

Abstract
Brain-derived neurotrophic factor (BDNF) has been shown to be necessary and sufficient for post-stroke recovery in rodents. From these observations, we and others have hypothesized that a common single nucleotide polymorphism (SNP) in the pro-domain of bdnf that leads to a methionine (Met) substitution for valine (Val) at codon 66 (Val66Met) will affect stroke outcome. Here we investigate the effect of the BDNF genetic variant on ischemic outcome by using mice with a genetic knock-in of the human BDNF variant in both alleles (BDNFMet/Met). Compared with wild-type mice, BDNFMet/Met mice exhibited reduced CNS BDNF levels without a discernable effect on infarct size. Diminished BDNF levels in BDNFMet/Met mice were associated with greater deficits in post-stroke locomotor functions. Additionally, the BDNFMet/Met mice showed reduced angiogenesis and elevated expression of thrombospondin-1 (TSP-1) and its receptor CD36, anti-angiogenic factors. To assess the functional role of CD36 in antagonizing angiogenic response in Met homozygosity at the BDNF locus, we crossed BDNFMet/Met mice with CD36 knock-out mice. The double-mutant mice rescued the angiogenic deficit associated with the BDNFMet/Met mice without alterations in BDNF levels, indicating that the behavioral deficit in BDNFMet/Met mice after stroke is partly related to an unfavorable balance in pro-angiogenic BDNF and anti-angiogenic TSP-1/CD36. The results suggest that CD36 inhibition may be a viable strategy to enhance angiogenesis and possible recovery in human stroke victims who are Met homozygotes at codon 66 of the BDNF locus.
Introduction
Unlike vasculogenesis, neoangiogenesis after stroke involves sprouting of new vessels from preexisting vessels (Hayashi et al., 2006). Enhanced angiogenesis in the ischemic penumbra is correlated with increased survival of stroke patients (Krupinski et al., 1994). Furthermore, the promotion of ischemia-induced angiogenesis within the ischemic boundary was suggested as a therapeutic strategy to improve stroke outcome (Rosell-Novel et al., 2004; Slevin et al., 2006).
Brain-derived neurotrophic factor (BDNF) promotes neuronal survival, differentiation, synaptic plasticity, and angiogenesis in normal and ischemic tissue (Donovan et al., 2000; Chao, 2003; Kermani et al., 2005; Wagner et al., 2005; Tongiorgi, 2008). BDNF expression is upregulated in the boundary of the infarct with much less expression in the infarct core (Kokaia et al., 1995, 1998). Whereas most studies indicate that enhanced BDNF availability and signaling ameliorate ischemic brain damage and functional recovery, some argue against the beneficial effect of BDNF (Gustafsson et al., 2003; Nygren et al., 2006).
A single nucleotide polymorphism (SNP) of the bdnf gene, which results in the substitution of a valine (Val) to a methionine (Met) in the prodomain of the BDNF protein, was identified. This exclusively human SNP occurs with relatively high frequency (Ventriglia et al., 2002; Egan et al., 2003; Itoh et al., 2004; He et al., 2007). Identified as the first genetic alteration in the neurotrophin system, the BDNF has been implicated in conferring susceptibility to various neuropsychiatric disorders and altered episodic memory in patients with psychiatric disease (Sklar et al., 2002; Egan et al., 2003; Sen et al., 2003). Although studies on the impact of the BDNF polymorphism in the outcome after ischemic stroke are limited, clinical studies suggest a correlation of this BDNF polymorphism with poor outcome in hemorrhagic stroke patients (Siironen et al., 2007; Vilkki et al., 2008).
Angiogenesis is tightly regulated by factors that promote as well as inhibit vessel formation. Whereas BDNF evokes pro-angiogenic responses in the ischemic hindlimb (Kermani et al., 2005; Kermani and Hempstead, 2007), ischemia also upregulates the angiostatic receptor CD36 in the post-ischemic brain (Cho et al., 2005). CD36 is expressed in the microvascular endothelium, and the interaction of CD36 with thrombospondin (TSP)-1 and TSP-2 mediates a signaling cascade that leads to endothelial cell apoptosis (Jiménez et al., 2000; Febbraio et al., 2001), offsetting compensatory angiogenesis-promoting cascades. Although CD36 expression is relatively low in the normal brain, increased expression of CD36 and TSPs has been reported after cerebral ischemia (Hayashi et al., 2003; Lin et al., 2003; Cho et al., 2005). The current study investigates the impact of BDNF SNP on ischemic outcome and angiogenic response using mice with a genetic knock-in of the human bdnf Val66Met variant. This study clarifies potential interactions between angiogenic and angiostatic factors to regulate angiogenesis in the post-ischemic brain. We report that the BDNF SNP contributed to reductions in stroke-induced BDNF release, poorer behavioral outcome, and deficits in angiogenic response. We also determined that the absence of CD36 can rescue the angiogenesis deficit in mice with the BDNF SNP.
Materials and Methods
Animals.
The use of animals and procedures performed were approved by the Institutional Animal Care and Use Committee of Weill Medical College of Cornell University. Experiments were performed in BDNF+/+ wild-type and BDNFMet/Met mutant mice generated and housed at Weill Cornell Medical College. These mice were backcrossed 10 times into the C57BL/6 strain, and the procedures for heterozygote breeding and genotyping were described previously (Chen et al., 2006). CD36 knock-out (KO) mice were generated by Dr. Maria Febbraio at Weill Cornell Medical College and backcrossed seven times into the C57BL/6 strain. The procedures for breeding and genotyping were described previously (Febbraio et al., 1999, 2000). BDNFMet/Met/CD36−/− double mutants (Dmt) (8× backcrossed C57BL/6) and background matching BDNFMet/Met/CD36+/+ controls (Dwt) were generated by crossing BDNFMet/Met mice with CD36 KO mice. All mouse lines were housed at the Burke Medical Research Institute.
Transient middle cerebral artery occlusion.
Procedures for middle cerebral artery occlusion (MCAO) were described previously (Cho et al., 2005, 2007). Both male and female mice were used to determine infarct volume and percentage hemispheric swelling. For the rest of the study, only male mice were used. Briefly, mice were anesthetized with a mixture of isoflurane/oxygen/nitrogen. A fiber optic probe was glued to the right parietal bone (2 mm posterior and 5 mm lateral to bregma) and connected to a laser-Doppler flowmeter (Periflux System 5010; Perimed) for continuous monitoring of cerebral blood flow (CBF) in the center of the ischemic territory. For MCAO, a 6-0 Teflon-coated black monofilament surgical suture (Doccol Co.) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the cerebral arterial circle to obstruct the origin of the MCA. The filament was left in place for 30 min and then withdrawn. Only animals that exhibited both a reduction in CBF >80% during MCAO and a recovery of CBF >80% by 10 min after reperfusion were included in the study. This procedure leads to similar blood flow reduction and reperfusion among the groups and to reproducible infarcts involving both the cerebral cortex and the striatum in the right hemisphere (Cho et al., 2005).
Behavior testing.
Functional outcome was assessed using a rotarod (Med Associates) and a Noldus Catwalk XT gait analysis system (Noldus Information Technology). These rotarod and Catwalk systems were used to assess functional recovery after stroke and other CNS injury (Hamers et al., 2006; Liauw et al., 2008; Wang et al., 2008; Neumann et al., 2009). For the rotarod test, the device was set to accelerate from 4 to 40 rpm over the course of 5 min. The animals were pretrained 6 consecutive days before ischemia, and post-stroke testing was performed 6 d after MCAO. The latency to fall from the rod (performance) was recorded and averaged for five trials. For the Catwalk gait analysis system, mice were pretrained daily for 2 weeks to cross an illuminated glass walkway three consecutive times and then tested at 1 d, 4 d, 7 d, 2 weeks, and 1 month after MCAO. The images from each trial that consist of three consecutive runs were converted into digital signals. After classification of each footprint, spatial parameters based on individual paws (maximum contact area and mean intensity), relative positions between paws [stride length and base of support (BOS)], temporal parameters (stand and swing speed), and parameter related to interlimb coordination (regularity index) were determined.
Tissue preparation.
Mice were killed 3 or 7 d after MCAO. Brains were excised, frozen, and sectioned using a cryostat. Because infarct typically spans ∼6 mm rostrocaudally starting from +2.8 mm from bregma and extending to −3.8 mm, we used an unbiased stereological sampling strategy to collect tissue in this region that included infarct. Tissue sections were collected serially for measurement of infarct volume/immunohistochemistry (20 μm thickness) and for gene expression and ELISA (100 μm) at a 600 μm intervals.
Measurement of infarct volume and swellings.
Infarct volume and percentage hemispheric swelling were determined in serial sections using Axiovision software (Carl Zeiss). The contribution resulting from swelling was corrected using a method described previously (Lin et al., 1993).
BDNF ELISA.
The BDNF protein concentrations were determined in the brain lysates using the BDNF Emax immunoassay system, recombinant mature BDNF as a standard according to the instructions of the manufacturer (Promega).
Immunohistochemistry.
Brain sections were completely air dried and subsequently fixed in −20°C methanol for 2–3 min or 4% paraformaldehyde for 15 min, then washed three times in PBS at pH 7.4, and incubated in 0.2% Triton X-100 for 5 min. After washing with PBS, slides were blocked in 1% BSA (Sigma) in PBS containing 5% normal goat serum for 1 h at room temperature, followed by overnight incubation with primary antibodies: Ki-67 (cell proliferation marker, 1:000; Abcam), CD31 (endothelial marker, 1:1000; Millipore Bioscience Research Reagents), CD36 (1:300; Santa Cruz Biotechnology), and glial fibrillary acidic protein (GFAP) (astrocytes marker, 1:3000; Millipore Bioscience Research Reagents). Sections were washed with PBS, incubated with secondary antibodies conjugated with Alexa Fluor 488 or 594 (1:200; Invitrogen) for 1 h at room temperature, and coverslipped to examine under a florescent microscope or laser scanning confocal microscopy (Carl Zeiss).
Assessment of proliferating endothelial cells.
Double-fluorescence immunohistochemistry of Ki-67 and CD31 was performed 3 d after ischemia. Using a laser scanning confocal microscope, the number of proliferating endothelial cells was identified by nuclear staining of Ki-67 juxtaposed CD31-immunoreactive endothelial cells in the entire infarct area. The density of proliferating endothelial cells was obtained from the total number of Ki-67/CD31-positive (CD31+) cells divided by the entire infarct area. A section at the level of striatum (+1.2 from bregma) was stained, counted, and averaged to represent the density for an animal. Values were presented as cell number per square millimeter.
Assessment of vessel density.
Double-fluorescence immunohistochemistry of GFAP and CD31 was performed 7 d after ischemia. To quantify vessel density, we modified our published nonbiased counting method (Cho et al., 1997). A 10 × 10 grid in a 0.1 mm2 frame was placed along the glial scar that is demarcated by GFAP immunoreactivity as shown in Figure 4G. For each animal, CD31+ vessels that cross grid lines were counted in a section (+1.2 mm from bregma). The number of intersections within a frame was counted and averaged from 7–10 randomly chosen inner and outer fields along the glial scar. The total number of intersections from the field was averaged to represent an inner and outer vessel density for each animal. Values were presented as number of intersections per frame area (0.1 mm2).
Real-time reverse transcription-PCR for CD36, CD47, and TSP-1/2 gene expression.
Relative mRNA levels of CD36, CD47, and TSP-1/2 were quantified with real-time quantitative reverse transcription-PCR using fluorescent TaqMan technology (Cho et al., 2007; Kim et al., 2008). Total RNA was extracted from brain tissues using TRI Reagent (Molecular Research Center). Total RNA from brain tissues was reverse transcribed using oligo-dT primers and the SuperScript First-Strand Synthesis System (Invitrogen), according to the protocol of the manufacturer. PCR primers and probes specific for CD47, CD36, and TSP-1/2 and β-actin were obtained as TaqMan predeveloped assay reagents for gene expression (Applied Biosystems). β-Actin was used as an internal control for normalization of samples. The PCR reaction was performed using FastStart Universal Probe Master (Roche) and Applied Biosystems 7500 Fast Real-Time PCR system, according to the instructions of the manufacturer. Reactions were performed in 20 μl total volume and incubated at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The results were analyzed by 7500 Fast Real-Time PCR System software.
Statistics.
Data are presented as mean ± SEM. All analyses were performed, and analyzed data were graphed using GraphPad Software Prism 4.0. Comparisons between two groups were statistically evaluated by the Student's t test. Multiple comparisons were made using two-way ANOVA. Differences were considered significant at p < 0.05.
Results
Mice with the BDNF SNP exhibit reduced stroke-induced BDNF levels without affecting injury size
To investigate the effect of the BDNF Val66Met SNP on ischemic outcome, we first determined whether the BDNF SNP has an effect on stroke-induced BDNF levels in the brain. Because ischemia upregulates BDNF transcript and protein expression (Kokaia et al., 1995; Krikov et al., 2008), we initially determined temporal changes in stroke-induced BDNF level using an entire hemisphere of C57BL/6 mice. Stroke-induced BDNF levels were increased at 3–7 d (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Using these time points, tissue was serially sectioned through the infarct territory (see Materials and Methods), and stroke-induced BDNF levels were compared. BDNFMet/Met mice exhibited lower BDNF levels relative to wild type at 3 and 7 d after ischemia, with a larger difference at 3 d (Fig. 1A,B). Infarct size and hemispheric swelling, assessed at 3 d after ischemia between two genotypes, were not different in both genders (Fig. 1C,D). No differences in cerebral blood flow reduction and reperfusion during and 10 min after ischemia suggests that the severity of ischemia was comparable between BDNF+/+ and BDNFMet/Met mice (Table 1). The dissociation between BDNF levels and ischemic outcomes indicates that the amount of BDNF in the post-ischemic brain is not a critical factor for acute infarct development.

Effect of BDNF Val66Met polymorphism on BDNF levels and ischemic outcome. Total BDNF levels from brain lysates were determined in BDNF+/+ and BDNFMet/Met mice using BDNF ELISA at 3 d (A) and 7 d (B) after stroke. Note that BDNF SNP resulted in a significant reduction of stroke-induced BDNF secretion. n = 5–10 per group. *p < 0.05 and ***p < 0.001 versus contralateral; ##p < 0.01 and ###p < 0.001 versus BDNF+/+ ipsilateral. Contl, Contralateral; Ipsl, ipsilateral. Infarct volume (Inf Vol) (C) and percentage hemispheric swelling (D) were determined 3 d after ischemia in male and female BDNF+/+ and BDNFMet/Met mice. No difference in ischemic outcomes was found between the genotypes. n = 11–15 per group.
Body weight, CBF reduction and reperfusion, and breakdown of the numbers of mice that were included in the study
Mice with the BDNF SNP exhibit poorer motor behavioral outcomes after stroke
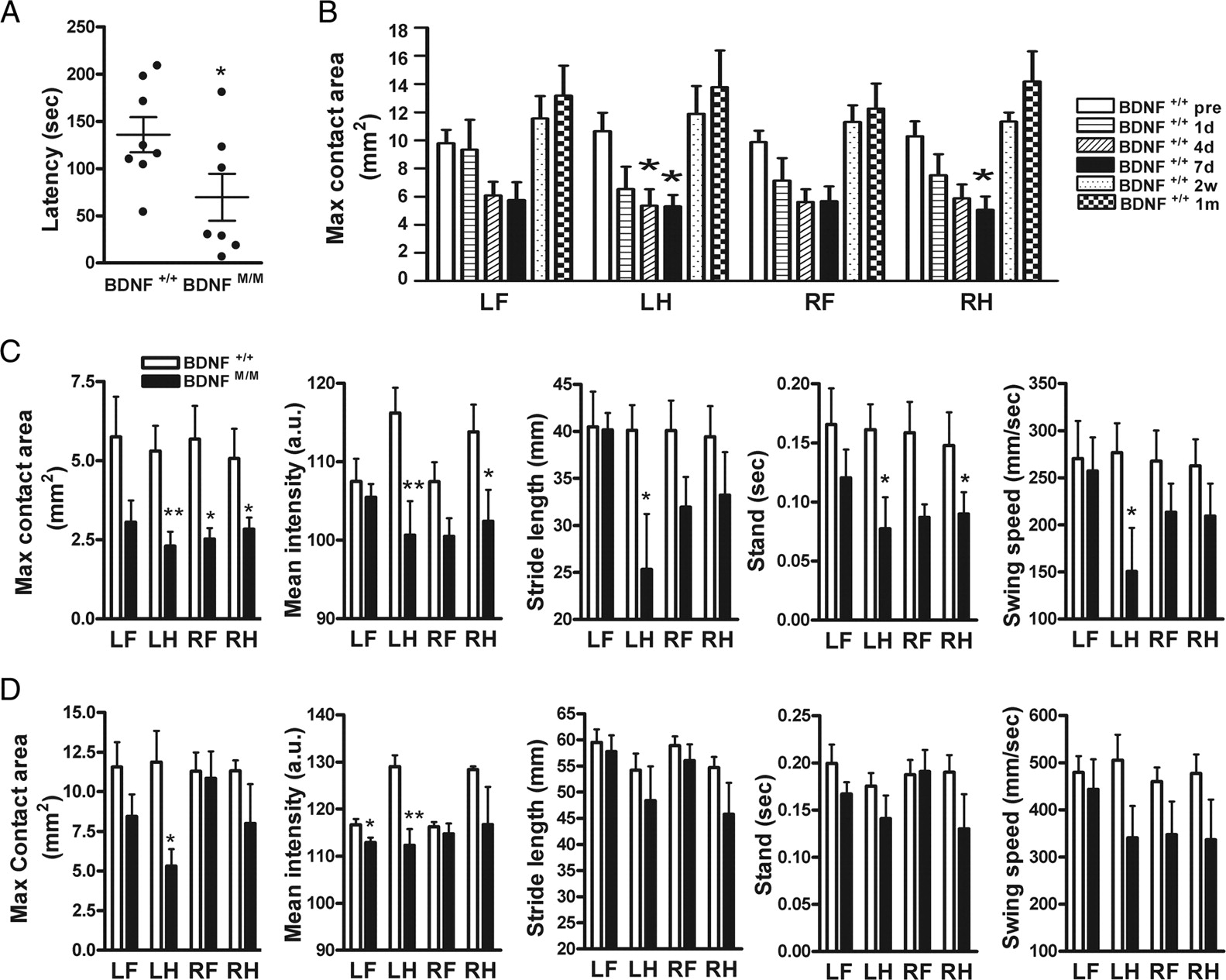
Post-ischemic motor function was assessed in BDNF+/+ and BDNFMet/Met mice. There was no difference between the genotypes in rotarod performance before ischemia. Compared with BDNF+/+ mice, BDNFMet/Met mice showed a significantly larger deficit 6 d after stroke (Fig. 2A). Temporal profiles of gait analysis in BDNF+/+ mice revealed that maximum contact area, a spatial parameter related to individual paws, showed a progressive decrease, with the greatest deficits at 7 d after ischemia in all four paws, and returned to baseline levels at 2 weeks and 1 month after ischemia (Fig. 2B). Other spatial and temporal parameters, including mean intensity, stride length, stand, swing speed, and regularity index, show a similar temporal pattern except BOS, which increases over 1 month after ischemia (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The gait function between the genotypes was compared at 7 d, 2 weeks, and 1 month after ischemia. Compared with BDNF+/+ mice, BDNFMet/Met mice showed greater deficits in maximum contact area in all paws at 7 d after ischemia. Reduced pressure exerted on the glass plate (mean intensity) in left and right hindpaws, shorter stride length in left hindpaws, shorter duration for the paw to contact the plate (stand) in left hindpaws and right forepaws, and the slower speed of the paw during swing (swing speed) in left hindpaws were found (Fig. 2C). Importantly, several BDNFMet/Met mice exhibited paw pressure below detection limit during the acute phase (<7 d), whereas all of BDNF+/+ mice were in detection range, indicating that BDNFMet/Met mice exhibited a greater deficit. Within BDNFMet/Met mice, we were unable to detect paw pressure in three of nine for forepaws and one of nine for hindpaws at the contralateral side at 7 d. This indicates greater deficits in the forepaw than hindpaw at this time. For data presentation, instead of arbitrarily assigning zero for these undetected animals, we performed statistical analysis only in animals that registered enough paw pressure. Thus, this quantitative comparison underestimates the deficit of forepaw. No difference was found in BOS (a parameter for the relative positions between paws) (BDNF+/+ vs BDNFMet/Met: forepaws, 5.8 ± 2.1 vs 7.2 ± 1.4 mm; hindpaws, 12.7 ± 4.2 vs 14.2 ± 2.7 mm; NS). Although BDNFMet/Met mice showed a reduced regularity index (a parameter for degree of interlimb coordination), it did not reach statistical significance (BDNF+/+ vs BDNFMet/Met: 67.2 ± 7.2 vs 40.6 ± 10.9%; p = 0.0515). All these deficits in the paws contralateral to the stroked side in BDNFMet/Met mice are indicative of unstable and impaired gait in the acute stage. Although BDNF+/+ mice returned to baseline at 2 weeks after ischemia, BDNFMet/Met mice showed deficits in maximum contact area in left hindpaw and mean intensity in left forepaw and left hindpaw (Fig. 2D). There were no behavior differences at 1 month after ischemia. These behavior results showed that BDNFMet/Met mice exhibited greater deficits in locomotor functions in an acute stage and delayed functional recovery compared with BDNF+/+ mice.

Effect of BDNF Val66Met polymorphism on motor behavior after stroke. Behavioral testing in BDNF+/+and BDNFMet/Met mice. All animals were trained for each testing before stroke. A, Mice were subjected to a rotarod, and latencies to fall were compared 6 d after ischemia. n = 7–8 per group. B, Gait changes were assessed by a computer-assisted Catwalk gait analysis system. Temporal changes of maximum contact area (B) in BDNF+/+ mice before ischemia (pre) and 1 d, 4 d, 7 d, 2 weeks, and 1 month after ischemia. C, Comparison between BDNF+/+and BDNFMet/Met mice in stroke-induced gait changes at 7 d after ischemia. Max contact area, The maximum area of a paw that comes into contact with the glass plate; Mean intensity, a measure of pressure (a.u., arbitrary unit) exerted on the glass plate; Stride length, the distance between consecutive steps with the same paw; Stand, a duration in seconds of contact of a paw with the glass plate; Swing speed, the speed (millimeters per seconds) of the paw during swing. D, Comparison between BDNF+/+and BDNFMet/Met mice in stroke-induced gait changes at 2 weeks after ischemia. n = 6–10 per group. *p < 0.05 versus BDNF+/+ mice before ischemia in B. *p < 0.05 and **p < 0.01 versus BDNF+/+ mice in C and D. LF, Left forepaw; LH, left hindpaw; RF, right forepaw; RH, right hindpaw.
Mice with BDNF SNP exhibit reduced proliferating endothelial cell and vessel density in the peri-infarct area
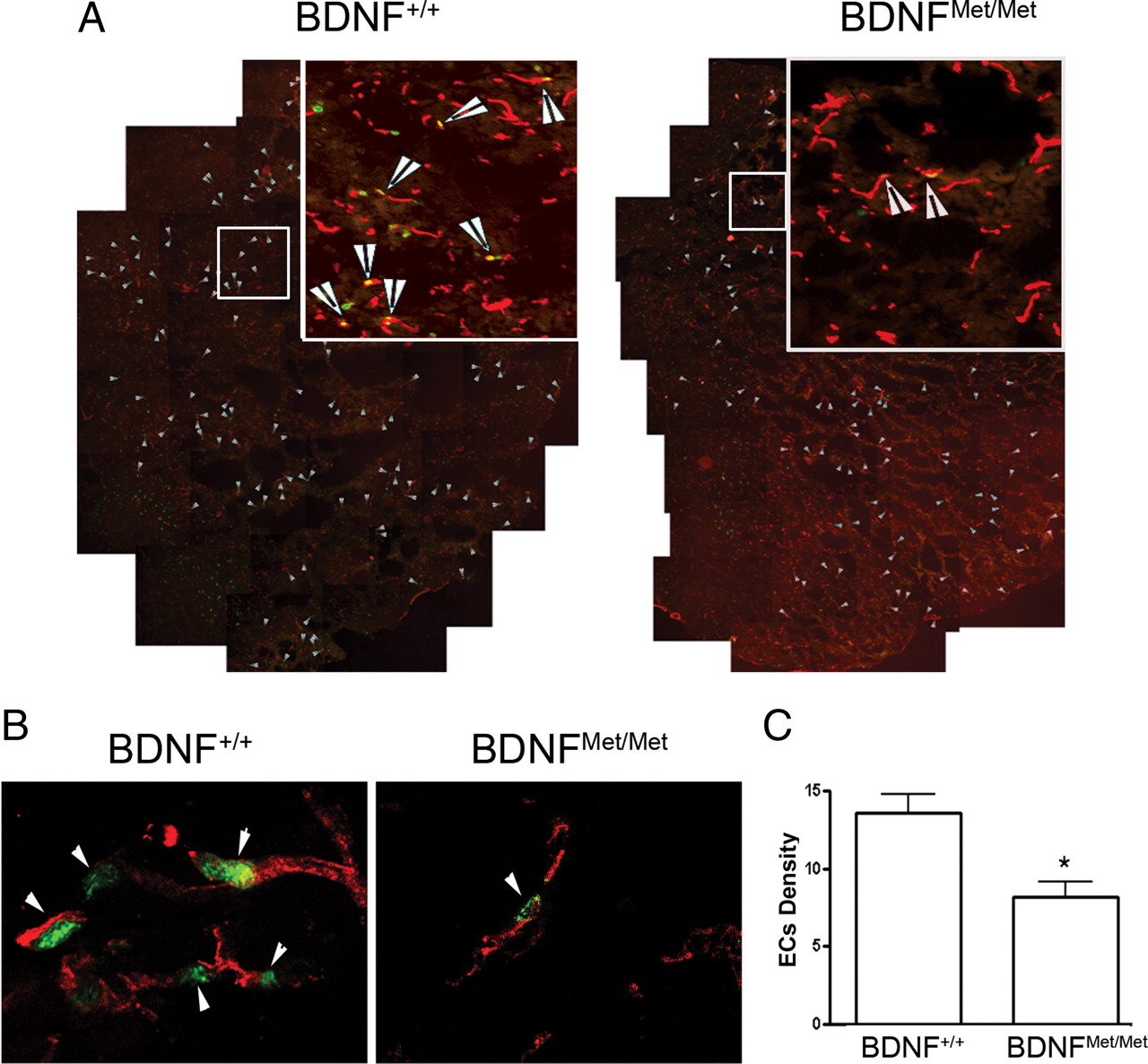
To assess the effect of BDNF SNP on stroke-induced angiogenesis, the density of proliferating endothelial cells was determined in the entire infarct area at 3 d after ischemia as an index of angiogenesis. Confocal images of double immunofluorescence showed that nuclear localization of Ki-67, a marker for proliferating cells, is juxtaposed to endothelial CD31+ cells (Fig. 3A,B, arrows). Quantitative analysis showed that the density of proliferating endothelial cells was significantly reduced in the infarct territory in BDNFMet/Met mice compared with BDNF+/+ mice (p < 0.05) (Fig. 3C).

Effect of BDNF Val66Met polymorphism on the proliferation of endothelial cells in the infarct territory. Proliferating endothelial cells were identified at 3 d after ischemia by double immunohistochemistry of Ki-67/CD31 in the infarct area. A, A composite of ipsilateral side of brain that shows Ki-67/CD31+ immunoreactive cells (indicated by arrows) in the infarct area. Images that show Ki-67/CD31+ immunoreactivity were taken to produce a composite in the infarct area. B, High magnification of confocal photomicrographs show the nuclear localization of Ki-67 (a proliferation marker, green) juxtaposed to endothelial CD31+ (red) cells. C, Quantification of proliferating endothelial cell density from BDNF+/+ and BDNFMet/Met mice. Note that there is a significant reduction in proliferating endothelial cells (EC) in BDNFMet/Met mice. Error bars indicate mean ± SEM; n = 3 per group. *p < 0.05 compared with BDNF+/+ mice.
To link the stroke-induced neoangiogenic response to vessel formation, vessel density was determined in the infarct core and inner and outer margin of the infarct border 7 d after ischemia (Fig. 4G). No difference in vessel density was found between genotypes in naive controls (data not shown) or the contralateral side 7 d after ischemia (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). There was a strong GFAP immunoreactivity in the outer margin of the infarct border representing a glial scar in the ischemic hemisphere (Fig. 4A,D,G). Vessel densities in the outer margin of the infarct were not different between genotypes (Fig. 4B,E,H). In contrast, the number of vessels in the inner margin of the infarct border were significantly lower in BDNFMet/Met mice compared with BDNF+/+ mice (Fig. 4B,E,H), whereas both genotypes showed similarly reduced vessel density in the core (Fig. 4C,F,H). The results show that ischemia induces angiogenesis primarily in the ischemic boundary zone, and BDNF Val66Met SNP influences ischemia-induced vascularization in this area.

Effect of BDNF Val66Met polymorphism on vessel density in the peri-infarct area. Vessel density was determined in the outer and inner margin of infarct border defined by glial scar 7 d after ischemia. A–G, Immunohistochemistry of GFAP (green) and/or CD31 (red) in BDNF+/+ (A–C) and BDNFMet/Met (D–F) mice. Note that there is strong GFAP immunoreactivity in the outer margin of infarct border indicated by dotted white lines (A, D). The inner margin of infarct border is indicated by dotted or solid white lines. G, Seven to 10 fields (shown as squares, area for each square is 0.1 mm2) from each section are chosen along the white infarct border. H, Quantification of vessel density in the core (C) and inner (I) and outer (O) margin of infarct border, and contralateral side (Contl) was compared between BDNF+/+ and BDNFMet/Met mice. n = 4–5 per group. *p < 0.05 versus BDNF+/+ mice inner margin. Scale bar, 300 μm.
Angiostatic factors CD36 and TSP-1 are upregulated in BDNFMet/Met mice in response to ischemia
New blood vessel growth is regulated by a well orchestrated balance between pro-angiogenic and anti-angiogenic factors. To assess the impact of BDNF Val66Met polymorphism on the expression of angiostatic factors, we determined the expression of CD47, CD36, and TSPs. Ischemia reduced CD47 gene expression at 3 and 7 d to a similar degree between genotypes (Fig. 5A,B). Unlike CD47, ischemia increased CD36 expression in both genotypes. Additionally, ischemia-induced CD36 mRNA levels in BDNFMet/Met mice were significantly elevated at 3 d compared with BDNF+/+ mice, and the increase was even more apparent at 7 d (Fig. 5C,D). The increased CD36 gene expression in the BDNFMet/Met mice was associated with the increased number of CD36-immunoreactive cells at 7 d after occlusion (Fig. 6A,B). Subsequent double immunofluorescence revealed that many of the CD36+ cells are endothelial cells in the post-ischemic brain (Fig. 6C–E).

Effect of BDNF Val66Met polymorphism on angiostatic receptor expression. The expression of CD47 (A, B) and CD36 (C, D) genes were determined in the BDNF+/+ and BDNFMet/Met brains at 3 d (A, C) and 7 d (B, D) after stroke. β-Actin was used for an internal control. n = 6–10 per group. *p < 0.05, **p < 0.01, and ***p < 0.001 versus contralateral; #p < 0.05 and ##p < 0.01 versus BDNF+/+ ipsilateral. Contl, Contralateral; Ipsl, ipsilateral.

Upregulation of CD36 protein in BDNFMet/Met endothelial cells. A, B, CD36 immunoreactivity is increased in BDNFMet/Met (B) mice compared with BDNF+/+ (A) mice at 7 d after ischemia. C–E, Confocal micrographs of double-fluorescence immunohistochemistry of CD36 and CD31. CD36+ cells (green) are colocalized with endothelial cell marker CD31 (red) in BDNFMet/Met brain (E, yellow). Arrows indicate cells in insets that are CD36+ endothelial cells in high magnification. Scale bar, 20 μm.
TSP-1 and TSP-2 show high affinity toward CD36, which mediates a signaling cascade that leads to endothelial cell apoptosis (Jiménez et al., 2000). Stroke induced TSP-1 mRNA levels were elevated in both genotypes at 3 d after ischemia (Fig. 7A). The increase of TSP-1 mRNA levels was sustained in BDNFMet/Met mice even at 7 d after ischemia, whereas the levels returned to baseline in BDNF+/+ mice (Fig. 7B). No significant differences were observed in TSP-2 mRNA levels between genotypes at these times (Fig. 7C,D). The covariance of reduced BDNF levels and increase of TSP-1/CD36 in BDNFMet/Met mice shows an unfavorable balance between pro-angiogenic and anti-angiogenic factors that may result in reduced angiogenesis in these mice.

Effect of BDNF Val66Met polymorphism on the expression of TSPs. TSP-1 (A, B) and TSP-2 (C, D) gene expression was determined in BDNF+/+ and BDNFMet/Met mice 3 d (A, C) and 7 d (B, D) after stroke. Note the elevated TSP-1 expression in BDNFMet/Met mice. β-Actin was used as an internal control. n = 6 per group. *p < 0.05 and ***p < 0.001 versus contralateral; #p < 0.05 versus BDNF+/+ ipsilateral. Contl, Contralateral; Ipsl, ipsilateral.
CD36 is critical to reduce stroke-induced angiogenesis in BDNFMet/Met mice
To determine whether upregulation of CD36 in BDNFMet/Met mice is critical to reduce stroke-induced angiogenesis, we generated 8× BDNFMet/Met/CD36 KO double mutant (Dmt) and the corresponding BDNFMet/Met /CD36 WT (Dwt) by crossing BDNFMet/Met (10× backcrossed with C57BL/6) with CD36 KO (7× backcrossed with C57BL/6). There was a small but significant increase (∼10%) in vessel density in the brain of Dwt mice relative to BDNFMet/Met mice. This may be attributable to a difference in their genetic background (8× vs 10× backcrossed to C57BL/6) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Stroke-induced BDNF levels were not different between Dmt and Dwt mice 7 d after stroke (Fig. 8D). Dwt mice showed a significant reduction in vessel density in the inner margin of the infarct border but not in the contralateral side or the outer margin of the infarct border (Fig. 8A,B). Notably, Dmt mice rescued the reduced vessel density observed in this region in the Dwt mice (Fig. 8A–C), suggesting that CD36 is sufficient to overcome the angiogenic deficit associated with BDNF SNP.

Effect of CD36 on post-ischemic vessel density in BDNFMet/Met mice. A, B, Photomicrographs showed immunohistochemistry of GFAP (green) and CD31 (red) in Dwt and Dmt mice. C, Quantification of vessel density. Vessel density was determined in the outer (O) and inner (I) margin of the infarct border defined by glial scar (dotted lines) and contralateral side (Contl). D, Total BDNF levels from brain lysates in Dwt and Dmt mice were measured by ELISA at 7 d after ischemia. n = 4–5 per group. ***p < 0.001 versus Dwt outer margin; ###p < 0.001 versus Dwt inner margin. Contl, Contralateral; Ipsl, ipsilateral side. Scale bar, 300 μm.
Discussion
The BDNF Val66Met polymorphism has been linked to human psychiatric diseases. The variant affects the anatomy of the hippocampus and prefrontal cortex, and the carriers of the polymorphism exhibit deficits in hippocampus-dependent memory tasks (Egan et al., 2003; Hariri et al., 2003; Pezawas et al., 2004). Because stroke is a leading cause of disability and >20% of Caucasians and up to 70% of the Asian population carry the BDNF SNP, understanding the impact of the genetic variant of BDNF on stroke pathology may offer significant insights into therapeutics to improve recovery for the patients that have this polymorphism.
A mouse line with the BDNF Val66Met allele knocked in was generated that reproduces the phenotypic traits of humans with the variant (Chen et al., 2006). By using a transient focal ischemia in these mice, the present study showed that BDNF levels were much lower in BDNFMet/Met relative to the BDNF+/+ brain (Fig. 1). Because the tissue used for BDNF protein measurement in the ipsilateral side of the brain included normal non-injured, peri-infarct, and infarct area, the differences in the ischemic infarct territory between the genotypes may be even larger. This finding is interesting in light of previous in vitro studies of the variant BDNF demonstrating defects in activity-dependent BDNF trafficking and secretion (Chen et al., 2004, 2005, 2006). Thus, reduced BDNF levels in the BDNFMet/Met brains after ischemia may reflect the defect in activity-dependent secretion and intracellular trafficking at the site of the stroke.
BDNF exists as pro and mature forms that use distinct receptors to mediate divergent neuronal actions. Mature BDNF exerts its biological functions via activation of TrkB, leading to endothelial cell survival/proliferation, whereas proBDNF binds to p75NTR and can initiate apoptosis through sortilin, a co-receptor of p75NTR (Nykjaer et al., 2004; Teng et al., 2005). Although our study does not distinguish pro from mature BDNF produced in post-ischemic brain, the positive link of total BDNF levels to angiogenesis between the genotypes suggests the presence of a sufficient amount of mature BDNF to elicit the biological function in the post-ischemic brains.
The role of BDNF in cerebral ischemia is controversial. Whereas decreased expression of BDNF in haploinsufficient BDNF+/− mice is associated with better motor performance and increased neuroblast number after experimental stroke (Nygren et al., 2006), enhanced BDNF availability and/or downstream signaling resulted in reduction or no effect in infarct size but promoted functional recovery (Endres et al., 2000; Kurozumi et al., 2004; Schäbitz et al., 2004, 2007). Our study also shows no discernable differences in infarct size and swelling between the two genotypes. However, the reduced BDNF levels in BDNFMet/Met mice were associated with greater motor deficits and gait impairment in acute ischemia. In BDNF+/+ mice, the several deficits exhibited during the acute period (<7 d) returned to baseline at 2 weeks. However, the deficits in their paw pressure were still present in BDNFMet/Met mice at that time, indicating delay in functional recovery (Fig. 2). This result is consistent with the report that the presence of BDNF SNP is associated with poor outcome among survivors of hemorrhagic stroke in human study (Siironen et al., 2007). Despite the dissociation of BDNF levels with infarct size, the positive correlation of BDNF levels with locomotor function indicates the presence of underlying BDNF-mediated events after stroke.
In response to ischemic insult and subsequent pathological events, regenerative and repair processes occur in affected tissues (Fan and Yang, 2007). Because enhanced angiogenesis in the ischemic penumbra is correlated with longer survival in stroke patients (Krupinski et al., 1994), the impact of BDNF Val66Met polymorphism on angiogenesis in this region was investigated. The findings that BDNFMet/Met mice have reduced BDNF levels, less proliferating endothelial cells, and reduced vessel density in the penumbra relative to BDNF+/+ mice suggests a causal link between BDNF levels and its functional role in stroke-induced angiogenesis.
The current study shows an involvement of CD36 in the underlying mechanism(s) by which angiogenic deficits in BDNFMet/Met occurs. CD36 has exhibited anti-angiogenic effects of TSP-1 in the corneal neovascularization assay, and blocking CD47 has been shown to increase tissue perfusion (Dawson et al., 1997; Isenberg et al., 2007). We found the suppression of the CD47 gene expression in the post-ischemic brain of BDNFMet/Met along with wild type, consistent with the report by Jin et al. (2009). The similar degree of reduction in CD47 mRNA levels between BDNF+/+ and BDNFMet/Met mice indicates a minimal involvement of CD47 in regulating BDNF-induced angiogenesis after stroke. In contrast, we showed that ischemia not only induced the expression of CD36 and TSP-1, as reported by others (Hayashi et al., 2003; Lin et al., 2003; Cho et al., 2005), but also that the increase was further potentiated in the BDNFMet/Met brain. The relevance of the upregulation of CD36 in stroke pathology is its anti-angiogenic nature, which offsets compensatory angiogenesis-promoting cascades in response to tissue ischemia. The reciprocal expression of angiogenic and angiostatic factors found in BDNF+/+ and BDNFMet/Met mice after ischemia thus suggests a possible BDNF-mediated inhibition of the CD36 pathway. The involvement of the CD36 pathway is also supported by the findings that there was a sustained elevation of TSP-1 gene expression in BDNFMet/Met mice up to 7 d after ischemia (Fig. 7B). Lack of increase in TSP-2 in these mice (Fig. 7C,D) may be attributable to its delayed expression (Lin et al., 2003). It has been shown that TSP-1 and TSP-2 are also involved in other long-term recovery functions such as synaptogenesis and axonal sprouting (Liauw et al., 2008). These studies showed a distinct temporal and spatial expression of early TSP-1 and delayed TSP-2 expression after stroke. Consistent with the literature, our study also shows a selective increase of TSP-1 and localization of CD36 in the endothelial cell in the BDNFMet/Met mice 3–7 d after ischemia. Therefore, it is conceivable that early TSP-1 expression may be primarily involved in inhibiting angiogenesis via CD36 during an acute post-stroke but TSP-2 in regulating synaptic plasticity in the long-term recovery through CD36 or other membrane receptors, including integrins, fibronectin, and gabapentin receptor α2δ-1 (Adams and Lawler, 2004; Bornstein, 2009; Eroglu et al., 2009).
The sufficiency to inhibit angiogenesis in BDNFMet/Met mice by elevated CD36 was further addressed. Dmt mice showed a complete rescue in vessel densities of Dwt mice without altering BDNF levels (Fig. 8). The finding that the absence of CD36 does not influence BDNF levels in the post-ischemic brain suggests that the inhibition of CD36 may act downstream of BDNF. Because of an intrinsic phenotype associated with CD36 deficiency, the behavior outcome in Dmt mice was not addressed. Unlike BDNF+/+ and BDNFMet/Met mice that show no discernable difference in infarct volume, CD36 KO mice exhibit a significantly smaller infarct size, and the neuroprotection resulted in better behavior outcome (Cho et al., 2005). In addition to its role in inhibiting angiogenesis, CD36 mediates free radical production, inflammation, and innate immunity that would influence the development of brain injury and behavior outcome. Thus, angiogenic rescue alone may not reflect behavior outcome in Dmt mice. Despite these caveats, the findings define a clear mechanistic insight by which elevated CD36 expression in BDNFMet/Met mice is critical to inhibit stroke-induced angiogenesis.
In summary, using knock-in mouse models of the human genetic variant of the BDNF polymorphism, the present study demonstrates that the BDNF Val66Met polymorphism negatively influences post-stroke motor function, reduces angiogenesis, and elevates angiostatic factors CD36 and TSP-1. Deletion of CD36 in BDNFMet/Met mice rescues the acute angiogenic deficits. This study links the Val66Met polymorphism to diminished stroke recovery and provides support for the notion that BDNF may influence recovery partly through affecting angiogenesis. It provides a potential stroke therapy that aims at promoting angiogenesis via targeting the CD36 pathway for BDNF Val66Met polymorphism carriers.
Footnotes
This work was supported by National Institutes of Health Grants HL82511 (S.C.), MH088814 (F.S.L., S.C.), and NS052819 (F.S.L.) and the Burke Foundation.
- Correspondence should be addressed to Dr. Sunghee Cho, Department of Neuroscience, Weill Cornell Medical College at Burke Medical Research Institute, 785 Mamaroneck Avenue, White Plains, NY 10605. suc2002{at}med.cornell.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}