

Abstract
GABAergic interneurons in prefrontal cortex (PFC) play a critical role in cortical circuits by providing feedforward and feedback inhibition and synchronizing neuronal activity. Impairments in GABAergic inhibition to PFC pyramidal neurons have been implicated in the abnormal neural synchrony and working memory disturbances in schizophrenia. The dopamine D4 receptor, which is strongly linked to neuropsychiatric disorders, such as attention deficit–hyperactivity disorder (ADHD) and schizophrenia, is highly expressed in PFC GABAergic interneurons, while the physiological role of D4 in these interneurons is largely unknown. In this study, we found that D4 activation caused a persistent suppression of AMPAR-mediated synaptic transmission in PFC interneurons. This effect of D4 receptors on AMPAR-EPSC was via a mechanism dependent on actin/myosin V motor-based transport of AMPA receptors, which was regulated by cofilin, a major actin depolymerizing factor. Moreover, we demonstrated that the major cofilin-specific phosphatase Slingshot, which was activated by calcineurin downstream of D4 signaling, was required for the D4 regulation of glutamatergic transmission. Thus, D4 receptors, by using the unique calcineurin/Slingshot/cofilin signaling mechanism, regulate actin dynamics and AMPAR trafficking in PFC GABAergic interneurons. It provides a potential mechanism for D4 receptors to control the excitatory synaptic strength in local-circuit neurons and GABAergic inhibition in the PFC network, which may underlie the role of D4 receptors in normal cognitive processes and mental disorders.
- interneuron
- dopamine D4 receptor
- AMPA receptor
- trafficking
- synaptic transmission
- actin
- myosin
- cofilin
- Slingshot
- calcineurin
Introduction
The neocortical areas, including prefrontal cortex (PFC), are composed of two major neuronal populations: glutamatergic pyramidal neurons and GABAergic interneurons. It has been found that GABAergic inhibition plays a key role in the regulation of working memory, a major cognitive process subserved by PFC (Rao et al., 2000; Constantinidis et al., 2002). Selective alterations in GABAergic local-circuit neurons, GABA synthesis enzyme GAD67, GABA content, and GABAA receptors have been discovered in PFC of schizophrenic patients (Akbarian et al., 1995; Woo et al., 1998; Volk et al., 2000, 2002; Hashimoto et al., 2003), suggesting that impairments in GABAergic inhibition in the PFC network might contribute to the aberrant neuronal activity and disrupted working memory in schizophrenia (Lewis et al., 2005).
Cortical GABAergic interneurons are heterogeneous, and they can be distinguished from pyramidal neurons by their distinct morphological features, circuit connections, electrophysiological characteristics, and protein expression patterns (Markram et al., 2004). For example, cortical GABAergic interneurons receive stronger and more frequent innervations from thalamus than pyramidal neurons; thus they are more strongly activated by excitatory inputs (Cruikshank et al., 2007). Moreover, the functional properties and subunit expression of AMPA receptors are distinct in cortical principal neurons and interneurons (Hestrin, 1993; Geiger et al., 1995). In principal neurons, AMPARs show relatively slow gating and low Ca2+ permeability, which is correlated with the abundant expression of both GluR1 and GluR2 subunits. In interneurons, AMPARs show faster gating and higher Ca2+ permeability, which is correlated with the lower expression of GluR2 subunits (Geiger et al., 1995). In addition, many calcium-binding proteins, such as parvalbumin, calbindin, and calretinin, are highly expressed in cortical GABAergic interneurons (Kawaguchi and Kubota, 1993, 1997). These proteins serve as calcium buffers, which effectively limit the spatial and temporal domains of calcium signaling. A functional consequence of the higher calcium buffering capacity in GABAergic interneurons may be the utilization of calcineurin, a protein phosphatase that can be catalytically activated by a low concentration of calcium.
Functions of PFC neurons are strongly influenced by dopamine signaling through D1 and D2 receptors (Sawaguchi and Goldman-Rakic, 1991; Williams and Goldman-Rakic, 1995; Wang et al., 2004). Anatomic studies have found that D4 receptors are enriched in PFC GABAergic interneurons (Mrzljak et al., 1996), while the physiological role of D4 receptors in these interneurons is largely unknown. The D4 receptor has been implicated in schizophrenia because of its high affinity to the uniquely effective antipsychotic drug clozapine (Van Tol et al., 1991; Kapur and Remington, 2001). Moreover, a D4 gene polymorphism that causes D4 receptor dysfunction is strongly linked to attention deficit–hyperactivity disorder (ADHD) (LaHoste et al., 1996; Rowe et al., 1998). Preclinical studies have suggested that D4 receptors are essential for the expression of hyperactivity and impulsive behaviors in a mouse model of ADHD (Avale et al., 2004). Since disturbances in synaptic transmission are the core pathophysiological feature of mental disorders (Lewis and Gonzalez-Burgos, 2006), it is conceivable that D4 receptors may be a key element in regulating the balance between glutamatergic excitation and GABAergic inhibition. In this study, we found that activation of D4 receptors induced a persistent depression of AMPAR transmission in PFC GABAergic interneurons via a mechanism involving calcineurin. Thus, D4 activation dampens the excitatory synaptic strength in GABAergic interneurons, which would lead to decreased GABAergic inhibition in the PFC circuit.
Materials and Methods
Electrophysiological recording.
To measure AMPAR-mediated synaptic transmission in PFC slices, we used the standard whole-cell recording techniques (Yuen et al., 2005, 2007). Rat (3–4 weeks old) PFC layer I GABAergic interneurons (Zhou and Hablitz, 1996) and GAD-positive interneurons at deep layers (II–V) of PFC from GAD-GFP transgenic mice (4–5 weeks old, The Jackson Laboratory) were used for recordings. In the GIN (GFP-expressing inhibitory neurons) transgenic mouse line, EGFP is expressed in a subpopulation of somatostatin-containing GABAergic interneurons in the hippocampus and neocortex, and the somata of EGFP-expressing interneurons are largely restricted to layers II–IV and upper layer V in the neocortex (Oliva et al., 2000). Patch pipettes (5–8 MΩ) were filled with the internal solution containing the following (in mm): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 10 HEPES, 5 EGTA, 2.2 QX-314, 12 phosphocreatine, 5 MgATP, 0.5 Na2GTP, 0.1 leupeptin, pH 7.2–7.3, 265–270 mOsm. PFC slices (300 μm) were perfused with oxygenated artificial CSF (ACSF) containing GABAAR antagonist bicuculline (10 μm) and NMDAR antagonist d-aminophosphonovalerate (APV, 25 μm). To record mEPSC, slices were perfused with modified ACSF containing a low concentration of MgCl2 (1 mm) and TTX (1 μm). Neurons were observed with a 40× water-immersion lens and illuminated with near infrared (IR) light, and the image was captured with an IR-sensitive CCD camera. Recordings were performed in neurons (held at −70 mV) using a Multiclamp 700A amplifier (Axon Instruments). Tight seals (2–10 GΩ) were obtained by applying negative pressure. The membrane was disrupted with additional suction and the whole-cell configuration was obtained. The access resistances ranged from 13 to 18 MΩ and were compensated 50–70%. EPSCs were evoked by stimulating the neighboring PFC neurons with a bipolar tungsten electrode (FHC). Data analyses were performed with the Clampfit software (Axon Instruments). Spontaneous synaptic events were analyzed with Mini Analysis Program (Synaptosoft). Statistical comparisons of the amplitude and frequency of mEPSC were made using the Kolmogorov–Smirnov (K–S) test.
Agents such as PD168077, GBR-12909, sulpiride, SCH23390, L-74172, colchicines, taxol, latrunculin, phalloidin, FK506, rapamycin, cypermethrin, cyclosporine A, okadaic acid, U73122, edelfosine (ET-18-OCH3), 2APB, heparin, and forskolin were purchased from Tocris, Calbiochem, or Sigma. Stocks were made up as concentrated stocks in water or DMSO and stored at −20°C. Stocks were thawed and diluted immediately before use. Antibodies used included anti-myosin V (Chemicon), anti-myosin VI (Santa Cruz), and anti-Slingshot1 (ECM Bioscience). As controls, antibodies were inactivated by heating in boiling water for 30 min before being used.
Immunocytochemistry.
Rat PFC cultures were prepared by modification of previously described methods (Yuen et al., 2005). To label surface GluR1 subunit, cultures (DIV 21–30) were fixed (4% paraformaldehyde, 30 min) without permeabilization, and then blocked (5% BSA, 1 h) to remove nonspecific staining. Cells were incubated with anti-GluR1 (N-terminal, 1:500, Upstate) at 4°C overnight. After three washes in PBS, cells were incubated with an Alexa 594 (red)-conjugated secondary antibody (1:200, Molecular Probes) at room temperature (RT) for 1 h. After washing, cells were permeabilized (0.2% Triton, 20 min), followed by incubation with anti-GAD (1:1000, Chemicon) for 2 h (RT) and Alexa 488 (green)-conjugated secondary antibody (1:200, Molecular Probes) for 1 h (RT). For p-cofilin staining, neurons (permeabilized) were incubated with anti-p-cofilin (1:500, Cell Signaling) for 2 h (RT). Coverslips were mounted on slides with Vectashield mounting media (Vector Laboratories).
Fluorescence images were detected using a 100× objective with a CCD camera mounted on a Nikon microscope. The surface GluR1 clusters in GAD-positive neurons were analyzed with ImageJ software. All specimens were imaged under identical conditions and analyzed with identical parameters. Several (3–4) dendritic segments (50 μm) with an equal distance away from the soma were selected on each neuron. For each coverslip, four to six individual neurons were chosen. Primary and secondary dendrites were analyzed separately. Clusters were detected with a threshold corresponding to twofold to threefold intensity of the diffuse fluorescence on the dendritic shaft. Three to four independent experiments were performed. Quantitative analyses were performed blindly without knowledge of experimental conditions.
Small interfering RNA.
The small interfering RNA (siRNA) for silencing Slingshot (SSH1L) was as follows (sense): 5′-UCGUCACCCAAGAAAGAUA-3′ (Nishita et al., 2004; Wang et al., 2005). The SSH siRNA or a scrambled siRNA was transfected into cultured cortical neurons (21 DIV) using Lipofectamine 2000. To validate the gene silencing efficiency of Slingshot siRNA, costaining of Slingshot1 and MAP2 was performed using anti-Slingshot1 (1:500, ECM Bioscience) and anti-MAP2 (1:500, Santa Cruz). Immunocytochemical experiments for surface GluR1 or p-cofilin staining were performed 2–3 d after transfection.
Results
Activation of D4 receptors suppresses AMPAR-mediated synaptic transmission in PFC GABAergic interneurons
Layer I of neocortex, which receives dopaminergic innervations (Smiley et al., 1992; Sesack et al., 1995), contains exclusively GABAergic interneurons (Gabbott and Somogyi, 1986; Winer and Larue, 1989; Prieto et al., 1994) that innervate deep layers with extensive axonal plexus (Zhou and Hablitz, 1996), so we first examined whether D4 receptors might regulate AMPARs in this neuronal population. As shown in Figure 1A, bath application of PD168077 (40 μm, 10 min) induced a significant reduction in the amplitude of AMPAR-EPSC in PFC layer I GABAergic interneurons (53.1 ± 3.0%, n = 6), while the current was stable in control neurons when no PD168077 was administrated.

D4 activation suppresses AMPAR-EPSC in PFC GABAergic interneurons. A, B, Plot of normalized peak AMPAR-EPSC showing the effect of the D4 agonist PD168077 (40 μm) in a rat PFC layer I interneuron (A) or a PFC layer V GABAergic interneuron from a GAD-GFP transgenic (GIN) mouse (B). C, D, Plot of normalized peak AMPAR-EPSC showing the effect of PD168077 (40 μm, C) or the DAT inhibitor GBR-12909 (10 μm, coapplied with 5 μm D2/3 antagonist sulpiride and 10 μm D1/5 antagonist SCH23390, D) in the absence or presence of the D4 antagonist L-74172 (10 μm) in PFC layer V GABAergic interneurons from GIN mice. w/o, Without. Inset (D): Representative current traces at time points denoted by #. Calibration: 25 pA, 10 ms. E, Bar graphs (mean ± SEM) showing the percentage reduction of AMPAR-EPSC by PD168077 or GBR-12909 (coapplied with sulpiride and SCH23390) in the absence or presence of L-74172 in PFC interneurons from GIN mice. *p < 0.001, ANOVA. F, Cumulative plot of the distribution of mEPSC amplitudes before (control) and after PD168077 application in a PFC interneuron from a GIN mouse. Inset: Representative mEPSC traces. Calibration: 10 pA, 1 s. G, Bar graphs (mean ± SEM) showing the percentage reduction of mEPSC amplitude or frequency by PD168077 in PFC interneurons from GIN mice. *p < 0.001, ANOVA.
To further understand the role of D4 receptors in PFC interneurons, we examined interneurons in other layers (II–V) using GAD-GFP transgenic mice, in which green fluorescent protein is expressed under the control of the GAD67 promoter that directs its specific expression in GABAergic interneurons (Oliva et al., 2000). As shown in Figure 1B, application of PD168077 (40 μm, 10 min) significantly decreased AMPAR-EPSC in GFP-positive neurons from GAD-GFP transgenic mice (52.4 ± 3.9%, n = 9) (Fig. 1E). A lower dose of PD168077 (20 μm) also produced a significant reduction of AMPAR-EPSC (21.7 ± 2.0%, n = 6), albeit to a smaller extent. Application of the specific D4 antagonist L-74172 (10 μm) had little effect on AMPAR-EPSC (2.3 ± 0.9%, n = 7), but significantly blocked the effect of PD168077 (9.7 ± 1.7%, n = 7) (Fig. 1C,E), suggesting the mediation by D4 receptors.
To determine whether endogenous activation of D4 receptors also affects AMPARs in PFC interneurons, we applied the specific dopamine transporter (DAT) inhibitor GBR-12909 (10 μm) to block the reuptake of dopamine. The D2/3 antagonist sulpiride (5 μm) and D1/5 antagonist SCH23390 (10 μm) were coapplied to block the activation of non-D4 dopamine receptors. As shown in Figure 1, D and E, GBR-12909 significantly reduced AMPAR-EPSC in GFP-positive neurons from GAD-GFP transgenic mice (40.8 ± 4.4%, n = 12), and this effect was largely blocked by the D4 antagonist L-74172 (8.2 ± 1.9%, n = 9). Application of sulpiride plus SCH23390 alone had no effect on AMPAR-EPSC (data not shown). These data suggest that activation of D4 receptors, either by exogenous agonist or endogenous dopamine, induces a persistent reduction of synaptic AMPA responses in PFC GABAergic interneurons.
To distinguish whether the D4 reduction of AMPAR-mediated synaptic transmission in interneurons results from presynaptic or postsynaptic modification, we recorded miniature EPSC (mEPSC), a response from quantal release of single glutamate vesicles. As shown in Figure 1, F and G, PD168077 (40 μm, 10 min) markedly reduced the amplitude but not frequency of mEPSC (amp: 28.8 ± 4.0%; freq: 5 ± 2.1%, n = 7) in GFP-positive neurons from GAD-GFP transgenic mice, suggesting that a postsynaptic mechanism underlies the D4 regulation of AMPAR transmission in PFC interneurons.
To examine whether the D4 reduction of AMPAR-EPSC in PFC interneurons is due to the change in AMPAR trafficking, we costained surface GluR1 and GAD in cultured cortical neurons. Dendritic segments with similar distances away from the soma were compared between control and D4 agonist-treated neurons. Segments from primary and secondary dendrites were analyzed separately. As shown in Figure 2, A and C, PD168077 treatment (40 μm, 10 min) significantly decreased the cluster density (# clusters/50 μm dendrite) of surface GluR1 on the primary dendrites of GAD-positive neurons (control: 30.9 ± 3.2, n = 11, PD: 15.6 ± 2.6, n = 15; p < 0.001, ANOVA). The fluorescence intensity of surface GluR1 clusters (normalized to GAD immunofluorescence) was also significantly reduced by PD168077 (control: 1.1 ± 0.06, n = 11, PD: 0.62 ± 0.04, n = 15; p < 0.001, ANOVA). The size (μm2) of clusters was slightly but not significantly decreased (control: 0.31 ± 0.04, PD: 0.26 ± 0.37, n = 14). Similar reductions on surface GluR1 cluster density and intensity were found on secondary dendrites of GAD-positive neurons treated with PD168077 (Fig. 2C). No change was found on GAD immunofluorescence with PD168077 treatment.

D4 activation reduces surface AMPAR clusters in PFC GABAergic interneurons. A, B, Immunocytochemical images of surface GluR1 in cultured PFC interneurons (GAD+) treated without (control) or with PD168077 (40 μm, 10 min) in the absence or presence of L-74172 (10 μm). Enlarged versions of the boxed regions of dendrites are also shown. C, Quantitative analysis of surface GluR1 clusters (density, intensity, and size) along primary and secondary dendrites in PFC interneurons treated without (−) or with PD168077 in the absence or presence of L-74172. *p < 0.001, ANOVA.
To confirm that D4 receptors mediate the effect of PD168077 on surface GluR1 expression, we pretreated PFC cultures with the D4 antagonist L-74172. As shown in Figure 2, B and C, application of L-74172 (20 μm) alone did not change the surface expression of GluR1, but blocked the effect of PD168077 on surface GluR1 cluster density on the primary dendrites of GAD-positive neurons (L-74: 32.2 ± 4.3, n = 10, L-74+PD: 29.3 ± 4.8, n = 12). The effect of PD168077 on GluR1 fluorescence intensity was also blocked by L-74172 (L-74: 122.6 ± 13.7, n = 13, L-74+PD: 123.1 ± 11.5, n = 13). Similar blocking was found on secondary dendrites of GAD-positive neurons (Fig. 2C). These data suggest that D4 activation reduces the number of AMPARs at the plasma membrane of PFC GABAergic interneurons.
The D4 reduction of AMPAR-EPSC is through a mechanism involving actin/myosin motor-based transport of AMPA receptors
We then examined the potential mechanism by which D4 reduces AMPAR-mediated transmission in PFC GABAergic interneurons. It has been shown that AMPAR trafficking can be regulated by actin and microtubule (MT) cytoskeleton (Kim and Lisman, 2001; Zhou et al., 2001; Derkach et al., 2007). To test whether D4 suppresses AMPAR-EPSC in PFC GABAergic interneurons via a cytoskeleton-dependent mechanism, we applied agents that perturb F-actin or MT stability. As shown in Figure 3A, bath application of the F-actin destabilizer latrunculin (10 μm) caused a gradual decline of AMPAR-EPSC (53.0 ± 3.5%, n = 8), and occluded the reducing effect of subsequently applied PD168077 (5.4 ± 2.5%, n = 5) (Fig. 3C). In contrast, application of the microtubule destabilizer colchicine (30 μm) reduced AMPAR-EPSC (50.3 ± 4.7%, n = 7), but failed to occlude the effect of PD168077 (47.0 ± 5.4%, n = 5) (Fig. 3C). Conversely, application of the F-actin stabilizer phalloidin (10 μm) decreased AMPAR-EPSC (48.8 ± 4.4%, n = 7) (Fig. 3B), consistent with a previous report (Kim and Lisman, 2001), and blocked the PD168077-induced reduction of AMPAR-EPSC (3.2 ± 1.5%, n = 5) (Fig. 3B,C). In contrast, the microtubule stabilizer taxol (10 μm) failed to alter the effect of PD168077 (44.1 ± 4.6%, n = 8) (Fig. 3B,C). These results suggest that the dynamics of actin cytoskeleton is important for D4 regulation of AMPAR transmission in PFC GABAergic interneurons.

The D4 suppression of AMPAR-EPSC in PFC GABAergic interneurons involves the transport of AMPA receptors on actin. A, B, Plot of normalized peak AMPAR-EPSC showing the effect of PD168077 in layer I interneurons treated with the actin destabilizer latrunculin (5 μm, A) versus the microtubule destabilizer colchicine (30 μm, A), or dialyzed with the actin stabilizer phalloidin (10 μm, B) versus the microtubule stabilizer taxol (10 μm, B). C, Bar graphs (mean ± SEM) showing the percentage reduction of AMPAR-EPSC by PD168077 in the absence or presence of various agents that alter actin or microtubule-based transport. *p < 0.001, ANOVA.
Because of the actin dependence of the D4 action in PFC GABAergic interneurons, we would like to know whether actin-based motor proteins are also involved. Myosin Va and Vb have been implicated in GluR1 receptor trafficking in hippocampal neurons (Lisé et al., 2006; Correia et al., 2008). Since the cortex has a large population of interneurons expressing homomeric GluR1 (Geiger et al., 1995), we speculate that D4 may suppress AMPAR-EPSC in PFC GABAergic interneurons by perturbing the actin/myosin V-based delivery of GluR1.
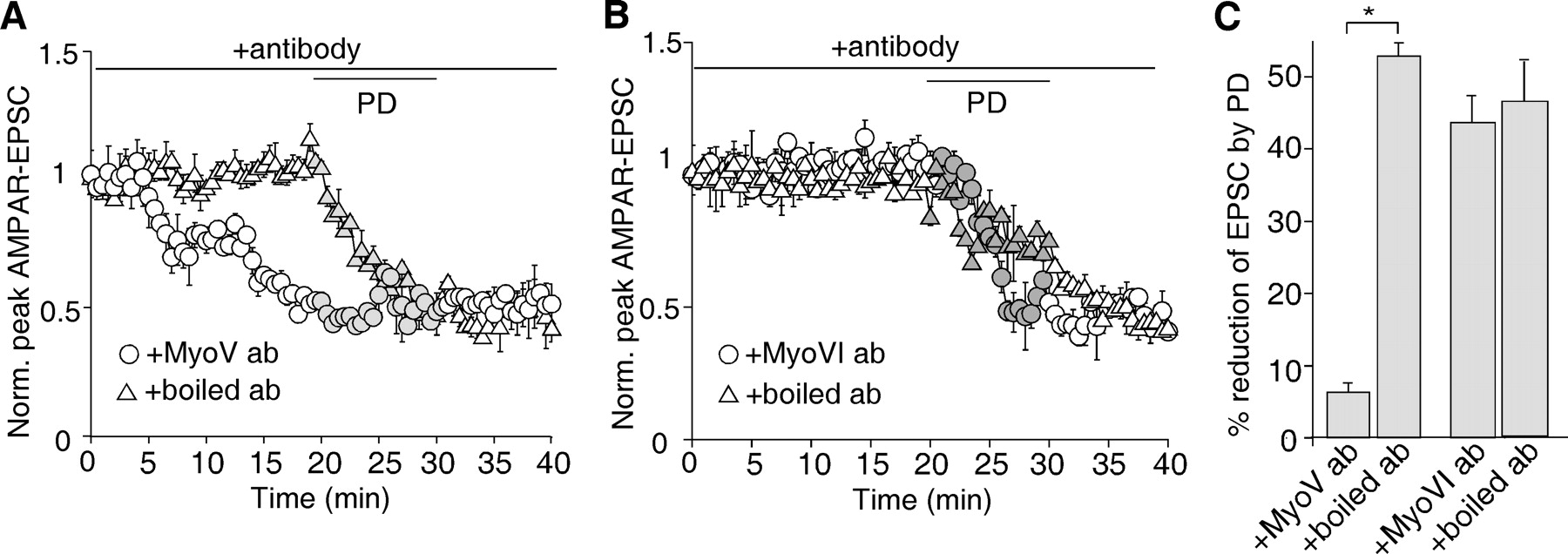
To test this, we dialyzed neurons with an antibody against myosin V to inhibit endogenous myosin V activity. As shown in Figure 4A, the myosin V antibody (10 μg/ml) produced a gradual decrease of AMPAR-EPSC in layer I interneurons (51.5 ± 6.4%, n = 5). Moreover, subsequent application of PD168077 failed to decrease AMPA-EPSC further (6.5 ± 2.6%, n = 7) (Fig. 4C), indicating that the effect of D4 receptors was occluded. The specificity of the myosin V antibody was further confirmed with the heat-inactivated myosin V antibody, which caused little change in basal AMPAR-EPSC and was ineffective in altering the reducing effect of PD168077 (52.0 ± 2.5%, n = 5) (Fig. 4A,C).

The actin motor, myosin V, is involved in D4 regulation of AMPAR-EPSC in PFC GABAergic interneurons. A, B, Plot of normalized peak AMPAR-EPSC showing the effect of PD168077 (40 μm) in layer I PFC interneurons dialyzed with a myosin V antibody (10 μg/ml) versus the boiled control antibody (A), or a myosin VI antibody (10 μg/ml) versus the boiled control antibody (B). C, Bar graphs (mean ± SEM) showing the percentage reduction of AMPAR-EPSC by PD168077 in the presence of different antibodies. *p < 0.001, ANOVA.
Myosin VI is another myosin that has been implicated in AMPAR trafficking (Wu et al., 2002; Osterweil et al., 2005). To test the potential involvement of myosin VI, we dialyzed neurons with an antibody against myosin VI to inhibit endogenous myosin VI activity. As shown in Figure 4B, the myosin VI antibody (10 μg/ml) failed to alter basal AMPAR-EPSC and the reducing effect of PD168077 in PFC interneurons (43.8 ± 4.2%, n = 5) (Fig. 4C). Together, these results suggest that the D4-induced suppression of AMPAR-EPSC in PFC GABAergic interneurons involves the myosin V-mediated transport of AMPARs along actin.
Cofilin, the major actin-depolymerizing protein, is involved in D4 regulation of glutamatergic transmission in PFC GABAergic interneurons
The functionality of the actin cytoskeleton depends on a dynamic equilibrium between filamentous and monomeric actin, thus, we speculate that D4 might regulate AMPAR trafficking and glutamatergic transmission by affecting actin dynamics. The dynamics of actin assembly is regulated by several important factors, one of which is the cofilin protein, a major actin depolymerizing factor (dos Remedios et al., 2003). The activity of cofilin is mainly regulated by phosphorylation at a single site (Ser 3). Dephosphorylation at this site greatly increases the actin-depolymerizing activity of cofilin (Morgan et al., 1993, Agnew et al., 1995).
To examine the involvement of cofilin in D4 regulation of AMPAR-EPSC in PFC GABAergic interneurons, we dialyzed neurons with two peptides consisting of 1–16 residues of cofilin with or without Ser3-phosphorylated (Aizawa et al., 2001; Zhou et al., 2004). The p-cofilin peptide (MASPGVAVSDGVIKVFN) serves as an inhibitor of endogenous cofilin, because it binds to cofilin phosphatases and thus prevents the dephosphorylation and activation of endogenous cofilin. The nonphosphorylated cofilin peptide serves as a negative control. As shown in Figure 5, A and B, dialysis with the p-cofilin peptide (50 μm) caused a reduction of AMPAR-EPSC (49.3 ± 4.8%, n = 8) and abolished the effect of PD168077 in PFC GABAergic (GAD+) interneurons (5.4 ± 1.4%, n = 8). In contrast, the nonphosphorylated cofilin peptide (50 μm) failed to alter the basal AMPAR-EPSC (4.7 ± 1.9%, n = 7) and the reducing effect of PD168077 (38.9 ± 4.3%, n = 8). These data suggest that D4 might regulate actin-based AMPAR trafficking by interfering with cofilin-mediated F-actin depolymerization in PFC GABAergic interneurons.

The D4 regulation of AMPAR-EPSC in PFC GABAergic interneurons involves the actin-depolymerizing protein cofilin. A, Plot of normalized peak AMPAR-EPSC as a function of time and PD168077 application in PFC GAD+ neurons (from GIN mice) dialyzed with the p-cofilin peptide (50 μm) or cofilin peptide (50 μm). B, Bar graphs (mean ± SEM) showing the percentage reduction of AMPAR-EPSC by PD168077 in the absence or presence of different peptides. *p < 0.001, ANOVA. C, Immunocytochemical images showing the costaining of p-cofilin and GAD in cultured PFC neurons treated without or with PD168077 (40 μm, 10 min). D, Bar graphs showing the p-cofilin fluorescence intensity (normalized to GAD immunofluorescence) at the somata and dendrites of control or PD168077-treated GAD+ neurons. *p < 0.01, ANOVA.
Next, we used a specific Ser3phospho-cofilin antibody to directly examine whether D4 activation could affect the phosphorylation and activity of cofilin in PFC interneurons. As shown in Figure 5, C and D, PD168077 treatment (40 μm, 10 min) significantly decreased the Ser3-phosphorylated (inactive) cofilin level (normalized to GAD immunofluorescence) in cultured PFC GABAergic neurons (soma: 0.53 ± 0.04 of control, n = 9; dendrites: 0.52 ± 0.03 of control, n = 12). The total cofilin level was not altered by PD168077 (data not shown). It suggests that the actin-depolymerizing activity of cofilin is increased after D4 activation.
The cofilin phosphatase Slingshot links D4 receptors to actin-based AMPAR trafficking in PFC GABAergic interneurons
Dephosphorylation of cofilin enables its actin severing and depolymerizing activity (Huang et al., 2006). The major phosphatase that plays a pivotal role in actin dynamics by dephosphorylating and reactivating cofilin is Slingshot (Niwa et al., 2002). In mammalian cells, Slingshot (SSH) has three isoforms, each with long and short variants. We speculate that D4 activation might increase Slingshot-mediated cofilin dephosphorylation, leading to actin depolymerization and the suppression of glutamatergic transmission in PFC GABAergic interneurons. To test this, we dialyzed neurons with a peptide, SKCLVHC468KMGVSRSAS, derived from the active site sequence in the catalytic domain of Slingshot (Niwa et al., 2002) to block the phosphatase activity of endogenous Slingshot. The same peptide with a point mutation at the Cys residue in the catalytic pocket, C468S, was used as an inactive control (Niwa et al., 2002). Dialysis with the Slingshot peptide (100 μm) induced a gradual decline of AMPAR-EPSC (35.2 ± 9.6%, n = 5), and subsequent application of PD168077 failed to reduce AMPAR-EPSC further (8.3 ± 2.0%, n = 5) (Fig. 6A,C) in PFC layer I interneurons. In contrast, the inactive control peptide (100 μm) had no effect on basal AMPAR-EPSC, and failed to alter the effect of PD168077 on AMPAR-EPSC (41.0 ± 4.9%, n = 4) (Fig. 6C).

The cofilin phosphatase Slingshot (SSH) is involved in D4 regulation of AMPARs in PFC GABAergic interneurons. A, B, Plot of normalized peak AMPAR-EPSC as a function of time and PD168077 application in PFC interneurons dialyzed with a Slingshot peptide (100 μm, A) versus the C468S mutant peptide (100 μm, A), or a Slingshot1 antibody (10 μg/ml, B) versus the heat-inactivated antibody (B). C, Bar graphs (mean ± SEM) summarizing the percentage reduction of AMPAR-EPSC by PD168077 in the presence of different agents that affect Slingshot activity. *p < 0.001, ANOVA. D, Immunocytochemical images of surface GluR1 in cultured PFC interneurons (GAD+) transfected with a scrambled control siRNA or SSH1 siRNA either untreated (control) or treated with PD168077 (40 μm, 10 min). Costaining of Slingshot1 and MAP2 is also shown to illustrate the knockdown of Slingshot1 expression by SSH1 siRNA. E, Quantitative analysis of surface GluR1 clusters (density, intensity, and size) along dendrites in control or PD168077-treated PFC interneurons transfected with different siRNA. *p < 0.001, ANOVA.
To further examine the involvement of Slingshot, we dialyzed PFC interneurons with a specific Slingshot1 antibody to block the function of this endogenous phosphatase. Dialysis with the Slingshot1 antibody caused a reduction of AMPAR-EPSC (43.6 ± 6.2%, n = 7), and prevented PD168077 from reducing AMPAR-EPSC in PFC GABAergic (GAD+) interneurons (6.3 ± 1.8%, n = 9) (Fig. 6B,C). In contrast, the heat-inactivated Slingshot1 antibody did not alter the basal AMPAR-EPSC (4.6 ± 1.6%, n = 8) or the reducing effect of PD168077 (44 ± 6.1%, n = 7) (Fig. 6C).
Next, we examined the role of Slingshot in D4 regulation of AMPAR trafficking by knocking down Slingshot1 in PFC cultures with siRNA transfection. As shown in Figure 6D, the Slingshot1 siRNA caused a specific and effective suppression of the expression of this phosphatase, which is likely to induce its functional inactivation. In GAD-positive neurons transfected with a scrambled control siRNA (Fig. 6D,E), PD168077 treatment (40 μm, 10 min) significantly decreased the surface GluR1 cluster density (control: 29.6 ± 2.3 clusters/50 μm dendrite, PD: 16 ± 1.8 clusters/50 μm dendrite, n = 11; p < 0.001, ANOVA) and fluorescence intensity (normalized to GAD immunofluorescence) (control: 1.09 ± 0.1, PD: 0.71 ± 0.06, n = 11; p < 0.001, ANOVA). However, in GAD-positive neurons transfected with Slingshot1 siRNA (Fig. 6D,E), surface GluR1 showed significantly (p < 0.001, ANOVA) reduced cluster density (17.6 ± 2.8 clusters/50 μm dendrite, n = 12) and fluorescence intensity (normalized to GAD immunofluorescence) (0.66 ± 0.06, n = 12), and application of PD168077 had no further effect (cluster density: 17.4 ± 1.8 clusters/50 μm dendrite, n = 10; normalized cluster intensity: 0.68 ± 0.08, n = 10). Together, these data suggest that Slingshot, the major phosphatase regulating cofilin activity and ensuing actin dynamics, is a key molecule linking D4 receptors to actin-based AMPAR trafficking in PFC GABAergic interneurons.
D4 regulation of glutamatergic transmission in PFC GABAergic interneurons requires activation of calcineurin
Next, we examined the possible mechanism underlying D4 activation of Slingshot. Our previous studies show that D4 activation triggers the PLC/IP3/Ca2+ pathway (Gu and Yan, 2004; Gu et al., 2006). Since cortical GABAergic interneurons do not express CaMKII (Benson et al., 1992), the major substrate of Ca2+ is likely to be calcineurin. Calcineurin has been found to dephosphorylate Slingshot1 and increase the cofilin-phosphatase activity of Slingshot1 in cell-free assays (Wang et al., 2005).
To test the possibility that the actin-dependent D4 suppression of glutamatergic transmission in PFC GABAergic interneurons is through activation of calcineurin, we applied the calcineurin inhibitor FK506. Rapamycin, which is structurally similar to FK506 but does not inhibit calcineurin (Liu, 1993), was used as a control. As shown in Figure 7A, PD168077 failed to decrease AMPAR-EPSC in the interneuron loaded with FK506 (5 μm, 5.8 ± 1.8%, n = 5) (Fig. 7C), while the effect of PD168077 was intact in the interneuron dialyzed with rapamycin (10 μm, 42.6 ± 4.3%, n = 5) (Fig. 7C). To substantiate that the effect of FK506 on D4 regulation of AMPAR-EPSC is due to inhibition of calcineurin, we applied another specific calcineurin inhibitor, cyclosporine A. As shown in Figure 7B, cyclosporine A (20 μm) blocked the PD168077-induced reduction of AMPAR-EPSC (7.0 ± 2.6%, n = 6) (Fig. 7C), while internal application of the PP1/2A inhibitor okadaic acid (OA, 1 μm) was ineffective (43.6 ± 4.3%, n = 5) (Fig. 7C). Injecting cypermethrin (10 μm), which inhibits calcineurin via mechanisms distinct from that of FK506 (Enan and Matsumura, 1992; Liu, 1993), also blocked the reducing effect of PD168077 on AMPAR-EPSC (5.0 ± 3.5%, n = 5) (Fig. 7C). In contrast, dialysis with KN-93 (20 μm), an inhibitor for Ca2+/CaM-dependent protein kinases, failed to alter the effect of PD168077 (45.6 ± 4.0%, n = 4) (Fig. 7C). None of these inhibitors alone significantly affected the basal AMPAR-EPSC (FK506: 4.8 ± 3.2%, n = 5; rapamycin: 2.8 ± 1%, n = 6; cyclosporine A: 5.8 ± 2.4%, n = 6; OA: 8.8 ± 1.7%, n = 7; cypermethrin: 3.3 ± 1.2%, n = 5; KN-93: 5.0 ± 1.2%, n = 5). These results suggest that calcineurin is specifically involved in D4 regulation of glutamatergic transmission in PFC GABAergic interneurons.

Calcineurin activity is required for D4 regulation of AMPAR-EPSC in PFC GABAergic interneurons. A, B, Plot of normalized peak AMPAR-EPSC as a function of time and PD168077 application in PFC layer I interneurons dialyzed with the calcineurin inhibitor FK506 (5 μm, A) versus its inactive analog rapamycin (10 μm, A), or the calcineurin inhibitor cyclosporine A (20 μm, B) versus the PP1/2A inhibitor okadaic acid (OA, 1 μm, B). C, Bar graphs (mean ± SEM) summarizing the percentage reduction of AMPAR-EPSC by PD168077 in the presence of different phosphatase or kinase inhibitors. *p < 0.001, ANOVA. D, F, Immunocytochemical images showing the costaining of p-cofilin and GAD in cultured PFC neurons (D: untreated vs FK506-pretreated; F: scrambled siRNA-transfected vs Slingshot1 siRNA-transfected) treated without or with PD168077 (40 μm, 10 min). E, G, Bar graphs showing the p-cofilin fluorescence intensity (normalized to GAD immunofluorescence) at the dendrites of control or PD168077-treated interneurons (GAD+) under different conditions. *p < 0.01, ANOVA.
To provide more direct evidence about the involvement of calcineurin/Slingshot signaling in D4 regulation of AMPARs, we examined whether D4 affects the activity of cofilin in PFC interneurons through a mechanism depending on calcineurin and Slingshot. As shown in Figure 7, D and E, the reducing effect of PD168077 (40 μm, 10 min) on the level of p-cofilin (inactive) was blocked by pretreatment with the calcineurin inhibitor FK506 (2 μm, 30 min) in cultured PFC GABAergic (GAD+) neurons (untreated: 0.58 ± 0.03 of control, n = 10; FK506-treated: 0.96 ± 0.06 of control, n = 11). FK506 alone did not affect the level of p-cofilin (0.98 ± 0.13 of control, n = 11). Moreover, PD168077 significantly reduced the level of p-cofilin in neurons transfected with a scrambled siRNA (0.63 ± 0.07 of control, n = 9) (Fig. 7F,G), while neurons transfected with the Slingshot1 siRNA showed a significant increase in the level of p-cofilin (1.33 ± 0.05 of control, n = 11), and prevented PD168077 from decreasing it (1.4 ± 0.12 of control, n = 11) (Fig. 7F,G). These data suggest that D4 regulates actin dynamics in PFC interneurons through activation of Slingshot via calcineurin.
Finally, we examined whether the D4-triggered PLC/IP3/Ca2+ pathway is potentially responsible for the activation of calcineurin, which leads to the actin/cofilin-dependent suppression of glutamatergic transmission in PFC GABAergic interneurons. As shown in Figure 8A, injecting a high concentration (10 mm) of BAPTA, the potent Ca2+ chelator, blocked the PD168077-induced reduction of AMPAR-EPSC (low BAPTA: 52.8 ± 3.8%, n = 6; high BAPTA: 9.0 ± 2.6%, n = 5) (Fig. 8D). The effect of PD168077 was also largely blocked by two inhibitors of PLC (Fig. 8B), U73122 (10 μm, 11.2 ± 3.3%, n = 5) (Fig. 8D) and edelfosine (50 μm) (Powis et al., 1992) (6.3 ± 2.4%, n = 6) (Fig. 8D). Similarly, two inhibitors for IP3R, 2APB (15 μm) and heparin (1 mg/ml, Takei et al., 1998), almost abolished the PD168077-induced reduction of AMPAR-EPSC (2APB: 6.8 ± 2.4%, n = 5; heparin: 9.0 ± 1.8%, n = 6) (Fig. 8C,D). Since D4 can also be linked to the inhibition of PKA pathway (Wang et al., 2002, 2003; Gu and Yan, 2004), we also examined the role of PKA in D4 regulation of AMPAR-EPSC in PFC GABAergic interneurons. As shown in Figure 8C, the effect of PD168077 was intact in the presence of the PKA activator forskolin (10 μm, 50 ± 3.5%, n = 5) (Fig. 8D), which rules out the involvement of PKA.

The PLC/IP3/Ca2+ pathway is involved in D4 regulation of AMPAR-EPSC in PFC GABAergic interneurons. A–C, Plot of normalized peak AMPAR-EPSC in PFC interneurons injected with the internal solution containing a high (10 mm) or low (0.1 mm) concentration of BAPTA (A), the PLC inhibitor U73122 (10 μm, B) or edelfosine (50 μm, B), the IP3R inhibitor 2APB (15 μm, C) or heparin (1 mg/ml, C), or the PKA activator forskolin (10 μm, C). D, Bar graphs (mean ± SEM) showing the percentage reduction of AMPAR-EPSC by PD168077 in the presence of different agents. *p < 0.001, ANOVA.
Based on our results, we propose the following model that may underlie the mechanism for the D4 regulation of glutamatergic transmission in PFC GABAergic interneurons (Fig. 9). Activation of D4 receptors in these cells causes a strong increase of calcineurin (CaN) activity, presumably via the PLC/IP3/Ca2+ cascade. CaN activation results in the increased activity of Slingshot (SSH), a major cofilin-specific phosphatase (Niwa et al., 2002; Wang et al., 2005). SSH induces dephosphorylation and activation of cofilin, a major actin depolymerizing factor (dos Remedios et al., 2003; Huang et al., 2006), leading to the disruption of actin filament dynamics. Consequently, the myosin V motor-mediated transport of GluR1-containing vesicles along actin is interfered, and therefore the AMPAR-mediated synaptic current is significantly reduced.

A schematic model for the D4 regulation of AMPARs in PFC GABAergic interneurons. D4 receptors activate calcineurin (CaN), leading to the increased activity of Slingshot (SSH). SSH induces dephosphorylation and activation of cofilin, resulting in actin depolymerization. Consequently, the myosin V motor-mediated transport of GluR1-containing vesicles along actin is interfered, causing the reduction of surface AMPAR clusters and AMPAR-mediated synaptic currents.
Discussion
Although GABAergic interneurons only account for ∼20% of the cortical neuronal population (Hendry et al., 1987), they are critical elements of cortical circuits by providing feedforward and feedback inhibition and synchronizing cortical activity (Galarreta and Hestrin, 1999; Cruikshank et al., 2007). PFC GABAergic interneurons are crucial for the gamma oscillations, which reflect the synchronization of pyramidal neuron activity during working memory processes (Tallon-Baudry et al., 1998; Howard et al., 2003). Thus, the abnormal neural synchrony and working memory disturbances in schizophrenia may be attributable to impairments in PFC GABAergic inhibition (Spencer et al., 2003; Lewis et al., 2005). In the present study, we have demonstrated that the excitatory synaptic strength in PFC GABAergic neurons is downregulated by dopamine D4 receptors. While cortical interneurons of different classes are often modulated by neurotransmitters in distinct, subtype-specific manners (Bacci et al., 2005), this regulatory effect of D4 receptors on AMPAR-EPSC is similar in all the tested GABAergic interneurons located in layer I or deep layers (II–V) of PFC. It provides a mechanistic link between dopamine, glutamate, and GABA transmission in PFC, all of which are crucial for cognitive processes mediated by PFC (Lewis and Gonzalez-Burgos, 2006). The D4-induced suppression of the excitatory drive to GABAergic interneurons could lead to reduced feedforward inhibition to PFC pyramidal neurons. This D4 action, in combination with the D4-induced decrease of GABAergic transmission in PFC pyramidal neurons (Wang et al., 2002), could synergistically increase the excitatory output of PFC.
Our combined electrophysiological and immunocytochemical data suggest that the D4 reduction of AMPAR-EPSC in PFC interneurons is through changing the actin-based trafficking of AMPA receptors to the synaptic membrane. Actin filaments are enriched at synapses where they undergo dynamic polymerization and depolymerization. Depolymerization of F-actin leads to the redistribution of AMPAR clusters to nonsynaptic sites in GABAergic neurons of hippocampal cultures (Allison et al., 1998) and enhances the internalization of AMPA receptors (Zhou et al., 2001). Disruption of the interaction of GluR1 with the actin-binding protein 4.1N also causes a reduction of the surface AMPAR level (Shen et al., 2000). By using specific agents to perturb actin or microtubule cytoskeletons, we have identified actin as the basis of D4-regulated AMPAR trafficking in PFC interneurons. Interestingly, not only the actin destabilizer (latrunculin), but also the actin stabilizer (phalloidin), caused a gradual decline of AMPAR-EPSC and prevented the effect of subsequently activated D4 receptors. It suggests that the dynamics of actin cytoskeleton is critical for AMPAR trafficking. Changing the dynamic equilibrium between filamentous and monomeric actin in either direction will lead to the reduced functional AMPARs at the synaptic membrane.
Myosins are motor proteins that move on F-actin, which are essential for a multitude of cellular processes, including membrane trafficking, cell movements, and signal transduction (Mermall et al., 1998). Myosin V and myosin VI have been implicated in glutamate receptor trafficking. Myosin Va associates with AMPARs through its cargo-binding domain and mediates the CaMKII-triggered GluR1 translocation from dendritic shafts to spines (Correia et al., 2008), while overexpression of dominant negative myosin Vb reduces the surface expression of GluR1-containing AMPARs in cultured hippocampal neurons (Lisé et al., 2006). In addition, myosin VI exists in a complex with AMPAR GluR1 and the scaffolding protein SAP97 in rat brain (Wu et al., 2002; Osterweil et al., 2005). Myosin VI-deficient hippocampus exhibits synapse loss and a significant deficit in the stimulation-induced internalization of AMPARs (Osterweil et al., 2005). By using specific antibodies to block the function of endogenous myosins, we have identified myosin V as the motor involved in D4 regulation of AMPAR trafficking on actin.
Cofilin, the actin-binding protein that is essential for the high rates of actin filament turnover through regulation of actin polymerization/depolymerization cycles (dos Remedios et al., 2003), provides a phosphoregulatory mechanism for actin reorganization (Bamburg, 1999; Huang et al., 2006). It is inactivated by phosphorylation at Ser3, and reactivated by dephosphorylation of this site (Morgan et al., 1993, Agnew et al., 1995). Thus cofilin phosphorylation/dephosphorylation at the critical Ser3 residue acts as a switch for actin assembly (F-actin stabilization) and disassembly (F-actin severing). By using a Ser3-phosphorylated cofilin peptide (Aizawa et al., 2001; Zhou et al., 2004) to inhibit the activation of endogenous cofilin, we have demonstrated that the D4 regulation of AMPAR-EPSC requires cofilin. Consistently, using a Ser3-phospho cofilin antibody, we have shown that D4 receptors increase the actin-depolymerizing activity of cofilin in PFC GABAergic interneurons.
Slingshot has been identified as the major phosphatase that dephosphorylates and activates cofilin, therefore playing a pivotal role in actin dynamics (Niwa et al., 2002). By cofilin regulation, Slingshot induces highly motile growth cones and enhances neurite extension rates (Endo et al., 2003). Slingshot contains a protein phosphatase domain with a canonical catalytic HCxxGxxR sequence found in protein tyrosine phosphatases and dual-specificity phosphatases (Niwa et al., 2002). Using a peptide derived from this sequence to block the phosphatase activity of endogenous Slingshot, we have demonstrated the involvement of Slingshot in D4 regulation of AMPAR-EPSC. The Slingshot1 antibody data provide another piece of evidence to complement the peptide experiments. Moreover, we have shown that knockdown of Slingshot1 expression by RNA interference reduces AMPAR surface clusters and prevents the effect of D4 in PFC GABAergic interneurons, suggesting that Slingshot is a novel regulator of AMPAR trafficking in central neurons.
Slingshot phosphatase activity is regulated by multiple proteins, including phosphoinositide 3-kinase (Nishita et al., 2004), F-actin, and 14-3-3 (Nagata-Ohashi et al., 2004). Biochemical assays show that calcineurin dephosphorylates SSH1L and increases its phosphatase activity, suggesting that the Ca2+-induced cofilin dephosphorylation and actin depolymerization are mediated by calcineurin-dependent activation of Slingshot (Wang et al., 2005). Consistently, it has been found that calcineurin is enriched at synapses together with F-actin and mediates the rapid loss of F-actin puncta at synapses induced by NMDA treatment in cultured hippocampal neurons (Halpain et al., 1998). Using specific inhibitors, we have demonstrated that the D4 regulation of AMPAR-EPSC requires calcineurin, which is likely to be activated by the PLC/IP3/Ca2+ pathway (Gu and Yan, 2004; Gu et al., 2006). Because of its high affinity to calcium, calcineurin may play a particularly important role in cortical interneurons, which express calcium-binding proteins and Ca2+ permeable AMPARs (Kawaguchi and Kubota, 1993; Geiger et al., 1995). In agreement with this, infusion of a calcineurin inhibitor in rat PFC has been shown to significantly impair working memory (Runyan et al., 2005).
Together, our results indicate that activation of D4 receptors induces a marked depression of the actin-based AMPAR trafficking and excitatory transmission in PFC GABAergic interneurons via the calcineurin/Slingshot/cofilin pathway. It provides a potential mechanism for D4 receptors to regulate PFC inhibitory neurons, which is a critical element for controlling cortical functions (McBain and Fisahn, 2001; Lewis et al., 2005).
Footnotes
-
This work was supported by grants from National Institutes of Health (MH084233, NS048911) to Z.Y. We thank Xiaoqing Chen for her technical support.
- Correspondence should be addressed to Dr. Zhen Yan, Department of Physiology and Biophysics, School of Medicine and Biomedical Sciences, State University of New York at Buffalo, 124 Sherman Hall, Buffalo, NY 14214. zhenyan{at}buffalo.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}