bipolar cell samples was performed in final volumes of 50 and 20 μl, respectively. The samples contained 1× firststrand buffer (Invitrogen), 10 mm dithiothreitol (Invitrogen), 0.6 μm oligo-dT primer (Promega), 0.6 μm random primer (Promega), 0.5 mm of each dNTP (Eppendorf, Hamburg, Germany), and 0.8 U/μl RNasin ribonuclease inhibitor (Promega). After primer annealing for 5 min at 65°C, the samples were chilled on ice briefly and incubated for 10 min at room temperature and for an additional 2 min at 42°C before 10 U/μl Superscript II-RT (Invitrogen) was added. cDNA synthesis was performed at 42°C for 1 hr and stopped by incubation of the samples for 15 min at 70°C. cDNAs were stored at -20°C.
For the detection of Cx45 transcripts in mouse retina and bipolar cells, two specific sets of primers (MWG Biotech, München, Germany) were designed according to the mouse Cx45 coding sequence (cf. Hennemann et al., 1992) [National Center for Biotechnology Information (NCBI) accession number X63100]. The predicted size of the expected Cx45 fragments was 363 bp (primer set 1: USP1, 5′-TGT GTG CAA CAC AGA GCA GC-3′; DSP1, 5′-CCA TCC TCT CGA ATT CGT CG-3′) and 444 bp (primer set 2: USP2, 5′-TTC CAA GTC CAC CCA TTT TAT-3′, DSP2, 5′-ATC GTT CCT GAG CCA TTC TGA-3′). The expression of mGluR6 in mouse retina and ON bipolar cells was analyzed by one set of primers (MWG Biotech) (USP, 5′-CAT GGT CAC GTG TAC AGT GTA TGC-3′; DSP, 5′-CTT CTT GAG GCT GCG CTT CCG C-3′) designed according to the mGluR6 coding sequence of rat (cf. Nakajima et al., 1993) (NCBI accession number D13963). Hot-start PCRs were performed in a total volume of 25 μl, which included 2 μl of retinal or 6 μl of bipolar cell cDNA, 1× reaction buffer (Eppendorf), 0.2 mm of each dNTP (Eppendorf), 0.8 μm of each primer, and 1 U Taq polymerase (Eppendorf). Amplifications of Cx45 and mGlu6 transcripts were performed in an Eppendorf Mastercycler under the following conditions: (1) for the detection of the 363 and 444 bp Cx45 fragments, denaturation for 2 min at 95°C, the addition of Taq polymerase, 40 cycles each consisting of 30 sec at 95°C, 30 sec at 59°C, 30 sec at 72°C, and a final extension for 15 min at 72°C; (2) for the detection of the 298 bp mGluR6 transcript, denaturation for 2 min at 95°C, the addition of Taq polymerase, 35 cycles each consisting of 30 sec at 95°C, 30 sec at 63°C, 1.5 min at 72°C, and a final extension for 10 min at 72°C. Aliquots (20 μl) of the amplification products were analyzed on 2% agarose gels and visualized on a transilluminator after ethidium bromide staining. Fragments of the expected size were selected, isolated from the agarose gels with the use of a gel extraction kit (Nucleospin Extract, Macherey-Nagel), reamplified in second round PCRs with the same set of primers, and subjected to nucleotide sequencing (MWG Biotech or SeqLab, Göttingen, Germany). DNA similarities and identity scores were analyzed by matching the query sequence to database entries with BLASTN and FASTA algorithms.
Electroretinogram. Before an experiment the animals were dark adapted for at least 12 hr. Mice were anesthetized by intraperitoneal injections of xylazine (50 mg/kg) and ketamine (20 mg/kg), and the pupils were dilated with 1% atropine sulfate. Surgery and subsequent handling were done under dim red light. For electroretinogram (ERG) recordings the tip of a moistened Ag/AgCl cotton-wick electrode was lowered onto the center of the cornea with a micromanipulator, and a platinum needle serving as reference electrode was placed into the scalp. A grounding needle was inserted subcutaneously into the tail. The mouse was laid on its side and fixed with surgical tape. Electrical potentials were recorded, bandpass filtered (1-500 Hz), and averaged online with the PowerLab system equipped with the ML bioamplifier (AD Instruments, Hastings, UK).
Test lights were generated with a 150 W halogen lamp and focused onto the cornea. Intensities were adjusted with neutral-density filters, and test flash duration (20 msec) was controlled with an electromagnetic shutter. For experiments with background light a beam splitter was positioned into the light path, and constant background light from a second 150 W halogen source was mixed with the test light flashes. Background-intensity was adjusted with neutral-density filters. Interstimulus intervals were 10 sec for dark-adapted conditions and 1.87 sec for light-adapted conditions. At least 10 responses were averaged at each intensity. Corneal illuminance (lux) was measured with a calibrated luxmeter (Gossen, Nürnberg, Germany) at the position of the cornea.
Data analysis was done with Chart v5.1, using the evoked response extension for measuring amplitudes (AD Instruments) and with JMP v5 (SAS Institute, Cary, NC) for statistical analysis. a-Wave amplitudes were determined with respect to averaged baseline before light flash onset, and b-wave amplitudes were determined as peak-to-peak amplitudes. At high intensities this included oscillatory potentials riding on top of the b-wave (see Fig. 5A). To compare the results for Cx45- and Cx36-deficient mice from this paper and the paper by Güldenagel et al. (2001), we had to replot the data from Cx36 mice, because b-wave amplitudes reported by Güldenagel et al. (2001) (see Fig. 6) were measured with respect to baseline and not as peak-to-peak amplitudes. Implicit times were measured from light onset to maximum responses.

ERG of wild-type and Cx45-deficient mice (A-D) and comparison of Cx36- and Cx45-deficient mice (E, F). A, Example of scotopic ERGs in response to 20 msec white light flashes at 3800 lux. Short dashed lines indicate how response amplitudes of a-wave (a) and b-wave (b) were measured (long dashed line, baseline). B, Example of photopic ERGs in response to light flashes at 1450 lux; adapting light of 4 lux corneal illuminance. C, Scotopic intensity-response curves for the a-waves (squares) and b-waves (circles) of wild-type (open symbols) and mutant (filled symbols) Cx45 mice. The data points plot the mean ± SEM (n = 8). Wild-type mice showed significantly larger b-wave amplitudes for corneal illuminance in excess of 10 lux (Mann-Whitney test; *p < 0.05; **p < 0.01). D, Photopic intensity-response curves for the b-waves of wild-type (open circles; n = 6) and mutant (filled circles; n = 5) Cx45 mice under different levels of light adaptation, indicated by the numbers on the right. E, Comparison of normalized scotopic mean b-wave intensity-response curves of Cx36- and Cx45-deficient mice with their respective wild-type littermates. Peak-to-peak b-wave amplitudes were normalized to the maximum responses of the respective wild type. In the case of Cx36 the original data from Güldenagel et al. (2001) were reanalyzed to determine peak-to-peak b-wave amplitudes as shown in A (see also Materials and Methods). F, The relative differences between b-wave amplitudes of wild-type and Cx-deficient mice were very similar for Cx36 and Cx45 and yielded sigmoidally shaped intensity difference-response curves. Curves could be well fit with the following Hill equation: [V/Vmax = Lh/(Lh + L50h)], with V equal to relative difference-response, Vmax equal to maximal difference-response, L equal to corneal illuminance, h equal to Hill coefficient, and L50 equal to illuminance evoking the half-maximal response. The fits yielded the following values: for Cx36, Vmax = 0.49; h = 0.41; L50 = 5.7; R2 = 0.955; and for Cx45, Vmax = 0.40; h = 0.61; L50 = 5.6; R2 = 0.972.

ON bipolar to AII amacrine cell gap junctions. A, EGFP labeling was not present in injected AII amacrine cells. B, Isolated EGFP-positive cone ON bipolar cells (green) labeled with an antibody against Cx36 (red). Arrowheads point to Cx36-reactive puncta on the ON bipolar cell terminals. C, D, Stack projection of an EGFP-positive (blue) ON bipolar cell (type 5) filled with Alexa594-hydrazide (red). The pattern of Cx36 immunoreactivity is superimposed on the cell and appears in green. D, Enlarged view of the axon terminal of the cell in C. E, Single confocal section from the stack in D. Arrows point to Cx36-reactive puncta on the ON bipolar cell terminals, whereas arrowheads indicate Cx36 labeling in close vicinity of the axon terminal. Scale bars: A, 20 μm; B, 10 μm; C-E, 5 μm.
Results
Construction of the targeting vector and transgenic mice
Because general ablation of Cx45 resulted in early embryonic lethality at embryonic day 10.5 caused by cardiovascular abnormalities (cf. Krüger et al., 2000; Kumai et al., 2000), we generated mice in which cell type-specific deletion of Cx45 could be accomplished by Cre-recombinase activity, leading to the expression of the reporter gene EGFP. For construction of the targeting vector, exon3 of the connexin45 gene locus harboring the entire coding region was flanked by loxP sites (Fig. 1A). Additionally, a neomycin-resistance gene under the control of the phosphoglycerate kinase promoter (PGK-neo), which was surrounded by frt sites allowing Flp-recombinase-mediated deletion of the selection cassette, was introduced into the intron between exon2 and exon3 of the Cx45 gene. After transfection of the targeting vector pHB1 into ES cells (HM1 ES cells) and G418 selection, 224 ES cell clones were isolated and expanded; aliquots were subjected to Southern blot hybridization (results not shown). Six clones showed correct homologous recombination in the targeted Cx45 allele, yielding a recombination frequency of 2.7%. Five clones were used for blastocyst injection, leading to chimeric offspring. From these, three independent ES cell clones led to the birth of agouti-colored heterozygous offspring. Initially, detection of germline transmission was performed by discrimination between the presence or absence of the 3′ loxP site (Fig. 1D).
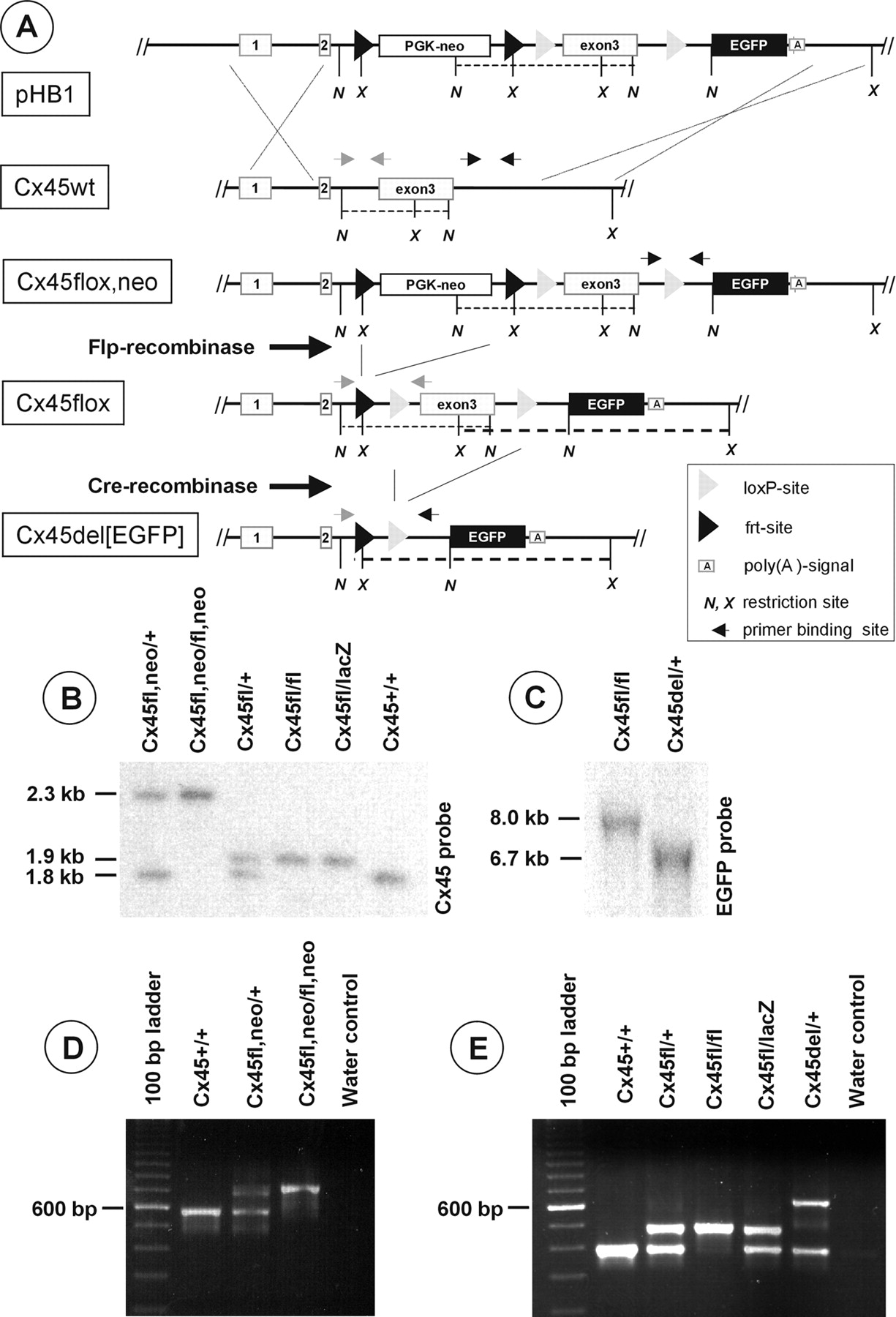
A, Targeting schematic and Flp-/Cre-mediated recombination of the Cx45 gene locus. The targeting vector pHB1 comprising a loxP-site-flanked Cx45 exon3 and a frt-site-flanked PGK-neo selection cassette was transfected into HM1 ES cells. Homologously recombined ES cell clones were tested by Southern blot analysis (data not shown) and injected into C57BL/6 blastocysts. Heterozygous mice showing germline transmission of the mutated Cx45fl, neo allele were mated first with Flp-recombinase-expressing mice to generate a Cx45fl allele with a deleted selection cassette. Then, heterozygous offspring were mated with Cre-recombinase-expressing mice to generate a Cx45del[EGFP] allele deficient for both the selection cassette and the Cx45 exon3 but expressing EGFP under the control of the endogenous Cx45 promoter. Restriction sites include the following: N, NcoI; X, Xba I. Black arrows indicate primer sites for testing for germline transmission; gray arrows show primer binding sites for detecting the presence of the PGK-neo resistance cassette. Bold dashed lines reflect XbaI fragments; thin dashed lines represent NcoI fragments. B, Correct genomic recombination after Flp-/Cre-mediated deletion was verified by Southern blot hybridization that used a 1.2 kb Cx45 PCR fragment as a radioactively labeled probe for the Cx45fl, neo (2.3 kb), Cx45wt (1.8 kb), or Cx45fl (1.9 kb) alleles after digestion by NcoI of liver DNA. C, Southern blot analysis of the Cx45del[EGFP] allele after digestion with XbaI, electrophoresis, and probing for EGFP. D, Agouti-colored offspring were tested for germline transmission of the Cx45fl, neo allele, using a two primer PCR. A 717 bp amplicon represents the mutated Cx45fl, neo allele, and a 579 bp amplicon represents the wild-type allele. E, A three-primer PCR discriminates among three different amplicons indicating the presence of the Cx45wt (389 bp), Cx45fl (473 bp), or Cx45del[EGFP] (620 bp) allele. Detection of the Cx45lacZ allele has been described previously (Krüger et al., 2000).
Transgenic animals with the Cx45fl,neo allele subsequently were bred to hACTB:FLPe mice (Rodríguez et al., 2000), resulting in Flp/frt-mediated removal of the PGK-neo selection cassette generating the Cx45fl allele (Fig. 1B,E). To generate theCx45del[EGFP] (Fig. 1C,E) allele, which represents an alternative Cx45 knock-out allele to that described by Krüger et al. (2000), we mated Cx45fl/fl mice to PGK-Cre mice (Lallemand et al., 1998). Heterozygous Cx45fl mice were backcrossed into aC57BL/6 genetic background and finally were mated to each other to generate homozygous Cx45fl/fl mice. Cell type-specific deletion was achieved by using Cx45fl/fl females that were bred with Cx45lacZ/+:Nes-Cre males. Because the inheritance of Cre transgenes from the mother, but not the father, had been reported to result in total ablation of the targeted gene [generation of a “del” allele by αMHC-Cre activity (cf. Eckardt et al., 2004) or PGK-Cre activity (Lallemand et al., 1998)], male Cre conductors have always been selected to avoid this phenomenon. The successful recombination events for generating the Cx45fl and Cx45del[EGFP] allele were tested by PCR and Southern blot analyses (Fig. 1B-D).
Cellular localization of Cx45
The retina is a highly organized and structured neuronal network with distinctly ordered layers. We first examined whether the deletion of Cx45 in Cx45fl/fl:Nes-Cre mice affected these characteristics. Nissl staining revealed that the retina was organized normally in layers of dimensions similar to the wild type (Fig. 2A,B). Cx45 deficiency did not affect the number, size, or density of the nuclei in the three nuclear layers, and mutant mice did not show gross morphological alterations at the light microscopic level.

Cellular localization of EGFP signals. Vertical sections of Cx45fl/fl (A) and Cx45fl/fl:Nes-Cre (B) retina counterstained with toluidine blue showed no obvious differences in their overall structure. C, EGFP label is present in bipolar, amacrine, and putative ganglion cells. Scale bars, 25 μm.
Commercially available antibodies against Cx45 were not suitable for immunohistochemistry in the retina, and we therefore were not able to compare directly the expression pattern of Cx45 in wild-type and transgenic animals. However, the EGFP signals overlapped with the lacZ signals in retinas from Cx45fl/lacZ:Nes-Cre animals, confirming an expression of the EGFP reporter gene corresponding to the one described for Cx45 by Güldenagel et al. (2000) (data not shown).
The expression of EGFP under the control of the Cx45 promoter in Cx45fl/fl:Nes-Cre mice revealed distinctly labeled neurons in the inner nuclear layer (INL) and in the ganglion cell layer (GCL), together with some blood vessels (Fig. 2C). The outer nuclear layer was completely void of labeling, excluding photoreceptors as Cx45-expressing cells. Within the INL bipolar cells and amacrine cells were easily discernible, based on their characteristic morphology. Labeled cell bodies in the GCL could represent either ganglion cells or displaced amacrine cells, which are quite frequent in the mouse retina (Jeon et al., 1998).
The different shapes and locations of the bipolar cells somata suggested that the EGFP-labeled cells comprised more than one subpopulation. In fact, closer examination revealed that some axons ramified within the distal inner plexiform layer (IPL) and some within the proximal part of the IPL, indicating that ON- and OFF-type bipolar cells were among the labeled cells. This finding, together with the relatively small number of labeled cells, most likely excludes rod bipolars from the labeled cells.
The population of labeled amacrine cells also appeared to comprise more than one subpopulation. One subtype had its soma located right at the INL/IPL border and several dendrites leaving its soma; another subtype had its soma farther away from the INL/IPL border and only one primary dendrite leaving its soma. The size of the somata also suggested that the labeled cells in the ganglion cell layer did not make up a homogenous population. There was a concentration of labeled dendrites within the outer and inner third of the IPL, whereas the middle third was almost void of labeling. Strong profiles irregularly crossing the inner retina at various locations are blood vessels; their smooth muscle cells are known to express Cx45 (Krüger et al., 2000).
Morphological classification of EGFP-positive bipolar cells
To identify the EGFP-positive bipolar cells morphologically, we performed intracellular dye injections. In the INL the EGFP-positive cells were identified by their green fluorescence and subsequently were injected with the red fluorescing dye Alexa594-hydrazide. We observed the filling of the cells by using epifluorescence. After injection the slices were immunostained for calretinin to reveal the stratification pattern of the injected cells (Haverkamp and Wässle, 2000). Their morphology was analyzed with confocal microscopy. We used the shape and level of stratification of the axon terminals in the IPL as the major criteria for classifying the injected cells according to the nomenclature for the mouse retina provided recently by Ghosh et al. (2004). According to this nomenclature, we identified EGFP-positive cells as cone ON and OFF bipolar cells.
Figure 3 shows four EGFP-positive Alexa-injected OFF bipolar cells. Each micrograph represents a different type of cell (Fig. 3A-D). The putative type 1 OFF bipolar cell (Fig. 3A) had a stout dendritic tree, and its cell body was located quite in the middle of the INL, as described by Ghosh et al. (2004). A type 2 bipolar cell is shown in Figure 3B with a dense plexus of varicosities in sublamina 1 of the IPL. Injecting EGFP-positive cells, we also found type 3 OFF bipolar cells with a delicate dendritic tree and an axon terminal that stratified to sublamina 2 in the IPL (Fig. 3C). Type 4 bipolar cells were EGFP-positive as well. Injections revealed the morphology of these cells characterized by a diffuse axon terminal with varicosities in both sublaminas 1 and 2 of the IPL (Fig. 3D).

EGFP-positive OFF bipolar cells (A-D) and ON bipolar cells (F-H). The injected cells are shown in red; the EGFP staining appears in green; and, as a reference for the stratification pattern, D and H show the calretinin immunolabeling in blue. A-D, Confocal fluorescence micrographs of four OFF bipolar cells representing the four different OFF bipolar cell types (BP 1-4) (Ghosh et al., 2004). Injection of EGFP-positive cells with Alexa594-hydrazide resulted in the yellow appearance of the cell body, revealing that all OFF bipolar cell types express Cx45. F-H, Confocal fluorescence micrographs of three ON bipolar cells representing the ON bipolar cell types 5-7 (Ghosh et al., 2004). The yellow staining of injected somata shows that the three major types of cone ON bipolar cells (type 5-7) express Cx45. We have indicated the stratification levels in the IPL as S1-S5. The corresponding Nomarski images of the vertical retina sections are shown in E and I. The retinal layers are indicated: ONL (outer nuclear layer), OPL (outer plexiform layer), INL, and IPL. Scale bars, 20 μm.
We found prominent EGFP expression in ON cone bipolar cells as well. Figure 3F--H shows three representative examples of ON cone bipolar cells, positive for EGFP and injected with Alexa594-hydrazide. Again, immunostaining of calretinin was used as a reference for the stratification levels of the axon terminals. Figure 3F shows an ON bipolar cell for which the axon terminal is restricted to sublamina 3 of the IPL. Therefore, this cell was classified as type 5, according to Ghosh et al. (2004). Figure 6C shows another EGFP-positive example of this cell type. In addition, we identified type 6 ON bipolar cells as EGFP-positive. The cell shown in Figure 3G had a rather delicate axon terminal that ended in layers 3 and 4 of the IPL. We also found EGFP expression in type 7 ON bipolar cells. Injections revealed the morphology of these cells with a narrowly stratified axon terminal just below sublamina 4 in the IPL (Fig. 3H).
These data suggested that ON- and OFF-type cone bipolar cells express Cx45. The expression of Cx45 in ON bipolar cells was analyzed further by using RT-PCR at the single-cell level. Cone and rod bipolar cells were isolated from the retina and identified according to their particular morphology. A characteristic feature of isolated ON bipolar cells is their relatively long axon and a rather stout axonal terminal (Fig. 4A). In general, the axon terminal and the somata of rod bipolar cells (cell 8) are larger than those of cone bipolar cells. Cells were patched, and the cytoplasm was harvested with the patch pipette. We performed RT-PCR on the samples with two different sets of primers to detect mRNA for Cx45 and mGluR6, the latter being expressed exclusively in ON bipolar cells (Masu et al., 1995). The corresponding amplicons (Fig. 4B) showed that all cells expressed mGluR6 and that cells 1-6 also expressed Cx45. Cells 8 and 9, which were identified as rod bipolar cells based on their morphology, did not express Cx45. This experiment was repeated with a second primer pair for Cx45, resulting in an amplicon of 363 bp and confirming the above results (data not shown). The molecular data are therefore in line with the histological data, demonstrating that ON cone bipolar cells express Cx45, but rod bipolar cells do not. We controlled this by immunolabeling rod bipolar cells with antibodies against PKC (Greferath et al., 1990) in vertical sections of Cx45fl/fl:Nes-Cre mice (Fig. 4C). The immunosignal for PKC indeed did not colocalize with the EGFP label in bipolar cells, confirming that rod bipolar cells do not express Cx45.
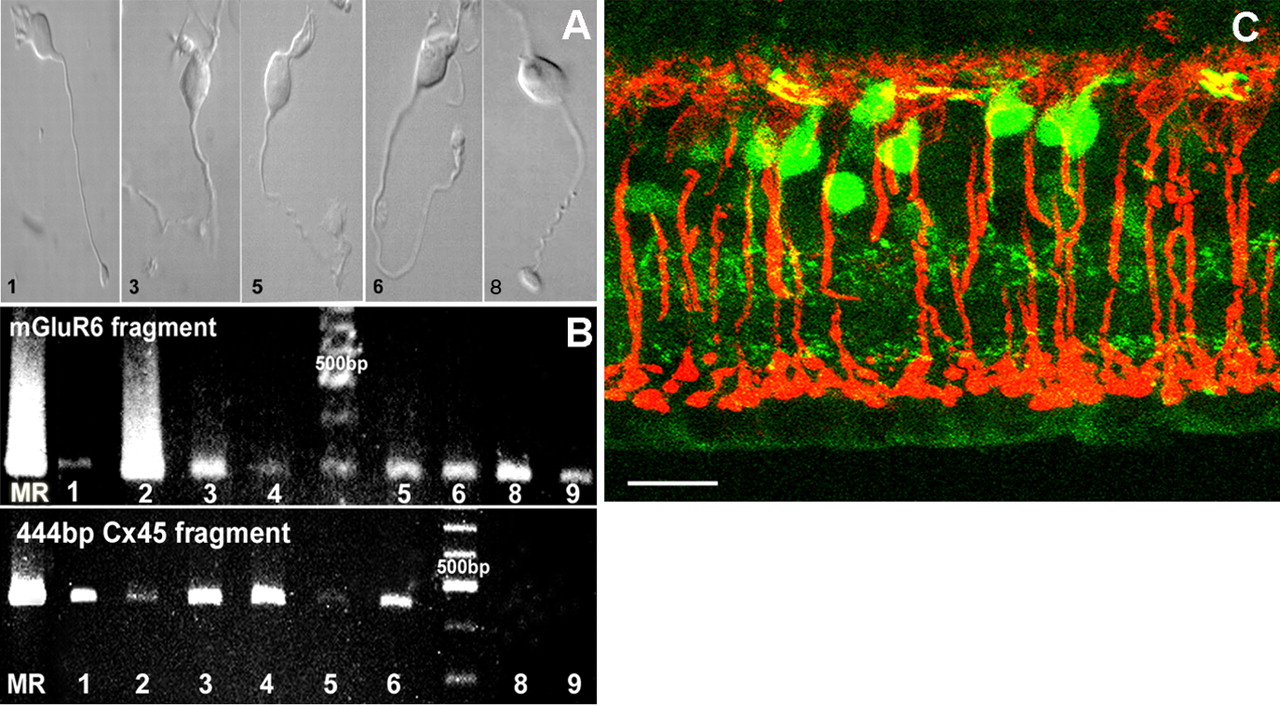
Detection of Cx45 in ON cone bipolar cells but not rod bipolar cells. A, Samples of ON cone bipolar cells (1, 3, 5, 6) and rod bipolar cells (8) subjected to RT-PCR for the detection of Cx45 and mGluR6 transcripts. B, Ethidium bromide-stained 2% agarose gels showing the detection of the 298 bp mGluR6 and 444 bp Cx45 transcripts in ON cone bipolar cells (1-6). No Cx45 transcript was found in rod bipolar cells (8, 9, bottom panel). C, PKC immunoreactivity in vertical sections of Cx45fl/fl:Nes-Cre mouse retina was localized exclusively in rod bipolar cells and showed no colocalization with EGFP-expressing cone bipolar cells. Scale bar, 20 μm.
ERG of Cx45-deficient mice
The presence of Cx45 in ON-type cone bipolar cells suggested that this connexin forms hemichannels in these cells and possibly forms the gap junctional channels with AII amacrine cells (Feigenspan et al., 2001). If this were the case, then one would expect an impairment of the rod pathway in Cx45-deficient animals similar to the one that has been described for Cx36-deficient mice (Güldenagel et al., 2001; Deans et al., 2002). We therefore tested the ERG of Cx45fl/fl:Nes-Cre animals under conditions similar to those used for the Cx36-deficient animals (Güldenagel et al., 2001).
Although the exact origin and generation of the different ERG waves still are not understood completely, it has become clear that the negative a-wave mainly originates in photoreceptor cells and the positive b-wave principally arises from depolarizing bipolar cells (Knapp and Schiller, 1984; Stockton and Slaughter, 1989; Steinberg et al., 1991; Masu et al., 1995). The oscillatory potentials riding on top of the b-wave are suggested to originate mainly in the inner retina (Wachtmeister and Dowling, 1978).
Scotopic ERG recordings from dark-adapted Cx45-expressing mice designated as wild-type (for example, Cx45fl/fl or Cx45fl/+ or Cx45fl/+:Nes-Cre) and Cx45-deficient mice (Cx45fl/fl:Nes-Cre or Cx45fl/lacZ:Nes-Cre) that followed a 20 msec white light flash showed both wave components (Fig. 5A). We quantified the positive and negative components by measuring their maximal amplitudes, including possible oscillations riding on top of the b-wave (Feigenspan et al., 2001). Although the a-wave did not differ significantly, the b-wave amplitudes of Cx45-deficient mice were considerably smaller than in wild-type animals (Fig. 5C). Because the b-wave has a lower threshold than the a-wave, both components were recorded as a function of illumination intensities. In wild-type mice the b-wave appeared ∼2 log units earlier than the a-wave and increased with light intensity. Although the threshold of the b-wave of the mutant animals was similar, the amplitudes were decreased significantly. The threshold of the a-wave was similar in both types of animals, and the amplitude gradually increased with higher intensities. The implicit time functions for both waves were identical in the two animal groups (data not shown).
Next we analyzed the cone pathway by recording light-adapted ERGs (Fig. 5B). Light flashes were superimposed on background illumination of increasing intensities. With a low background of 0.37 lux, wild-type b-wave amplitudes were, on average, still larger than in Cx45-deficient mice, although this difference was no longer significant (Fig. 5C). Increasing background illumination further reduced this difference, and at the highest background intensity that was used (400 lux), b-wave response amplitudes became identical (Fig. 5D). Over the tested range of background illumination, increment threshold functions were identical for wild-type and Cx45-deficient mice (data not shown).
The effects seen in the scotopic ERGs were compared with those that we reported for Cx36-deficient animals (Güldenagel et al., 2001). To do this, we normalized the peak-to-peak b-wave amplitudes to the wild-type maximal amplitudes, because absolute amplitudes differed between the mouse strains and recording conditions were not completely identical. Figure 5E shows that the relative reduction of these peak-to-peak amplitudes (including oscillations) from wild-type to deficient mice was very similar for Cx45- and Cx36-deficient animals. For both mouse populations the difference in the relative b-wave amplitudes between wild-type and deficient mice was plotted against corneal illuminance in Figure 5F. The resulting difference curves were nearly identical and showed a sigmoidal dependence on corneal illuminance, which could be well fit with Hill equations.
AII amacrine-ON cone bipolar cell coupling is disrupted in Cx45-deficient mice
The similarity of the difference curves hinted to a defect in the rod pathway (Güldenagel et al., 2001) in the Cx45-deficient animals. This defect does not result from a deficiency on the AII amacrine cell side, because AII amacrine cells do not express Cx45 (Fig. 6A), but more likely from a defect on the ON bipolar side, where Cx45 is expressed. To see whether Cx36 is in close association with axon terminals of Cx45-expressing ON bipolar cells, we tested for Cx36 expression in Cx45fl/fl:Nes-Cre mice and immunostained vertical retinal slices and isolated bipolar cells. Figure 6B shows two representative isolated cone ON bipolar cells that are EGFP-positive. Their axon terminals are decorated with puncta indicating Cx36 (arrowheads), whereas the cell body and the proximal axon display virtually no Cx36 staining. Figure 6C shows an Alexa594-injected cell (type 5 ON bipolar) positive for EGFP and superimposed with the immunostaining against Cx36. The picture represents a stack of 50 optical sections, 0.2 μm each, projected onto a single plane. Colors were changed (Alexa-injected, red; EGFP, blue; Cx36 staining, green) to ensure better visualization of Cx36-positive puncta. Soma and dendrites, as well as the proximal part of the axon, displayed no overlap with Cx36 puncta. However, in the region of the axon terminal Cx36-positive puncta appeared to be localized on or in close vicinity to the ON bipolar cell terminal. At higher magnification (Fig. 6D,E) Cx36-positive puncta could be observed decorating individual dendrites within the terminal structure.
Is the localization of Cx36-positive puncta on EGFP-labeled profiles indicative of Cx36/Cx45 gap junctions at these sites? It has been shown that the coupling between AII amacrine cells and ON cone bipolar cells forms the basis of neurotransmitter coupling (Vaney et al., 1998). Cone bipolar cells show an elevated level of glycine that is not derived from high-affinity uptake or de novo synthesis but is obtained by neurotransmitter coupling, in particular through gap junctions with the glycinergic AII amacrine cells, and is reduced strongly in Cx36-deficient mice (Güldenagel et al., 2001). In the case of heterologous Cx36/Cx45 gap junctions one would expect a similarly reduced level of glycine in ON cone bipolar cells in Cx45-deficient animals. We used an antibody against glycine to compare neuronal glycine levels in wild-type and homozygous Cx45-deficient mice (Fig. 7A,B) as well as in heterozygous animals (Fig. 7D). The number of heavily labeled amacrine cell somata was approximately the same in all three populations; the number of labeled cone bipolar cells, however, was reduced strongly in Cx45-deficient animals. The presence of EGFP labels (Fig. 7C) demonstrated that cone bipolar cells were still present in Cx45fl/fl:Nes-Cre mice but indeed void of glycine. It also demonstrated that glycinergic amacrine cells, including AII amacrine cells, do not express EGFP label, confirming that AII amacrine cells do not express Cx45. In heterozygous animals we observed some bipolar cells that were EGFP-positive and positive for glycine. This confirms that glycine-positive bipolar cells indeed express Cx45 (Fig. 7D-F).

Neurotransmitter coupling between ON bipolar cells and AII amacrine cells. Immunostaining of wild-type (A), Cx45fl/fl:Nes-Cre (B), and Cx45fl/+:Nes-Cre (D) mouse retina with polyclonal antibodies against glycine. In wild-type and heterozygous Cx45-deficient animals the glycinergic amacrine cells were labeled strongly, and cone bipolar cells showed a weaker but still significant labeling (A, D). Immunoreactivity of amacrine cells was similar in homozygous Cx45-deficient mice, whereas only very few bipolar cells appeared to be immunoreactive for glycine, although the number of cone bipolar cells as revealed by the EGFP expression (C) was not reduced. E, EGFP expression in the Cx45fl/+:Nes-Cre mouse retina. F, Overlay of the two fluorescent images. Arrowheads point to bipolar cells that are positive for both EGFP and glycine. Scale bars: A-C, 40 μm; D-F, 25 μm.
Discussion
In this study neuron-directed replacement of the Cx45 gene by the reporter gene EGFP was used to examine the role of Cx45 in visual transmission. The strategy circumvented embryonic lethality caused by heart failure and allowed for the identification of Cx45-expressing cells together with functional analysis. Diverse populations of retinal neurons, including almost every type of cone bipolar cell, displayed Cx45 expression. Coupling between AII amacrine cells and ON cone bipolar cells was disrupted and the b-wave of the scotopic ERG reduced, indicating that Cx45 is required in the rod signaling pathway.
Cx45 expression in the retina
Cx45 is an essential protein for the development of the cardiovascular system, and its deletion results in early embryonic death (Krüger et al., 2000; Kumai et al., 2000; Nishii et al., 2003). We avoided embryonic lethality by using the Cre/loxP system with Nes-Cre, which led to deletion of Cx45 in neuronal cells and expression of the EGFP reporter gene. Some peripheral blood vessels also expressed EGFP, possibly because of ectopic expression of Nes-Cre in smooth muscle cells. The transgenic animals developed normally and showed no apparent abnormalities. Their retinas displayed all nuclear and interplexiform layers with no morphological changes. In line with the study by Güldenagel et al. (2000) demonstrating expression of Cx45 based on a lacZ reporter gene, an abundance of neurons within the INL was labeled with EGFP and identified as bipolar and amacrine cells. We identified four types of OFF cone bipolar cells (types 1-4) (Ghosh et al., 2004) and three types of ON cone bipolar cells (types 5-7) (Ghosh et al., 2004) as EGFP-positive and thus Cx45-expressing. Bipolar cells identified immunocytochemically and morphologically as rod bipolar cells never showed EGFP labeling. Single-cell RT-PCR confirmed the expression of Cx45 in cone ON bipolar cells. A recent histological analysis involving several transgenic mouse models (Feigenspan et al., 2004) localized Cx36 to the dendrites of cone OFF bipolar cells. Our finding that all four classes of OFF cone bipolar cells express Cx45 opens the possibility that both Cx36 and Cx45 are expressed by the same bipolar cell. Because the subcellular localization of Cx45 is not known, a functional role for Cx45 in OFF bipolar cells is, however, difficult to assign.
Cx45 in the rod pathway
A key element of the rod pathway in the inner retina is gap junctional coupling between AII amacrine cells and ON cone bipolar cells. Several studies identified Cx36 as the channel-forming protein on the amacrine cell side (Feigenspan et al., 2001; Güldenagel et al., 2001; Mills et al., 2001; Deans et al., 2002), but the identity of the connexin involved on the bipolar side remained controversial.
Two lines of physiological evidence presented in this study indicate that Cx45 is expressed in ON bipolar cells. First, neurotransmitter coupling between AII amacrine cells and cone bipolar cells (Vaney et al., 1998) was disrupted in Cx45-deficient animals, and, second, these deficient animals showed ERG abnormalities that were very similar to those observed in Cx36-deficient mice. Whereas the a-wave was not affected, the b-wave revealed a significant reduction in amplitude, indicating a functional deficit in the ON pathway downstream of the photoreceptors (Peachey et al., 1993; Masu et al., 1995). The sigmoidal shape of the intensity difference-response curves (Fig. 5F) is in line with the assumption that the disrupted transmission from AII amacrine cells to cone ON bipolar cells is, as in the Cx36-deficient animals, mainly responsible for the ERG differences between wild-type and Cx45-deficient mice. If one accepts that the ERG b-wave arises mainly from depolarization of ON bipolar cells, the intensity dependence of the difference should resemble the rod intensity-response curves multiplied by unknown nonlinear factors, reflecting the coupling strengths between AII and cone ON bipolar cells and between rods and cones. Indeed, comparing the intensity difference-response curves from Figure 5F with reported intensity-response curves from rat rod photoreceptors suggests that this is the case (Nakatani et al., 1991).
Although the ERGs of the Cx36- and Cx45-deficient animals were mostly similar, several differences were found that can be explained by the different cellular localizations of the two connexins. First, oscillatory potentials (OPs) survived in Cx45-deficient mice both in the dark- and slightly light-adapted states (Fig. 5A,B). This was not the case in Cx36-deficient mice, which showed very small if any OPs (Güldenagel et al., 2001). This is a further indication that OPs are generated by the AII network that per se is uncoupled in Cx36-deficient mice but should be intact in Cx45-deficient animals. Second, Cx36-deficient mice showed an unexpected decrease in b-wave sensitivity under photopic conditions compared with wild-type, which was not seen in Cx45-deficient mice. This supports the speculation by Güldenagel et al. (2001) that “leaky” hemichannels in ON cone bipolar cells of Cx36-deficient animals might have led to this decreased sensitivity, because hemichannels should not exist in ON cone bipolars of Cx45-deficient mice. In addition, the ERG b-wave of Cx45-deficient mice became more and more indistinguishable from the wild-type ERG with increasing light adaptation (Fig. 5B,D). This can be explained by assuming that wild-type animals approach the functional state of the Cx45-deficient mice by increasingly closing the gap junctions between AII and ON cone bipolar cells at higher light intensities.
Because AII amacrine cells do not express Cx45, our data strongly suggest that Cx36 and Cx45 form heterotypic and maybe even heteromeric gap junctions between AII amacrine cells and ON bipolar cells. Are there any physiological data that support or contradict this view? Teubner et al. (2000) did not detect transfer of microinjected Neurobiotin between Cx36- and Cx45-transfected HeLa cells. However, this negative result could be attributable to low expression of the corresponding connexins in these HeLa transfectants. Alternatively, essential cofactors for modification of these connexins might be expressed only in retinal neurons. Veruki and Hartveit (2002) characterized the electrical synapse between AII amacrine cells and ON bipolar cells in the rat retinal slice by using dual whole-cell recordings. Although they measured symmetrical junctional conductances in these gap junctions, they found a strong asymmetry of coupling coefficients, indicating that transmission will be more effective from AII amacrine cells to ON bipolar cells than in the other direction. This functional asymmetry agrees with morphological data from Strettoi et al. (1992), who reported a structural asymmetry of these gap junctions. Mills and Massey (1995) showed that larger biotinylated tracers pass rather poorly through gap junctions between AII amacrine cells and bipolar cells, whereas they pass easily through homotypic gap junctions between AII amacrine cells. However, tracer coupling seems to be bidirectional between AII amacrine and ON bipolar cells in rabbit retina (Trexler et al., 2001), in agreement with physiological data from Xin and Bloomfield (1999), who demonstrated that cone signals, presumably transmitted by means of the ON bipolar/AII amacrine cell gap junctions, can be measured in AII amacrine cells. Thus physiological data neither support nor contradict conclusively the possibility that Cx36 and Cx45 form the heterotypic gap junctions between AII amacrine and ON bipolar cells. In expression systems the channels formed by Cx36 or Cx45 show different transjunctional voltage dependences, and Cx36 channels are considerably less sensitive to voltage changes than are Cx45 channels (Steiner and Ebihara, 1996; Srinivas et al., 1999). The voltage difference between AII amacrine and ON cone bipolar cells certainly will vary, depending on the ambient light conditions, and one might speculate that the different transjunctional voltage properties of the two connexins will support the light-dependent opening state of this gap junction (Mills and Massey, 1995).
So far, the functional data referring to connexins in the retina highlight their importance in the rod pathway. However, the patterns of expression of known connexins in the mouse retina also strongly indicate an involvement in the cone pathway and in multiple functional aspects. Because not only do connexins act as channel-forming proteins in gap junctions but at least some connexin isoforms may form hemichannels (Kamermans et al., 2001; Goodenough and Paul, 2003; Pottek et al., 2003), this involvement might be far more complex.
Footnotes
Work in the Bonn laboratory was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 400-E3 and Wi 270/22-3/4 to K.W.). Work in the Oldenburg laboratory was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 517-A2 to R.W. and JA 854/1-1 to U.J.-B.), the European Community (Cortical Visual Neuroprosthesis for the Blind to J.A.), and the Stiftung Volkswagenwerk (to R.W.). We thank Gerda Hertig and Ina Fiedler (Bonn) as well as Bettina Kewitz and Susanne Wallenstein (Oldenburg) for technical assistance and Jennifer Shelley for reading this manuscript.
Correspondence should be addressed to Dr. Reto Weiler, Department of Neurobiology, University of Oldenburg, D-26111 Oldenburg, Germany. E-mail: reto.weiler{at}uni-oldenburg.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250566-10$15.00/0
↵* S.M. and K.D. contributed equally to this work





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}