

Abstract
Postsynaptic responses to presynaptic stimulation are used regularly to assess the amount of transmitter released from presynaptic release zones. At climbing fiber-to-Purkinje cell synapses, the number of vesicles released per active zone follows changes in release probability such that, normally, more than one vesicle is released per presynaptic action potential. This leads to high occupation of postsynaptic AMPA receptors by glutamate and thus may render the postsynaptic response relatively insensitive to changes in release. We find that paired-pulse depression of presynaptic release is not accurately represented by postsynaptic responses because of receptor saturation. By lowering vesicular glutamate concentrations or by using nonsaturated Bergmann glial AMPA receptors to monitor presynaptic release, we find that presynaptic depression of release is much greater than suggested previously. In addition, densely expressed glutamate transporters can shield Bergmann glial AMPA receptors and presynaptic metabotropic glutamate receptors from activation.
Introduction
At excitatory synapses in the CNS, vesicular release of glutamate into the small volume of the synaptic cleft results in the rapid activation of postsynaptic ionotropic glutamate receptors. Diffusion of glutamate into the extrasynaptic space, along with binding to glial and neuronal transporters, rapidly reduces the cleft glutamate concentration (Barbour et al., 1994; Bergles and Jahr, 1997; Bergles et al., 1997;Clark and Barbour, 1997). The glutamate concentration transient in the synaptic cleft after exocytosis of single vesicles is thought to occupy approximately one-half or fewer of the postsynaptic AMPA receptors, on average (Clements et al., 1992; Diamond and Jahr, 1995; Choi et al., 2000; McAllister and Stevens, 2000). Thus, AMPA receptor (AMPAR)-mediated EPSCs should follow variations in the size of the glutamate transient. However, if postsynaptic AMPARs are close to fully occupied by glutamate, as they are thought to be at climbing fiber–Purkinje cell (CF–PC) synapses because of high release probability (Pr) (Dittman and Regehr, 1998; Silver et al., 1998) and multivesicular release (Wadiche and Jahr, 2001), the EPSC will not be an accurate monitor of the glutamate transient. In the extreme case of receptor saturation, the size of the EPSC will reflect the number of receptors and not the amount of transmitter released into the cleft. In this condition, using the EPSC to estimate changes in presynaptic release will underestimate changes in release Pr during short trains or paired stimulation of the presynaptic element.
Very large synaptic glutamate transients resulting from multivesicular release may also temporarily overwhelm clearance mechanisms and allow activation of extrasynaptic receptors. Because release probability and therefore multivesicular release are diminished by repetitive stimulation, clearance may become faster and limit extrasynaptic receptor activation. Thus, actions of receptors located outside the cleft may occur only during low-frequency presynaptic activity at climbing fiber synapses.
In this study, we decreased synaptic vesicle glutamate concentrations using inhibitors of the vacuolar type ATPase, bafilomycin A1 (Baf) and concanamycin A (Conca), to study paired-pulse depression at the CF–PC synapse with a reduced synaptic glutamate transient to determine the effect of receptor occupancy on synaptic depression. We also used extrasynaptic AMPARs expressed by Bergmann glial cells (BGs) as nonsaturable sensors of release as a comparison. In addition, we examined the ability of glutamate transporters to shield Bergmann glial AMPARs and presynaptic metabotropic glutamate receptors (mGluRs) from transmitter at low and high release probability.
Materials and Methods
Slices and solutions. Parasagittal slices of 8- to 16-d-old rat cerebella were cut at a thickness of 250–300 μm with a vibrating microtome (VT 1000S; Leica, Wetzlar, Germany) in ice-cold solution containing the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose (saturated with 95% O2–5% CO2). The slices were incubated in the same solution at 34°C for 15–30 min and then stored at room temperature. During recording, slices were superfused with the above solution with the addition of 100 μm picrotoxin. Outside-out patch experiments were performed with an external solution containing the following (in mm): 140 NaCl, 1.8 CaCl2, 1.3 MgCl2, and 5 HEPES, pH of 7.4. For synaptic experiments in Purkinje cells, the pipette solution contained the following (in mm): 130 CsMeSO4, 4 NaCl, 1 MgCl2, 10 HEPES, and 10 EGTA, pH of 7.2. The sources of the chemicals are as follows: concanamycin A, bafilomycin A1, picrotoxin, monosodium glutamate, MgCl2, HEPES, EGTA, CoCl2, and CaCl2 were from Sigma (St. Louis, MO); (±)-amino-4-carboxy-methyl-phenylacetic acid (MCPG), (RS)-α-cyclopropyl-4-phosphonophenyl-glycine (CPPG), 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), threo-β-benzyloxyaspartate (TBOA), cyclothiazide (CTZ), and D-600 were from Tocris Cookson (Ellisville, MO); NaCl, KCl, NaH2PO4, NaHCO3, and glucose were from Mallinckrodt (Paris, KY); and pardaxin and tetrodotoxin were from Alomone Labs (Jerusalem, Israel).
Recording and perfusion techniques. Bergmann glia were identified by their size and location using a 40× water-immersion objective on an upright microscope (Axioskop FS; Zeiss, Oberkochen, Germany) equipped with infrared–differential interference contrast optics. Bergmann glial currents were recorded at their resting potential. Purkinje cell bodies were identified by their large size, layered arrangement, and large dendritic tree. Evoked CF–PC EPSCs were recorded at −10 to −20 mV; miniature EPSCs (mEPSCs) were recorded at −70 mV in the presence of tetrodotoxin (1 μm). To isolate mEPSCs resulting from CF release, slices from postnatal day 8 (P8) to P10 rats were used because functional parallel fiber synapses are not usually present at this age (Yamada et al., 2000); we were unable to evoke EPSCs in Purkinje cells by stimulation in the molecular layer in these slices. Climbing fibers were stimulated with a theta glass pipette pulled to a 5–10 μm tip and filled with external solution. The stimulating electrode was placed in the granule cell layer ∼10–50 μm from the Purkinje cell layer. A constant-voltage isolated stimulator (Digitimer, Hertfordshire, UK) was used to supply a 20–100 μsec pulse of 10–99 mV. Pipette position and stimulus intensity were adjusted until the current necessary to produce an all-or-none response was minimized. Climbing fiber failures were complete, indicating that there was no contamination by parallel fiber synapses. Synaptic currents were filtered at 2 kHz and digitized at 10 kHz, and patch currents were filtered at 5 kHz and digitized at 30–50 kHz. Synaptic and patch currents were recorded with an Axopatch 1C (Axon Instruments, Foster City, CA). A four-barreled glass pipette mounted on a piezoelectric bimorph (Morgan Matroc, Bedford, OH) was used for agonist application to outside-out patches (Tong and Jahr, 1994). Data were acquired using Axobasic software (Axon Instruments) and Igor Pro software (Wavemetrics, Lake Oswego, OR). Data analysis was performed using Axograph 4.6, and results were compiled and statistical analysis was performed using Microsoft Excel (Microsoft, Redmond, WA) and GraphPad (GraphPad Software, San Diego, CA). All experiments were performed at 34–36°C using an in-line heating device (Warner Instruments, Hamden, CT). Values are given as mean ± SEM, and confidence limits were determined using Student's t tests.
Results
Bafilomycin A1 and concanamycin A reduce the frequency and amplitude of PC mEPSCs
If high Pr (Dittman and Regehr, 1998; Silver et al., 1998) results in high occupancy of AMPARs at CF–PC synapses, then AMPAR-mediated EPSCs should be relatively insensitive to small changes in the glutamate concentration transient in the synaptic cleft. We used two blockers of the vacuolar-type ATPase, Baf and Conca (Drose and Altendorf, 1997), to reduce glutamate concentrations in synaptic vesicles (Zhou et al., 2000) by abolishing the proton and voltage gradient required by vesicular glutamate transporters (Carlson and Ueda, 1990; Maycox et al., 1990). First, we tested the efficacy of Baf or Conca to reduce the glutamate concentration in CF vesicles by comparing the amplitude and frequency of mEPSCs recorded in PCs in control P8–P10 cerebellar slices with those that had been incubated in Baf (2 μm) or Conca (5 μm) for ≥1 hr. Because the frequency of mEPSCs in PCs is very low, 1 μmpardaxin (Bergles et al., 2000) was used to induce asynchronous release. Both Baf and Conca resulted in a significant decrease in pardaxin-induced mEPSC amplitude as demonstrated by a leftward shift in the cumulative amplitude distribution (Fig.1A,B). Incubation with Baf or Conca also dramatically decreased pardaxin-stimulated mEPSC frequency (Fig.1A,C) (control, 4.86 ± 0.74 Hz vs Baf, 0.426 ± 0.084 Hz; n = 3;p < 0.0001; control, 3.03 ± 0.59 Hz vs Conca, 0.38 ± 0.055 Hz; n = 4; p < 0.003), consistent with the majority of mEPSC amplitudes decreasing to undetectable levels (Zhou et al., 2000).

Bafilomycin A1 reduces the amplitude and frequency of induced mEPSCs from Purkinje cells. A, Representative consecutive traces from a cerebellar Purkinje cell, showing pardaxin- (1 μm) induced mEPSCs, in control (A1) and in slices pretreated with bafilomycin A1 (2 μm) for ≥1 hr (A2 ). Recorded in the presence of TTX (1 μm) and picrotoxin (100 μm). Holding potential, −70 mV. B, Cumulative probability distribution of mEPSC amplitude before and after bafilomycin A1 treatment. C, Frequency of mEPSCs before and after bafilomycin A1 treatment.
Bafilomycin A1 and concanamycin A increase paired-pulse depression at CF–PC synapses
Paired-pulse depression at CF–PC synapses has been shown to result from a lower Pr after the second stimulus than that after the first stimulus (Dittman and Regehr, 1998; Hashimoto and Kano, 1998;Silver et al., 1998). If AMPAR occupancy follows Pr (Silver et al., 1998; Wadiche and Jahr, 2001), then we would expect that occupancy during the first EPSC (EPSC1) would be higher than that during the second EPSC (EPSC2) and thus that EPSC1 may be less sensitive to changes in the glutamate concentration transient than EPSC2. Addition of Baf (2 μm) or Conca (5 μm) to the perfusate resulted in a reduction in the amplitudes of both EPSCs over time (Fig. 2) (interstimulus interval, 50 msec). However, EPSC2 decreased sooner and to a greater extent than EPSC1 (Fig. 2B,C) (time to 10% reduction in amplitude in Baf, 320 ± 20 sec EPSC1 vs 183 ± 14 sec EPSC2;p = 0.005; n = 3; in Conca, 535 ± 44 sec vs 451 ± 34 sec; p = 0.006;n = 5), and, thus, Baf and Conca application caused a decrease in the paired-pulse ratio (EPSC2/EPSC1; control, 0.72 ± 0.03 vs Baf, 0.36 ± 0.005; n = 3; control, 0.74 ± 0.01 vs Conca, 0.32 ± 0.04; n = 5). This differential inhibition suggests that AMPARs are highly occupied after the first stimulus. The much larger paired-pulse depression seen after 30 min in Baf or Conca indicates that presynaptic depression of release is far greater than predicted from the paired-pulse ratio of EPSCs in control conditions. In addition to the effects on paired-pulse depression, Baf treatment tended to accelerate the decay of EPSC1 (control, 3.17 ± 0.51 msec vs Baf, 2.27 ± 0.20 msec, weighted decay time constants;p = 0.07; n = 3), suggesting that the large bolus of glutamate released by the first stimulus temporarily overwhelms clearance mechanisms.

Bafilomycin A1 reduces climbing fiber-evoked EPSC amplitude and increases paired-pulse depression in Purkinje cells. A, Sample traces of CF-evoked EPSCs at 0, 20, 25, and 30 min after addition of bafilomycin A1 (2 μm) to perfusate. Each trace is an average of at least five sweeps. Holding potential, −20 mV. B, Recordings in A scaled to the peak of the first EPSC at time 0. C, Time course of the effect of bafilomycin A1 on the normalized amplitude of EPSC1 (filled circles) and EPSC2 (open circles).
AMPARs expressed by Bergmann glial cells are more sensitive to changes in Pr than those of PCs
Climbing fiber synapses are surrounded by sheets of BG membrane (Palay and Chan-Palay, 1974), which express both AMPARs (Baude et al., 1994) and glutamate transporters (Rothstein et al., 1994; Chaudhry et al., 1995). Stimulation of CFs activates currents in BGs mediated by both of these glutamate receptors (Bergles et al., 1997). Because the peak concentration of glutamate reaching the BG AMPARs is ∼200 μm (Dzubay and Jahr, 1999), it seems unlikely that receptor saturation occurs. Thus, BG AMPARs may be more sensitive to changes in the amount of glutamate released from CF synapses than those in the PC postsynaptic membrane. To test this, responses of both cell types to CF stimulation were compared in control and in the presence of 700 μm Co2+ to inhibit presynaptic voltage-dependent Ca2+currents and thereby decrease multivesicular release and the resulting glutamate concentration transients. Co2+decreased the CF–PC EPSC by only 7.4 ± 2.5% (n= 5), whereas the CF–BG AMPAR component (in CTZ, 200 μm and TBOA, 50 μm; see below) was inhibited by 49.7 ± 7.2% (n = 7) (Fig. 3A). Because BG AMPARs but not those of PCs are calcium permeable (Geiger et al., 1995), the differential sensitivity could result from channel block of BG AMPARs by Co2+. However, AMPAR currents evoked in outside-out patches from BG by 10 mm glutamate were not significantly altered by 700 μmCo2+ (Fig. 3B). These results, like those with Baf or Conca, are consistent with the proposal that multivesicular release creates a large transient concentration of glutamate in the cleft that results in the occupation of practically all postsynaptic PC AMPARs. Because glutamate diffuses to BG AMPARs, dilution in the extrasynaptic space is apparently sufficient to decrease the glutamate concentration transient to nonsaturating levels.

Reduction of climbing fiber release probability has different effects on response amplitude of Purkinje cells and Bergmann glia cells. A, Average reduction in response amplitude by CoCl2 (700 μm) in Purkinje cells and Bergmann glia cells. Inset, Recordings from a Purkinje cell and a Bergmann glial cell before and during CoCl2 (700 μm) application. Recordings are averages of at least five sweeps. Bergmann glial responses were recorded in the presence of CTZ (200 μm) and TBOA (50 μm). Holding potential, −80 mV for the Bergmann glial cell and −20 mV for the Purkinje cell. *p < 0.05; **p < 0.005. B, Addition of CoCl2 (700 μm) had no significant effect on Bergmann glial outside-out patch responses to 10 mmglutamate in 200 μm CTZ.
Paired-pulse depression is larger in BGs than in PCs
If BG glutamate receptors are more sensitive monitors of changes in presynaptic release from CFs than PCs, we would expect them to report greater paired-pulse depression than PCs in normal conditions. In fact, paired-pulse depression (150 msec interstimulus interval) was dramatically greater in BGs than in PCs (Fig. 4A,D). In these recordings, the two components of the CF–BG response were separated pharmacologically. The AMPAR-mediated component was isolated by subtracting the synaptic transporter current (STC) (10 μm NBQX) (Fig. 4B) from the control currents (Bergles et al., 1997; Dzubay and Jahr, 1999). The paired-pulse ratio of the CF–BG AMPAR-mediated current was 0.14 ± 0.01 (Fig. 4A) (n = 12). This is a sixfold larger depression than that of the CF–PC EPSC (Fig.4D) (0.83 ± 0.025; n = 5).

Climbing fiber stimulation elicits both AMPA receptor and glutamate transporter currents in Bergmann glial cells.A, Comparison of the dual-component Bergmann glial response (Control) and the isolated AMPA receptor-mediated response (Subtracted) by subtraction of the recording in B (interstimulus interval, 150 msec). B, CF-evoked transporter current from the same cell as in A in the presence of NBQX (10 μm). C, Current remaining after the addition of TBOA (50 μm). D, CF–PC EPSCs evoked by stimuli at a 150 msec interval. All traces are averages of at least five sweeps. Holding potential, −80 mV for the Bergmann glial cell and −20 mV for the Purkinje cell.
The CF–BG STC (Fig. 4B) accounted for less of the first CF–BG current than the second (16 ± 2 and 39.5 ± 3.5%, respectively), indicating that the conductances mediated by BG AMPA receptors and transporters do not have the same sensitivity to the same presynaptic release events. In part, this may be the result of differential access of glutamate to AMPA receptors and transporters and to nonlinear dose–response relationships (see below). The paired-pulse ratio of the CF–BG STCs, as measured by their charge transfer, should not be affected by a nonlinear dose–response relationship, because the stoichiometry of uptake is fixed in the absence of permeable anions; each glutamate molecule taken up results in two charges crossing the membrane (Zerangue and Kavanaugh, 1996). Thus, the integral of the current in time should represent the number of glutamate molecules taken up by the BG. When expressed as charge transfer, the paired-pulse ratio for the CF-BG STC was 0.59 ± 0.06 (range, 0.34–0.91;n = 9), falling between the paired-pulse ratio of the CF–PC EPSC (0.83) and the CF–BG AMPAR response (0.14). The paired-pulse depression of the CF–BG STC amplitudes (0.51 ± 0.03; n = 9) was greater than that of charge transfer because the decay time of the first STC (8.5 ± 0.7 msec) was longer than that of the second STC (6.6 ± 0.7; n= 7; p < 0.01). The difference in these decay times suggests that the larger glutamate transient after the first stimulus takes longer to clear than that after the second stimulus and also suggests that clearance mechanisms are taxed more by large glutamate transients (see below).
Cyclothiazide decreases paired-pulse depression
The surprisingly large paired-pulse depression of the CF–BG AMPAR response may result not only from depression of presynaptic release but also from the nonlinear dose–response relationship of these AMPARs (Dzubay and Jahr, 1999). CTZ (200 μm), a blocker of BG AMPAR desensitization that also increases the apparent affinity of AMPARs (Patneau et al., 1993; Yamada and Tang, 1993; Partin et al., 1994, 1996; Koh et al., 1995; Dzubay and Jahr, 1999), significantly increased the first and second CF–BG AMPAR responses to paired stimulation by 6.43 ± 0.25 and 8.89 ± 0.42-fold, respectively, as well as the paired-pulse ratio (0.18 ± 0.01;n = 12). This effect is consistent with the action of CTZ to potentiate AMPAR responses evoked by lower concentrations of glutamate more than AMPAR responses evoked by higher concentrations (Dzubay and Jahr, 1999). In addition, if AMPAR desensitization contributed to paired-pulse depression at an interstimulus interval of 150 msec, then CTZ would inhibit this effect.
Glutamate transporters shield BG AMPARs
The paired-pulse depression of the CF–BG AMPAR response is also influenced by surrounding glutamate transporters. In the presence of CTZ, inhibition of glutamate transporters (TBOA, 50 μm) increased the amplitude and slowed the decay phase of both the first and second CF–BG AMPAR responses (Fig.5A). However, the amplitude of the second response was increased more (57%) than the first (23%), resulting in an additional decrease in paired-pulse depression (control, 0.20 ± 0.017 vs TBOA, 0.26 ± 0.023;p = 0.003; n = 9) (Fig. 5). This result suggests that transport is overwhelmed by the large glutamate transient resulting from the first CF stimulus and that this allows activation of more BG AMPA receptors. The depressed release after the second CF stimulus results in a smaller glutamate transient that is more effectively curtailed by transport and causes less BG AMPA receptor activation.
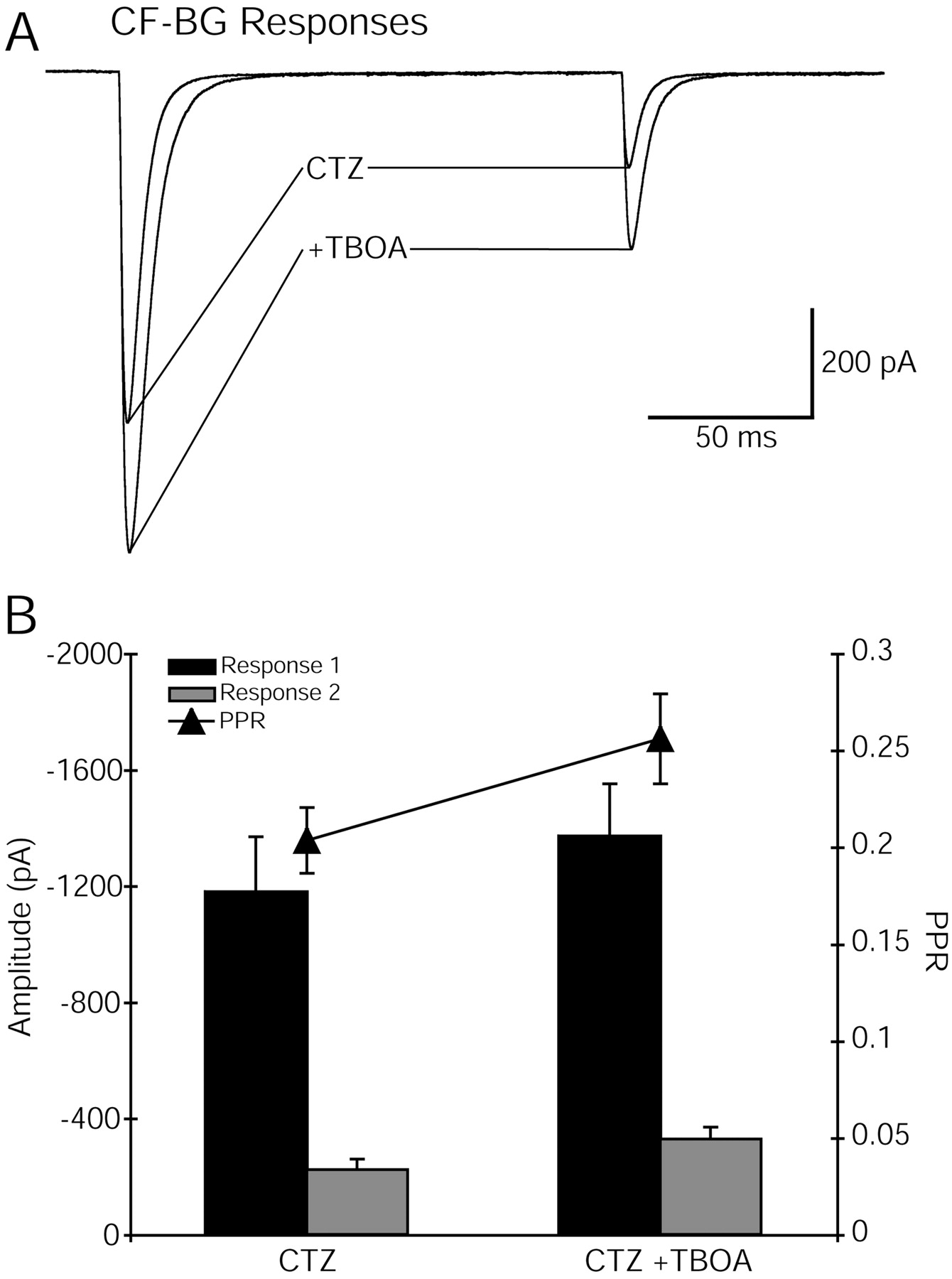
Inhibition of glutamate transporters decreases paired-pulse depression of CF–Bergmann glial responses.A, Paired-pulse depression of CF-evoked Bergmann glial responses was decreased by TBOA (50 μm) in the presence of CTZ (200 μm). Holding potential, −78 mV.Traces are an average of at least five sweeps.B, Average effects of TBOA on peak amplitudes and paired-pulse ratio (PPR; response 1/response 2) of six Bergmann glial cells.
Glutamate transporters shield presynaptic metabotropic glutamate receptors
The fact that transporters apparently shield BG AMPARs suggests that they might shield other extrasynaptic glutamate receptors (Brasnjo and Otis, 2001) that could alter paired-pulse depression. Activation of mGluRs by exogenous agonists suppresses transmission at CF synapses by a presynaptic mechanism (Hashimoto and Kano, 1998). However, these presynaptic mGluRs are not activated by release of endogenous glutamate (Hashimoto and Kano, 1998). In keeping with these results, CF–BG responses recorded in the presence of CTZ (200 μm) were not altered by inhibitors of mGluRs (Fig.6A). However, if glutamate transporters were first inhibited by TBOA, then paired-pulse depression was decreased by mGluR antagonists at an interstimulus interval of 150 msec but not 40 msec (Fig. 6B) (control, 0.36 ± 0.04 vs mGluR antagonist, 0.43 ± 0.04;p < 0.0001; n = 5). The response to the first stimulus was not altered by mGluR antagonists, indicating that basal levels of glutamate are too low to cause significant mGluR activation. However, with glutamate transport inhibited, the transient elevation of glutamate after release is sufficient to activate presynaptic mGluRs, causing a decrease in subsequent release. The linkage between mGluRs and their effects on the release process apparently requires >40 msec.

Glutamate transporters shield presynaptic metabotropic glutamate receptors. A, mGluR antagonists (500 μm CPPG and 1 mm MCPG) had no effect on the paired-pulse ratio (PPR) at an interstimulus interval (ISI) of 40 msec in the presence (filled circles; n = 5;n = 2 for washout) or absence (open circles; n = 5) of TBOA (50 μm). CTZ at 200 μm was present in both conditions. Insets, Superimposed average currents, with or without TBOA (50 μm), in the absence and presence of mGluR antagonists. B, mGluR antagonists (500 μm CPPG and 1 mm MCPG) had no effect on the paired-pulse ratio at an interstimulus interval of 150 msec in the absence (open circles; n = 5) of TBOA (50 μm), but, in the presence of TBOA, the paired-pulse ratio was significantly increased (filled circles; n = 5; n = 2 for washout). *p < 0.05. Insets, Superimposed average currents, with or without TBOA (50 μm), in the absence and presence of mGluR antagonists. Recordings from the same cells as in A. Eachtrace is an average of at least five sweeps.
Discussion
AMPA receptor saturation at the climbing fiber–Purkinje cell synapse
The frequency-dependent depression of CF–PC EPSCs has been attributed to presynaptic reduction of transmitter release (Dittman and Regehr, 1998; Hashimoto and Kano, 1998; Silver et al., 1998). Although this is clearly true, our results show that paired-pulse depression of release is greater than that reported by the EPSCs. This underestimate of depression results from the high occupancy of postsynaptic AMPARs caused by multivesicular release (Wadiche and Jahr, 2001). In these conditions, the number of postsynaptic receptors, rather than the size of the glutamate concentration transient in the cleft, limits the size of the EPSC. Subsequent stimuli result in less multivesicular release, lower receptor occupancy, and therefore a more accurate measure of the glutamate concentration transient by the AMPAR EPSCs.
Multivesicular release overwhelms clearance mechanisms
The results of several experiments suggest that multivesicular release results in such a large glutamate transient that clearance from climbing fiber synapses is retarded. This is particularly apparent in comparisons of the characteristics of EPSCs evoked by paired stimuli 40–150 msec apart. First, in control conditions, the decay time course of the first EPSC is slower than that of the second, and this cannot be attributed to voltage-clamp errors (Wadiche and Jahr, 2001). Second, as Baf washed into the slice, the decay phase of EPSC1 tended to become faster, because the quantity of glutamate in each vesicle presumably decreases. Similar effects are found by lowering release probability by decreasing external calcium (Wadiche and Jahr, 2001). Third, low-affinity competitive antagonists of AMPARs, which are more effective at blocking low rather than high concentrations of glutamate, speed the decay of the first EPSC (Wadiche and Jahr, 2001). Fourth, Bergmann glial AMPAR responses to paired climbing fiber stimuli are differentially altered by blocking transporters. The amplitude of the first response is increased only slightly, whereas the second is increased by >50%. This suggests that transporters are overwhelmed by the first glutamate transient, whereas the transient resulting from the second stimulus is readily curtailed by transporters. This finding is corroborated by the comparison of paired-pulse depression of the Bergmann glial AMPARs and the STC. Whereas the former displays very large depression, the depression of the STC is less. Because all glutamate released must eventually be taken up, the Bergmann glial transporter current may offer a less biased measure of presynaptic function. Indeed, the decay time constant of the first STC of a pair is longer than that of the second, indicating that clearance can be overwhelmed if enough glutamate is released.
In addition to shielding AMPARs expressed by Bergmann glia, glutamate transporters apparently protect presynaptic mGluRs from released transmitter. Similar results indicate that postsynaptic mGluRs are also protected by glutamate transporters at parallel fiber synapses (Brasnjo and Otis, 2001). Exogenous agonists of mGluRs have been shown to decrease release from climbing fibers (Hashimoto and Kano, 1998), but physiological activation of these receptors has not been shown. In the presence of glutamate transporter blockers, we show that paired-pulse depression of the BG AMPAR response is decreased by metabotropic receptor antagonists without affecting the amplitude of the first response. This indicates that the glutamate released by the first stimulus is responsible for this effect rather than an elevation of ambient glutamate. Because this phenomenon was observed with interstimulus intervals of 150 msec but not 40 msec, it is consistent with activation of a G-protein-coupled receptor, because the downstream effectors of such receptors require a variable period after receptor binding to assert their actions. Although the decrease of paired-pulse depression within 150 msec is rapid, it is in line with βγ subunit-linked effects (Hille, 2001). Whether these mGluRs are ever activated in vivo remains to be determined.
Physiology of receptor saturation
We suggest that multivesicular release results in the near-saturation of AMPA receptors expressed by Purkinje cells at climbing fiber synapses. What is the consequence of this seemingly inappropriate waste of transmitter? By increasing receptor occupancy, multivesicular release will augment charge transfer in the Purkinje cell after climbing fiber stimulation and ensure that the Purkinje cell will fire a complex spike. However, complex spikes generally are evoked by climbing fiber action potentials within a few tens of milliseconds of previous climbing fiber spikes, when release probability is far lower. Because the frequency of climbing fiber action potentials reaches only 8 Hz (125 msec period), the safety factor at this synapse is rather high. Indeed, it may be that the usefulness of multivesicular release and the resulting underreporting of presynaptic depression is to keep this safety factor high. Although presynaptic release is dramatically lower at 150 msec after a previous stimulus, as judged by paired-pulse depression in Bergmann glia, the Purkinje cell EPSC in control conditions is decreased by only ∼20%. This nonlinear relationship between release and postsynaptic response ensures the fidelity of transmission at this synapse, which is thought to provide essential error correction capabilities to the motor system.
Whether the postsynaptic responses at other synapses similarly underestimate changes in presynaptic release is uncertain. However, recent reports that the incidence of multivesicular release follows Pr at hippocampal excitatory synapses (Oertner et al., 2002) suggest that, even at synapses that have much lower Pr than those of the climbing fibers, postsynaptic responses may not faithfully report presynaptic release. In addition, at some inhibitory synapses, both multivesicular release (Auger et al., 1998) and high postsynaptic receptor occupancy (Auger and Marty, 1997; Auger et al., 1998; Hajos et al., 2000) can occur, the same conditions that lead to underreporting changes in release at the climbing fiber–Purkinje cell synapse.
Footnotes
This work was supported by National Institutes of Health Grant NS40056 and Human Frontier Science Program Grant 119/2000. We thank the members of the Jahr laboratory for discussions and helpful comments.
Correspondence should be addressed to Craig E. Jahr, Vollum Institute, Oregon Health and Science University, L474, 3181 S.W. Sam Jackson Park Road, Portland, OR 97201-3098. E-mail: jahr{at}ohsu.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}