

Abstract
Low-dose intravenous cocaine administration to pregnant rabbits causes permanent structural alterations in dopamine-rich cerebral cortical areas, substantially reduced dopamine D1 receptor coupling to Gs-protein, and deficits in cognitive function. The developmental influences of reduced D1–Gs coupling and the underlying cellular basis are unknown. Using primary neuronal cultures derived from the medial frontal cortex and striatum of in utero saline- and cocaine-exposed embryos, spontaneous neurite outgrowth of in utero-exposed cortical neurons was greater than in control neurons. In contrast, striatal neurons exposed to cocaine in utero exhibited an entirely opposite adaptive response, with diminished spontaneous neurite outgrowth compared with saline-exposed controls. Control neurons isolated from the two structures also exhibited opposite regulatory responses to the D1 receptor agonist SKF38393 (1-phenyl-2,3,4-5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrochloride), inhibiting outgrowth in cortical cultures and stimulating outgrowth in striatal cultures. The agonist was ineffective in modulating neurite outgrowth of neurons from either structure isolated from cocaine-exposed fetuses, reflecting the reduced D1–Gs coupling. Total D1 receptor number was indistinguishable in neurons from the cocaine- and saline-exposed animals, but cell imaging and receptor binding of differentially isolated membranes showed that the lack of responsiveness was because of greatly reduced cell-surface localization of D1 receptors. These data suggest that prenatal exposure to cocaine causes a novel, long-lasting adaptive response in the subcellular distribution of D1 receptors, resulting in alterations in signaling capacity that have developmental and behavioral consequences.
Introduction
Agonist-induced receptor internalization is an important homeostatic regulatory process to protect cells from abnormally high levels of stimulation (Tsao and von Zastrow, 2000; Bernard et al., 2006). This process is induced rapidly after administration of an exogenous agonist or in the context of high levels of the endogenous neurotransmitter. Internalization often involves phosphorylation of the receptor and usually reverses within minutes of stimulus removal. In the mature organism, cortical and striatal D1 receptor activity modulates a variety of neural functions, including motor activity, reward, and cognition (Arnsten, 1997; Goldman-Rakic et al., 2004). After acute administration of D1-like (D1, D5) receptor agonists, D1 receptors are phosphorylated on serine/threonine residues and internalized through the actions of arrestins and G-protein receptor kinases (Bernard et al., 2006). The degree to which there is modulation of receptor trafficking because of atypical ligand exposure during development is unknown.
Prenatal exposure to cocaine in a rabbit intravenous model leads to permanent alterations in the dendritic length and shape of pyramidal neurons in dopamine-rich areas of the cerebral cortex, defects in interneuron differentiation, cognitive deficits, and a severe attenuation of D1 receptor-mediated responses through Gs (for review, see Stanwood and Levitt, 2004). Although initial experiments in the rabbit model used injections [4 mg/kg, twice per day (b.i.d.)] from embryonic day 8 (E8) to E29, we have demonstrated that a lower exposure (3 mg/kg, b.i.d.) during a discrete sensitive period (E16–E25) is sufficient to produce the neuroanatomical and behavioral alterations (Stanwood et al., 2001; Stanwood and Levitt, 2003; Thompson et al., 2005). Alterations in D1 receptor tone in the medial frontal cortex (MFC) have been linked previously to attentional and cognitive dysfunctions (Arnsten, 1997; Arnsten and Li, 2005), which also have been reported in children exposed prenatally to cocaine (Richardson et al., 1996; Schroder et al., 2004; Singer et al., 2004) and in animal models (Romano and Harvey, 1996; Garavan et al., 2000; Gendle et al., 2004; Thompson et al., 2005). Also, D1 null mice express neuroanatomical alterations similar to the changes observed in the rabbit prenatal cocaine model (Stanwood et al., 2005), suggesting that the cocaine-induced reduction in D1 signaling may represent the primary defect. In the prenatal cocaine model, D1 receptors appear to be expressed at normal levels but are constitutively hyperphosphorylated (Zhen et al., 2001).
D1 receptor activation modulates neurite outgrowth in a cell context-dependent manner (Reinoso et al., 1996; Schmidt et al., 1996, 1998). Here, we examined the differential impact of prenatal cocaine exposure on cortical and striatal neuronal growth and tested the hypothesis that altered growth properties may be attributable to reduced D1 receptor coupling that is caused by the altered subcellular distribution of the receptor, thus preventing normal coupling.
Materials and Methods
Animals
Proven breeder Dutch-belted rabbits from Myrtle’s Rabbitry (Thompsons Station, TN) were housed individually in a 12 h light/dark cycle with ad libitum access to food and water. The day of breeding was designated as E0, and injections of cocaine hydrochloride (3 mg/kg; National Institute on Drug Abuse, Rockville, MD) or saline were given intravenously through the marginal ear vein twice daily from E16 to E21 (culture experiments) or from E16 to E25 (postnatal experiments). Maternal weight gain, kit weight, and litter sizes were normal, as described previously (Stanwood et al., 2001; Thompson et al., 2005). Rabbits were born on E30–E31, and the day of birth was designated postnatal day 0 (P0). At P60–P65, which corresponds to periadolescence in rabbits, offspring were anesthetized with isoflurane and decapitated. Brains were removed and dissected or frozen in isopentane. In some models using in utero treatments, it has been demonstrated that the responses of animals within a litter are less variable than those across litters (Spear and File, 1996). Using statistical analysis of neuroanatomical and behavioral parameters, we have demonstrated that within- and across-litter variances do not differ significantly in this model (Thompson et al., 2005); nevertheless, no more than one measurement was made per litter in the current experiments.
Tissue culture
Preparation of neuronal cultures.
Primary neuronal cultures were prepared from the presumptive MFC, visual cortex, and striatum of saline- and cocaine-exposed rabbit embryos at E22 using slight modifications of published methods (Reinoso et al., 1996; Jones et al., 2000). This corresponds to the latter period of neurogenesis in the rabbit (Stensaas, 1967a,b). The MFC includes both medial prefrontal and anterior cingulated cortical fields, both of which receive a dense dopamine input and exhibit the same dendritic alterations in adult rabbits exposed to cocaine in utero. For dissections, the midline, rostrum, genu, and splenium of the corpus callosum and the lateral ventricles were used as landmarks. Tissues were cut into small pieces (∼2 mm2) in Earle’s balanced salt solution and incubated in 0.35% collagenase-dispase for 30 min at 37°C, and a cell suspension was prepared by repeated trituration using a fire-polished Pasteur pipette. Viability was assessed using trypan blue. Cells were plated at low density (5 × 104/cm2) on poly-l-ornithine-coated coverslips, incubated in FBS-supplemented DMEM/F-12 medium (Invitrogen, San Diego, CA) for 5 h to enhance attachment, and switched to N2-supplemented glia-conditioned DMEM/F-12 and maintained for 3–4 d in vitro (DIV). The glia-conditioned medium was harvested from neonatal rat cerebral cortical glial cultures (Reinoso et al., 1996) and was found to provide excellent neuronal survival and baseline growth of rabbit neuronal cultures, as described previously (Jones et al., 2000). Approximately 95% of the cells in these cultures are neurons (data not shown). When the effects of exogenous receptor agonists (SCH23390, quinpirole) were assessed, drugs were first added at the time of the 5 h medium switch and then reapplied during a half-volume medium change at 48 h.
Immunostaining.
Cultures were used for biochemical experiments or were fixed in 10% formalin for immunohistochemical staining with a polyclonal antibody against the somatodendritic neuronal marker microtubule-associated protein-2 (MAP-2) (1:400; gift from Itzhak Fischer, Drexel University, Philadelphia, PA) or a rat monoclonal antibody against the D1 receptor (1:500; Sigma, St. Louis, MO). Coverslips were washed and incubated in secondary anti-rabbit or anti-mouse IgG–cyanine 2 (Cy2) or Cy3 (1:1000; Jackson ImmunoResearch, West Grove, PA). Neurite lengths were measured on MAP-2-stained neurons as described below. For study of D1 receptor localization, epifluorescence images of MAP-2 and D1 receptor double-labeled cells were processed using AutoDeblur 9.0 (two-dimensional blind deconvolution; AutoQuant, Troy, NY). For each of these preparations, data were derived from at least three coverslips in each of three culture sessions per treatment group. Based on the variance of the assay and the effect size, we found that this sample size had sufficient power to detect statistical significance between treatment groups.
Neurite outgrowth.
To examine the effects of different substrates and culture conditions on neurite outgrowth, images of MAP-2-positive neurons were captured using an Axiocam high-resolution camera and software (Zeiss, Thornwood, NY). Neurite length, defined as the total extent of the longest neurite from the cell soma to the growth cone, excluding branches, was then quantified using the public-domain programs Image J or Scion (Frederick, MD) Image, as described previously (Eagleson et al., 2003). Cell sampling was done by making nonoverlapping horizontal sweeps of the coverslip to encounter neurons for measurement. In these neuronal cultures, we found that there is limited variation in the distribution of neurons on different coverslips. Measurements were performed only on those neurons for which processes were not contacted by processes of a neighboring neuron, thus reducing the risk of measuring neurites from two neurons simultaneously. In each culturing session, the length of the longest neurite of the first 20 cells encountered that met these criteria was measured on each of three coverslips prepared for each experimental condition. Three independent dissection and culturing sessions were performed, which served as our sample size for statistical testing; analyses thus indicate data derived from 180 neurons in each condition. Cumulative neurite length histograms showing the distribution of neurite lengths after each in utero and in vitro condition were generated and analyzed using a χ2 statistic (Excel; Microsoft, Redmond, WA) as described previously (Walicke, 1988; Spencer et al., 1996; Zhukareva et al., 1997; Eagleson et al., 2003). Data were also expressed as mean neurite length and analyzed by ANOVA, followed by post hoc Scheffé’s test, with p < 0.05 considered significant (SPSS, Chicago IL). The individuals capturing the images and measuring neurite length were blinded as to the experimental condition. There were no differences in the overall number of cells in each condition either at plating or after fixation at 3 DIV.
D1 receptor labeling.
Bodipy-SCH23390 (20–50 nm; Invitrogen) was used to label D1 receptors on cultured neurons (Vincent et al., 1993; Hoyt and Reynolds, 1996). Live cultures (3 DIV) were incubated with bodipy-SCH23390 in N2 medium for 1 h at 4°C. Cells were quickly washed three times with medium and fixed in 10% formalin for 10 min at 4°C. Plates were then washed with PBS. Nonspecific binding (NSB) was defined using 10 μm (+)-butaclamol. Coverslips were mounted onto slides using aqueous medium. Images were captured with a high-resolution camera and assessed by an observer blinded to the treatment. The experiment was repeated after three independent dissection sessions with similar results obtained each time.
In other experiments, an acid-wash procedure was used to quantify cell-surface D1 receptor expression on cultured neurons (Toews, 2000). [3H]-SCH23390 (3 nm) was diluted in N2 medium and added to live primary cells for 1 h at 4°C. Plates were then washed quickly three times with N2 medium. After the final wash, 0.5 ml of ice-cold 0.2 m acetic acid/0.5 m NaCl was added to each well, and cells were incubated for 5 min at 4°C. This caused the dissociation of any bound [3H]-SCH23390 from binding sites on the surface of the cells, and this solution was then collected for scintillation counting. Plates were then rinsed three additional times in medium, and 0.5 ml of 1 m NaOH was added to each well to dissolve any internalized [3H]-SCH23390. NSB was defined with 2 μm (+)-butaclamol. Data were derived from triplicate observations in three culture sessions per treatment group. Statistical significance was determined by unpaired t tests.
Offspring
[3H]-SCH23390 binding.
We used a method for receptor binding in tissue homogenates that separates plasma and light membrane fractions (Sibley et al., 1986). Striatal tissue from male peripubertal P60–P65 rabbits (n = 4/group) exposed to cocaine or saline prenatally as noted above was homogenized on ice using the following solution (in mm): 50 Tris, pH 7.4, 5 MgCl2, 5 KCl, 1 EDTA, 120 NaCl (binding buffer). The homogenate was centrifuged at 180 × g at 4°C for 3 min to remove debris, and the supernatant was incubated at 30°C for 5 min to degrade endogenous ligand. The sample was centrifuged at 20,000 × g at 4°C for 30 min. The pellet (plasma membranes) was resuspended in 4 ml of binding buffer and homogenized, and the supernatant was centrifuged at 100,000 × g at 4°C for 70 min. The final supernatant was discarded, and the pellet (light membranes) was resuspended in 1 ml of binding buffer and homogenized. Protein concentration was determined, and samples were stored at −80°C until assayed. Binding reactions occurred at room temperature for 75 min using 2 nm [3H]-SCH23390 in a 1 ml total reaction volume. A concentration of 2 μm (+)-butaclamol was used to define NSB. Total binding was assessed in triplicate, and NSB was assessed in duplicate, from each membrane preparation in each animal. Reactions were terminated by addition of 8 ml of ice-cold wash buffer, and samples were filtered through Whatman (Maidstone, UK) GF/B filters and rinsed twice with 8 ml of ice-cold wash buffer. Bound radioactivity was quantified by scintillation counting.
Receptor autoradiography.
For receptor autoradiography, frozen brains were sectioned (20 μm) on a cryostat, thaw mounted onto charged slides, and stored at −80°C. Before incubation with [3H]-SCH23390, tissue sections were thawed and preincubated for 30 min at room temperature in the following solution (in mm): 50 Tris, pH 7.4, 5 MgCl2, 5 KCl, 1 EDTA, 120 NaCl. D1 receptors were labeled with [3H]-SCH23390 (3 nm) in the same buffer for 75 min at room temperature. NSB was defined with 2 μm (+)-butaclamol. After incubation, slides were rinsed in 50 mm Tris, pH 7.4, at 4°C for 60 min, dipped in ice-cold doubly distilled H2O, and dried. Sections were apposed to [3H]-Hyperfilm (Amersham Biosciences, Arlington Heights, IL) for 2 weeks. Autoradiograms were digitized and analyzed using Image J. Optical density was converted to nanocurie per milligram of protein based on calibration curves generated from tritium standards (Stanwood et al., 2000).
Results
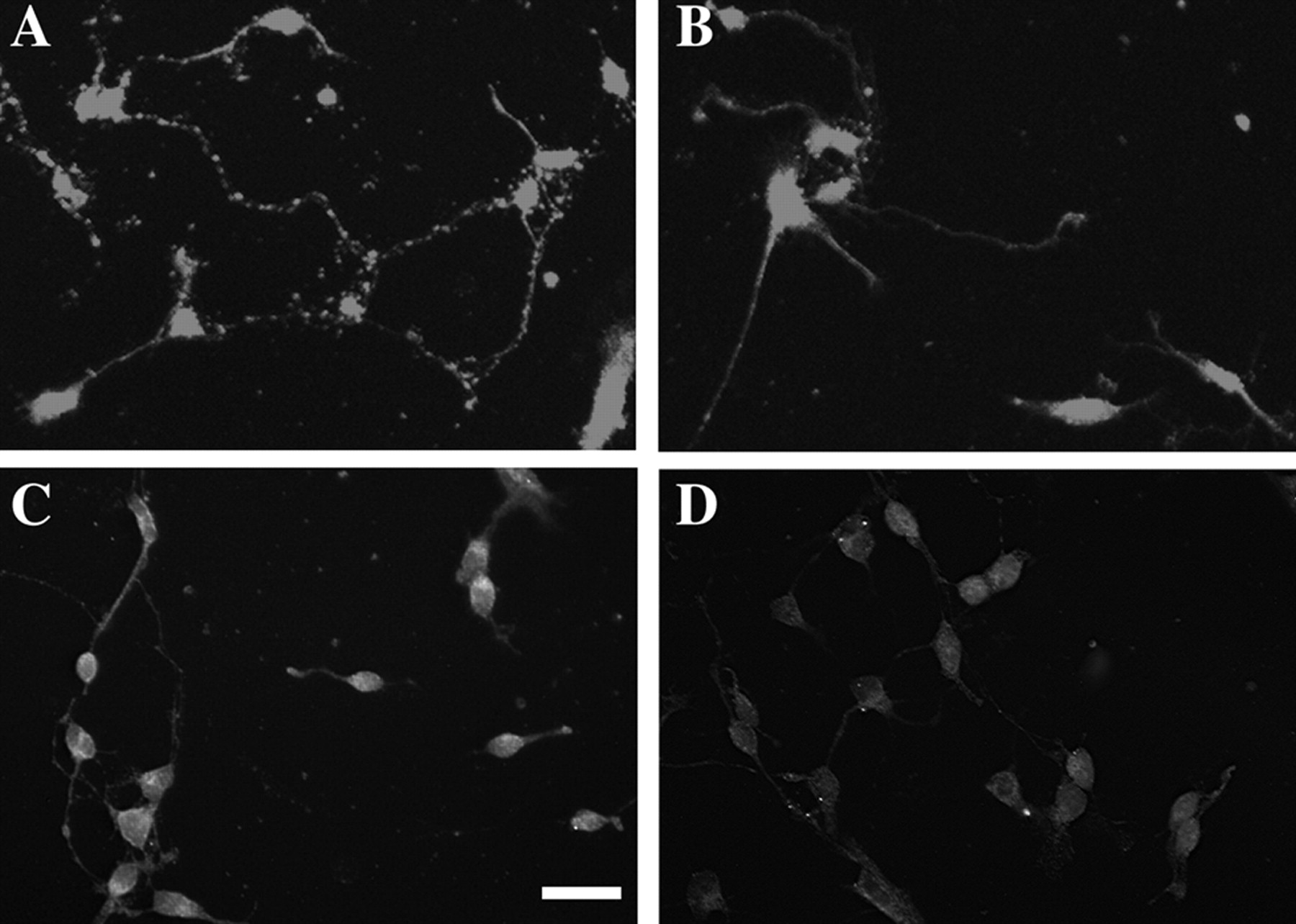
We reproduced our previous finding (Jones et al., 2000) of alterations in spontaneous neurite outgrowth here after a much more limited time of treatment (6 d in contrast to 14 d in the previous study) of prenatal cocaine in utero. MFC neurons harvested from drug-exposed fetuses exhibited longer neurites (Fig. 1B) compared with neurons harvested from saline-exposed animals (Fig. 1A) (χ2 test, p < 0.05). In contrast, striatal neurons exposed in utero to cocaine exhibited diminished neurite outgrowth (Fig. 1C,D) (χ2 test, p < 0.05). There were no differences in neurite outgrowth of cells harvested from the dopamine-poor visual cortex in saline- and cocaine-treated fetuses (Fig. 1E), consistent with our in vivo neuroanatomical studies showing that cocaine exposure alters selectively the growth properties of neurons in dopamine-rich regions. Similar patterns were revealed by analysis of mean neurite length by ANOVA; these data are displayed in Figure 1F.

A–D, Photomicrographs of MAP-2 immunostaining of neuronal cultures derived from the frontal cortex (A, B) and striatum (C, D) of in utero saline-exposed (A, C) and cocaine-exposed (B, D) animals. Scale bar, 50 μm. E, Histograms depict distributions of neurite outgrowth length (micrometers) in neuronal cultures derived from the MFC, striatum, or visual cortex of rabbits exposed to saline or cocaine. In utero cocaine leads to increased neurite outgrowth in the MFC cultures, decreased neurite outgrowth in the striatum, and no changes in the visual cortex. Data are derived from triplicate observations in three culture sessions (i.e., litters) per treatment group. The y-axis refers to the percentage of neurons with lengths greater than “X,” where X is the neurite length as displayed on the x-axis. F, Mean neurite lengths of in utero saline- or cocaine-exposed neurons derived from different brain regions ± SEM. Three separate experiments were examined for each experimental condition. Statistical analysis by ANOVA reveals the same changes evidenced in the plots of cumulative neurite length histograms displayed in E. *p < 0.05. Sal, Saline; Coc, cocaine; Str, striatum; VC, visual cortex.
Addition of the D1-like receptor agonist 1-phenyl-2,3,4-5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrochloride (SKF38393; 10 μm) produced a decrease in neurite outgrowth in cortical neuronal cultures derived from saline animals (χ2 test, p < 0.05) but failed to modulate neurite growth in cultures prepared from cocaine-exposed fetuses (Fig. 2A). This lack of responsiveness is consistent with diminished or absent D1 receptor signaling. In striatal cultures produced from saline animals, addition of SKF38393 (10 μm) increased neurite outgrowth. Similar to the cortical cultures, the agonist failed to modulate neurite growth in cells derived from fetuses exposed to cocaine in utero (Fig. 2B). The selectivity of the prenatal cocaine effect on D1 signaling was also demonstrated by the use of a D2-like receptor agonist. Quinpirole (3 μm) increased spontaneous neurite outgrowth in MFC cultures and decreased outgrowth of striatal neurons, regardless of whether the neurons were derived from saline- or cocaine-exposed fetuses (data not shown).

Histograms depict cumulative distributions of neurite outgrowth length (micrometers) in neuronal cultures derived from the MFC (A) and striatum (B). In vitro addition of the D1 receptor agonist SKF38393 (SKF; 10 μm) decreases spontaneous neurite outgrowth in control MFC cultures [saline (Sal); A], increases spontaneous neurite outgrowth in control striatal cultures (B), and has no effect on cocaine (Coc)-exposed cells from either region (A, B).
We measured total D1-like receptor expression after 6 d of in utero exposure to the reduced dosage of 3 mg/kg. Quantitative receptor autoradiography revealed no differences in total [3H]-SCH23390 binding in P60 offspring exposed to saline or cocaine from E16 to E25 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Scatchard analyses demonstrated high-affinity and saturable [3H]-SCH23390 binding (Kd = 0.14 ± 0.04 nm; Bmax = 2413 ± 331 fmol receptor/mg protein) in rabbit striatal membrane homogenates.
The presence of normal levels of D1 receptors, but an absence of a response to D1 receptor agonists, suggested to us that less of the receptor may be available for mediating ligand-induced signaling. Thus, we hypothesized that cocaine exposure prenatally might induce an adaptive cellular response in which levels of the D1 receptor are normal but are not expressed appropriately in the plasma membrane. To test this possibility, we used a live cell-labeling paradigm in striatal neurons, which express higher levels of the D1 receptor compared with cortical neurons. A fluorescently labeled D1 receptor ligand, bodipy-SCH23390, prominently labeled neurons harvested from controls with a punctate pattern (Fig. 3A). Neurons obtained from cocaine-exposed fetuses exhibited less punctate labeling, particularly on processes (Fig. 3B). Immunolabeling of D1 receptors was also consistent with decreased expression on the plasma membrane of neurons harvested from fetuses exposed to cocaine compared with controls (Fig. 3C,D). To verify these qualitative observations using a more quantitatively precise measure, we next used an acid-wash binding protocol to measure D1 receptor binding sites selectively on the plasma membrane of striatal neurons harvested from saline- and cocaine-exposed fetuses. Consistent with the receptor localization studies, [3H]-SCH23390 labeled specific cell-surface binding sites on the plasma membrane of control striatal neurons (120 ± 31.8 fmol/103 cells), but surface expression was greatly diminished in neuronal cultures prepared from cocaine-exposed fetuses (21.7 ± 16.7 fmol/103 cells; n = 3; p < 0.01). Similar alterations in D1 receptors were observed in cultures from the MFC (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
A, B, Photomicrographs depict bodipy-SCH23390 labeling of in utero saline-exposed (A) and cocaine-exposed (B) striatal neurons. C, D, Photomicrographs depict immunolocalization of D1 receptor by in utero saline-exposed (C) and cocaine-exposed (D) striatal neurons. Scale bar, 50 μm. An acid-wash radioligand binding technique quantitatively confirmed loss of D1 receptor binding sites from the cell surface (see Results).
Finally, we wanted to determine the extent to which this atypical distribution of D1 receptors was reflected long term in vivo. We performed binding studies on membrane fractions prepared from offspring exposed to cocaine or saline for only 6 d in utero. Plasma membrane expression of [3H]-SCH23390 binding sites was reduced significantly in the striatum of peripubertal cocaine-exposed offspring (Fig. 4). In contrast, light membrane fractions of cocaine-exposed rabbits expressed elevated levels of D1-like receptor binding sites (Fig. 4), consistent with altered subcellular distribution of the D1 (and perhaps D5) receptor. The plasma membrane fraction in controls contained ∼5.4-fold more D1-like receptors than the light membrane fraction (based on total protein levels), whereas cocaine-exposed offspring expressed fewer D1-like receptors in the plasma fraction compared with the light fraction (0.57-fold; p < 0.05).

The graph depicts [3H]-SCH23390 binding (mean ± SEM) in P60–P65 striatal membrane fractions. [3H]-SCH23390 binding sites were significantly decreased on the plasma membrane fraction of cocaine (COC)-exposed offspring, with a corresponding increase in light membrane fractions. Data are derived from triplicate observations in three animals per treatment group. *p < 0.05. SAL, Saline.
Discussion
The current study thus provides the first evidence of long-lasting, adaptive changes in the subcellular distribution of a neurotransmitter receptor caused by prenatal drug exposure. Moreover, the alteration in subcellular distribution, without changes in total receptor protein, highlights an adaptive strategy that may be uniquely developmental and that has long-lasting, potent effects on cell signaling via the dopamine D1 receptor. Our findings are consistent with data demonstrating reduced D1 receptor coupling to Gsα (Wang et al., 1995; Jones et al., 2000; Zhen et al., 2001) and hyperphosphorylation of the D1 receptor (Zhen et al., 2001).
Dopamine modulates several specific features of prenatal and postnatal brain development, including effects on the regulation of neurogenesis (Ohtani et al., 2003), differentiation (Reinoso et al., 1996; Schmidt et al., 1996; Porter et al., 1999), and circuit formation (Tang et al., 2001). Dopamine receptor activation has distinct consequences on differentiation in different neuronal types. D1 receptor activation, for example, is a negative modulator of neurite outgrowth in the MFC (Reinoso et al., 1996; Song et al., 2002) but a positive stimulus of growth in striatal neurons (Schmidt et al., 1996, 1998). The current study demonstrates that spontaneous neurite outgrowth is differentially altered in cortical and striatal neurons after in utero exposure to cocaine, likely caused by the common loss of D1 receptor signaling in both regions. Furthermore, the induction of phenotypic changes in dendritic growth patterns and reduction in D1 receptor signaling after exposure to cocaine can be induced with a relatively short duration exposure (E16–E21) in the rabbit, comparable in developmental epoch to E13–E16 in the mouse and the end of the first trimester in humans (Clancy et al., 2001). The defining features of this sensitive period are unknown but are likely common to both cortical and subcortical regions where the D1 receptor and dopamine afferents coexist.
Previous studies (Wang et al., 1995; Jones et al., 2000; Zhen et al., 2001) in which animals were exposed to a higher dose of cocaine (4 mg/kg) for a longer time period (E8–E29) demonstrated reduced D1 receptor coupling to Gsα in the context of a normal level of total D1-like receptor expression. Coupling of D2-like receptors to Gi and of muscarinic receptors to Gs appears normal, suggesting specific desensitization of the D1 (and perhaps D5) receptor. Here, we measured total D1-like receptor expression after just 6 d of exposure to the reduced dosage of 3 mg/kg. We detected no differences in [3H]-SCH23390 binding after prenatal cocaine, similar to previous reports. The anatomical and pharmacological characteristics of [3H]-SCH23390 binding in our assays were similar to previous descriptions in the rabbit (Reader et al., 1988, 1989).
A unique aspect of the current findings is the demonstration that prenatal exposure to cocaine can induce long-lasting changes in the subcellular distribution of dopamine D1 receptors. The loss of normal D1 receptor expression in the plasma membrane results in a dramatic decrease in D1 receptor-mediated intracellular signaling and likely contributes to the enduring changes in circuit formation, gene expression, and cognitive behavior (Jones et al., 2000; Stanwood et al., 2001; Stanwood and Levitt, 2003; Thompson et al., 2005). The adaptive downregulated signaling after prenatal exposure is distinct from changes that occur in the adult, in which sensitization after drug exposure is induced (Stanwood and Levitt, 2003; Robinson and Berridge, 2004; Anderson and Pierce, 2005). Our data are also consistent with the finding of Bloch and colleagues (Dumartin et al., 2000) in dopamine transporter (DAT) knock-out mice, in which plasma membrane D1 receptors are reduced and instead accumulate in the endoplasmic reticulum and Golgi apparatus. The developmental contribution to the alteration in D1 receptor subcellular distribution in that mutant is unknown, however, because of ongoing hyperactivation of the receptor by increased extracellular dopamine after constitutive deletion of DAT. In contrast, the prenatal cocaine-exposed offspring exhibit normal basal extracellular dopamine levels and DAT expression (Du et al., 1999). Moreover, long-lasting changes in subcellular distribution of the D1 receptor here occur after only 6 d of prenatal drug exposure. It will be very interesting to determine why D1 receptors specifically undergo such adaptive changes after limited developmental exposure to cocaine.
The abundance of receptors at the plasma membrane is a critical parameter in the regulation of neuronal sensitivity to neurotransmitters. Acute stimulation of membrane-bound G-protein-coupled receptors leads to internalization, typically by complex, receptor-specific mechanisms (Tsao and von Zastrow, 2000; Kieffer and Evans, 2002; Bernard et al., 2006). In some cases, receptor and ligand dissociate from one another in early endosomes, allowing recycling of the receptor and resensitization, as observed recently for D1 receptors exposed to 6-chloro-N-allyl-SKF-38393 hydrobromide (SKF82958) in striatal cultures (Martin-Negrier et al., 2006). In other cases, particularly after chronic stimulation, internalized receptors can be targeted to degradative pathways to produce receptor downregulation (Bernard et al., 2006). The effects after prenatal cocaine exposure may be uniquely developmental, in that the resultant receptor internalization is long lasting, but the total receptor number is not altered. Additional work is needed to determine the mechanism that underlies the permanent receptor-targeting defect. Friedman and colleagues (Zhen et al., 2001) have demonstrated previously that D1 receptors are constitutively hyperphosphorylated in this prenatal cocaine model. Ongoing experiments are assessing the dynamics of the trafficking defect, the specific domains controlling responsiveness in the D1 receptor protein, and the contribution of D1 receptor-interacting proteins. In theory, the restoration of proper D1 receptor localization and signaling could lead to strategies to normalize functioning in our animal model and, ultimately, if this occurs clinically, in children gestationally exposed to cocaine.
Footnotes
-
This work was supported by National Institute on Drug Abuse Grant DA11165 and National Institute of Child Health and Human Development Grant P30HD15052. Joshua Parlaman and Ricardo Washington provided excellent technical assistance and Drs. Chris Evans, Laura Lillien, Barbara Thompson, Kathie Eagleson, and BethAnn McLaughlin provided helpful discussions.
- Correspondence should be addressed to Gregg D. Stanwood, 8110A MRBIII, Nashville, TN 37232. gregg.stanwood{at}vanderbilt.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}