

Abstract
Murine cortical cultures containing both neurons and glia (daysin vitro 13–15) were exposed to periods of oxygen–glucose deprivation (5–30 min) too brief to induce neuronal death. Cultures “preconditioned” by sublethal oxygen–glucose deprivation exhibited 30–50% less neuronal death than controls when exposed to a 45–55 min period of oxygen–glucose deprivation 24 hr later. This preconditioning-induced neuroprotection was specific in that neuronal death induced by exposure to excitotoxins or to staurosporine was not attenuated. Neuroprotection was lost if the time between the preconditioning and severe insult were decreased to 7 hr or increased to 72 hr and was blocked if the NMDA antagonist 100 μm3-((d)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid was applied during the preconditioning insult. This was true even if the duration of preconditioning was increased as far as possible (while still remaining sublethal). A similar preconditioning effect was also produced by sublethal exposure to high K+, glutamate, or NMDA but not to kainate ortrans-1-aminocyclopentane-1,3-dicarboxylic acid.
Kitagawa et al. (1990) found that gerbils subjected to sublethal transient global ischemia exhibited reduced hippocampal CA1 neuronal death after a more severe ischemic insult 24–48 hr later, a phenomenon they called “ischemic tolerance.” Similar findings were reported by others (Liu et al., 1992; Simon et al., 1993; Gidday et al., 1994; Glazier et al., 1994;Chen et al., 1996; Cai et al., 1997). A neuroprotective effect was also induced in vivo by application of KCl (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996; Taga et al., 1997) or systemic injection of the mitochondrial inhibitor 3-nitroproprionic acid (3-NPA) (Riepe et al., 1997).
The mechanistic basis of ischemic tolerance has not been fully delineated. A role for NMDA receptor activation was suggested by Kato et al. (1992), because MK801 blocked the development of ischemic tolerance in gerbils (although as those authors pointed out, this treatment likely attenuated the preconditioning insult itself). In addition, Kasischke et al. (1996) made the curious observation that 3-NPA-induced preconditioning in hippocampal slices could be abolished by exposing the slices to an NMDA antagonist before hypoxic test challenge. Recently, Heurteaux et al. (1995) suggested that the activation of adenosine A1 receptors or KATP channels may be involved.
Alterations in gene expression are likely also required for the development of ischemic tolerance. Expression of c-JUN increases in CA1 neurons after preconditioning ischemia (Sommer et al., 1995), possibly contributing to the induction of other survival proteins such as HSP70 (Kirino et al., 1991; Kitagawa et al., 1991; Liu et al., 1993; Glazier et al., 1994; Chen et al., 1996; but see Abe and Nowak, 1996), superoxide dismutase (Toyoda et al., 1997), and bcl-2 (Shimazaki et al., 1994). Furthermore, ischemic stress enhances expression of the glucose transporter GLUT1 in microvessels and astrocytes (McCall et al., 1996), heme-oxygenase-1 in neurons and glia (Paschen et al., 1994;Geddes et al., 1996; Takeda et al., 1996), and growth factors in neurons, astrocytes, and endothelial cells (Takeda et al., 1993;Krupinski et al., 1996; Lee et al., 1996; Lin et al., 1997; Cobbs et al., 1998).
To gain leverage on elucidating underlying mechanisms, several investigators have modeled ischemic tolerance in vitro. Murine cortical cell cultures exposed to sublethal oxygen–glucose deprivation exhibited reduced neuronal death after subsequent more severe test challenge the next day (Grabb and Choi, 1995). Sakaki et al. (1995) described a neuroprotective effect of sublethal hypoxia on hypoxic neuronal death in young [days in vitro (DIV) 1–3] cortical cultures and correlated this effect with increased basic fibroblast growth factor expression. Recently, Bruer et al. (1997) described a neuroprotective effect of sublethal oxygen–glucose deprivation or Na+/K+ ATPase inhibition in cortical cultures. Neuronal death in this latter study predominantly exhibited apoptosis, perhaps reflecting the relatively young cultures used (DIV 10–12), a feature likely to attenuate excitotoxicity (Choi et al., 1987; Frandsen and Schousboe, 1990) and potentiate apoptosis (McDonald et al., 1997).
We report here a pharmacological characterization of ischemic tolerance in murine cortical cell cultures, aimed at testing the hypothesis that NMDA receptor activation is specifically required for its induction.
MATERIALS AND METHODS
Cortical cell culture preparation. Brain cortices were dissected from fetal Swiss Webster mice (14–16 d gestation) and plated at a density of 3.75 hemispheres per 24 well culture, on established glial monolayers (see below), as previously described (Rose et al., 1993). The plating medium consisted of Eagle’s Minimum Essential Medium (MEM; Life Technologies, Gaithersburg, MD) lacking bicarbonate and glutamine, supplemented with 5% fetal bovine serum, 5% horse serum (serum was obtained from HyClone, Logan, UT), 2 mm glutamine, 26.2 mm bicarbonate, and glucose added to bring the solution to a final glucose concentration of 20 mm. Cells were fed twice a week with MEM containing 5% horse serum, and at DIV 5–7 they were treated with cytosine arabinoside (10 μm) to eliminate the dividing microglia from the cultures. The cultures were used for experiments at 13–15 d in culture.
Glial cultures were prepared using the same procedure, except the brain cortices were dissected from mice, postnatal 1–3 d, in plating medium similar to above (with 10% horse serum, 10% fetal bovine serum, and 10 nm epidermal growth factor). Neurons were plated on glial cultures after the glia had become confluent, ∼2–4 weeks in culture.
All chemicals used were obtained from Sigma (St. Louis, MO) except where noted.
Oxygen–glucose deprivation. Mixed cortical cultures were subjected to oxygen–glucose deprivation injury using protocols previously described (Goldberg and Choi, 1993). Cultures were placed in an anaerobic chamber (Forma Scientific) and washed three times with balanced salt solution (BSS: 116 mm NaCl, 5.4 mm KCl, 0.8 mm MgSO4, 1.0 mm NaH2PO4, 26.2 mm NaHCO3, 1.8 mmCaCl2, 0.01 mm glycine, and 10 mg/l phenol red) lacking glucose and aerated with an anaerobic gas mix (85% N2/5% CO2/10% H2) to remove residual oxygen. Cell cultures were incubated in the solution at 37°C for a designated period to produce either mild or lethal oxygen–glucose deprivation. Control cell cultures not deprived of oxygen and glucose were placed in BSS containing 20 mm glucose, and not bubbled with anaerobic gas, during the experiment. To terminate the oxygen–glucose deprivation, cells were carefully washed with MEM (containing 2 mm glutamine, 20 mm glucose, and 10 μm glycine) and then removed from the anaerobic chamber.
For preconditioning, cortical cultures were deprived of oxygen and glucose for 5–30 min, an insult that did not induce neuronal death, as measured by lactate dehydrogenase (LDH) release or trypan blue staining. In some cultures, the NMDA receptor antagonist 3-((d)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (d-CPP, 100–300 μm; Research Biochemicals, Natick, MA) was used to block NMDA receptor activation during the preconditioning episode, and in a subset of these cultures, mild ischemia was extended to 40–50 min. At a designated period after the preconditioning stimulus (7, 24, 48, or 72 hr), the cultures were again deprived of oxygen and glucose, this time for a period long enough to cause ∼50% neuronal death in control cell cultures (45–55 min), where the longer deprivation periods were used with less dense cultures. We have previously noted that the neuronal death that occurs after oxygen–glucose deprivation varies somewhat as a function of neuronal density, with high-density cultures exhibiting greater vulnerability than low-density cultures (Goldberg and Choi, 1993; and unpublished observations). The cultures used within any given experiment (e.g., preconditioned vs control) were always sister cultures derived from a single plating and hence were exposed to a single insult duration.
Excitatory amino acid exposure. Mixed cortical cultures were subjected to excitotoxic injury by treatment with glutamate or the glutamate receptor agonists kainate and NMDA. To induce ∼50% neuronal death, brief exposure to glutamate (200 μm) or NMDA (300 μm) was performed at room temperature by rapidly exchanging the growth media with a HEPES-buffered balanced salt solution (HBBSS) containing 5.5 mm glucose (we lowered the glucose for 5–15 min exposures) and 10 μm glycine for 5 min. To terminate the exposure, cultures were washed by triple exchange of the media with MEM (containing glutamine and glycine). For lethal kainate exposures, 50 μm kainate and 10 μmMK801 (Research Biochemicals; used to block secondary NMDA receptor-induced neuronal death) were applied to the cells for the full 24 hr in an MEM-based solution.
Sublethal exposures of glutamate (10, 20, and 30 μm), NMDA (5, 7.5, and 10 μm), or kainate (10, 30, and 45 μm) were applied in a BSS solution containing 20 mm glucose for 30 min at 37°C, 24 hr before oxygen glucose deprivation. The highest concentrations of each agonist tested represented maximal sublethal exposures, i.e., the most intense exposures that did not alone induce neuronal cell death. In addition, the broad spectrum mGluR agonisttrans-1-aminocyclopentane-1,3-dicarboxylic acid (trans-ACPD, 100–200 μm; Research Biochemicals) was added to the cultures using the same paradigm.
Potassium-induced depolarization. HBBSS, containing 45 mm KCl and 76.4 mm NaCl, was added to cortical cultures at room temperature for 15 min. Cultures were then washed back into the MEM-based solution. The NMDA receptor antagonistd-CPP (100 μm) was used to block NMDA receptor activation during the preconditioning episode in a subset of cultures.
Staurosporine-induced apoptosis. Cell cultures were exposed to staurosporine (150 nm) in an MEM-based solution to induce apoptotic cell death in neurons (Koh et al., 1995). To confirm the neuronal death as apoptotic, the protein synthesis inhibitor cycloheximide (0.5 μg/ml) was administered with staurosporine in an additional set of cultured cells.
Quantitation of neuronal injury. Neuronal injury was assessed by examination of the neurons with phase-contrast microscopy and by measurement of LDH efflux into the bathing medium 24 hr after every sublethal and lethal insult (excluding neuronal apoptosis, which was assessed after 48 hr). It has previously been established that LDH release correlates linearly with the number of damaged or dying neurons after both excitotoxic (Koh and Choi, 1987) and apoptotic injuries (Koh et al., 1995). Sister cultures were treated with 300 μmNMDA for 24 hr to induce near complete neuronal death without glial death.
LDH values of maximally injured, preconditioned, and control cultures were compared in all experiments. In addition, cell death was confirmed in some experiments by the trypan blue exclusion assay.
RESULTS
As previously reported, cultures exposed to 45–55 min of combined oxygen–glucose deprivation developed widespread neuronal cell body swelling by the end of the exposure period and widespread neuronal degeneration without glial degeneration by 24 hr later (Goldberg and Choi, 1993). In contrast, cultures “preconditioned” by previous nonlethal exposure to 20–30 min oxygen–glucose deprivation on the previous day exhibited only approximately half as much neuronal death 24 hr after the same 45–55 min test challenge (Fig.1). This protective effect of preconditioning persisted when cultures were examined 48 hr after test challenge (data not shown). The protective efficacy of preconditioning exposure depended on both the duration of preconditioning exposure (Fig. 2; maximal protective effect was achieved with 30 min of preconditioning, because 40–50 min exposures became intrinsically damaging) and the interval between preconditioning exposure and test challenge. Specifically, no protective effect was seen with an interval between preconditioning and test challenge of 7 or 72 hr (Fig. 3).
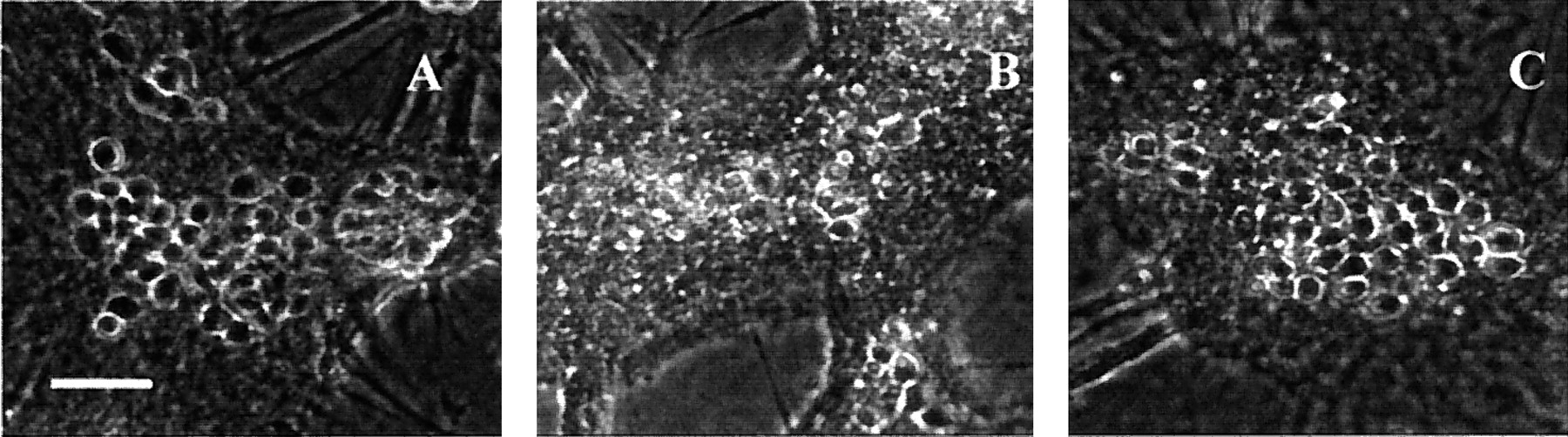
Morphological evidence for preconditioningin vitro. A, Phase-contrast micrograph of control cortical cultures 24 hr after sham wash. Scale bar, 50 μm.B, Same but 24 hr after 50 min oxygen–glucose deprivation. C, Sister cultures exposed to the same insult as in B, but preconditioned with a previous 30 min exposure to oxygen–glucose deprivation 24 hr before the test insult.

Neuroprotection is dependent on the duration of preconditioning exposure. Cortical cultures were preconditioned with 5–30 min of oxygen–glucose deprivation as indicated and then exposed 24 hr later to oxygen glucose deprivation for 45–55 min. Twenty-four hours later, resultant neuronal death was assessed by LDH release to the bathing medium, scaled to the release associated with near complete neuronal death without glial death (=100, induced by exposure to 300 μm NMDA for 24 hr). Mean LDH + SE is displayed;n = 8–20 cultures per condition pooled from five experiments. *p < 0.05 versus control (no preconditioning) by one-way ANOVA followed by Student–Newman–Keuls multiple comparisons test.

Neuronal protection is dependent on the time between the two episodes of oxygen–glucose deprivation (OGD). The timing between the preconditioning oxygen–glucose deprivation (10–30 min) and severe oxygen–glucose deprivation was varied from 7 to 72 hr. Mean + SE is displayed;n = 12–20 cultures per condition pooled from three to five experiments. *p < 0.05 versus control (no preconditioning) by one-way ANOVA followed by Student–Newman–Keuls multiple comparisons test.
To determine whether the same preconditioning stimulus could protect neurons from other forms of excitotoxic injury, cultures were exposed to toxic levels of NMDA (300 μm for 5 min), kainate (50 μm with 10 μm MK801 for 24 hr) or glutamate (200 μm for 5 min) after a 10–30 min preconditioning exposure 24 hr earlier. In each case, the excitotoxin exposure was chosen to produce ∼50% neuronal death in control cultures. This preconditioning exposure produced no significant changes in either rapidly triggered NMDA- or glutamate-induced excitotoxic neuronal death or slowly triggered kainate-induced excitotoxic neuronal death (Table1).
Ischemic preconditioning did not reduce subsequent excitotoxic death
We next examined whether preconditioning exposure to oxygen–glucose deprivation could attenuate neuronal vulnerability to apoptotic cell death. Control cultures exposed for 24 hr to the nonspecific kinase inhibitor staurosporine (150 nm) developed intermediate levels of neuronal apoptosis by the end of this exposure, characterized by cell body shrinkage, internucleosomal DNA fragmentation, and sensitivity to protein synthesis inhibitors or caspase inhibitors (Falcieri et al., 1993; Jacobson and Raff, 1995; Koh et al., 1995). Optimal preconditioning exposure (20–30 min 24 hr earlier) did not alter this staurosporine-induced apoptosis (Fig.4).
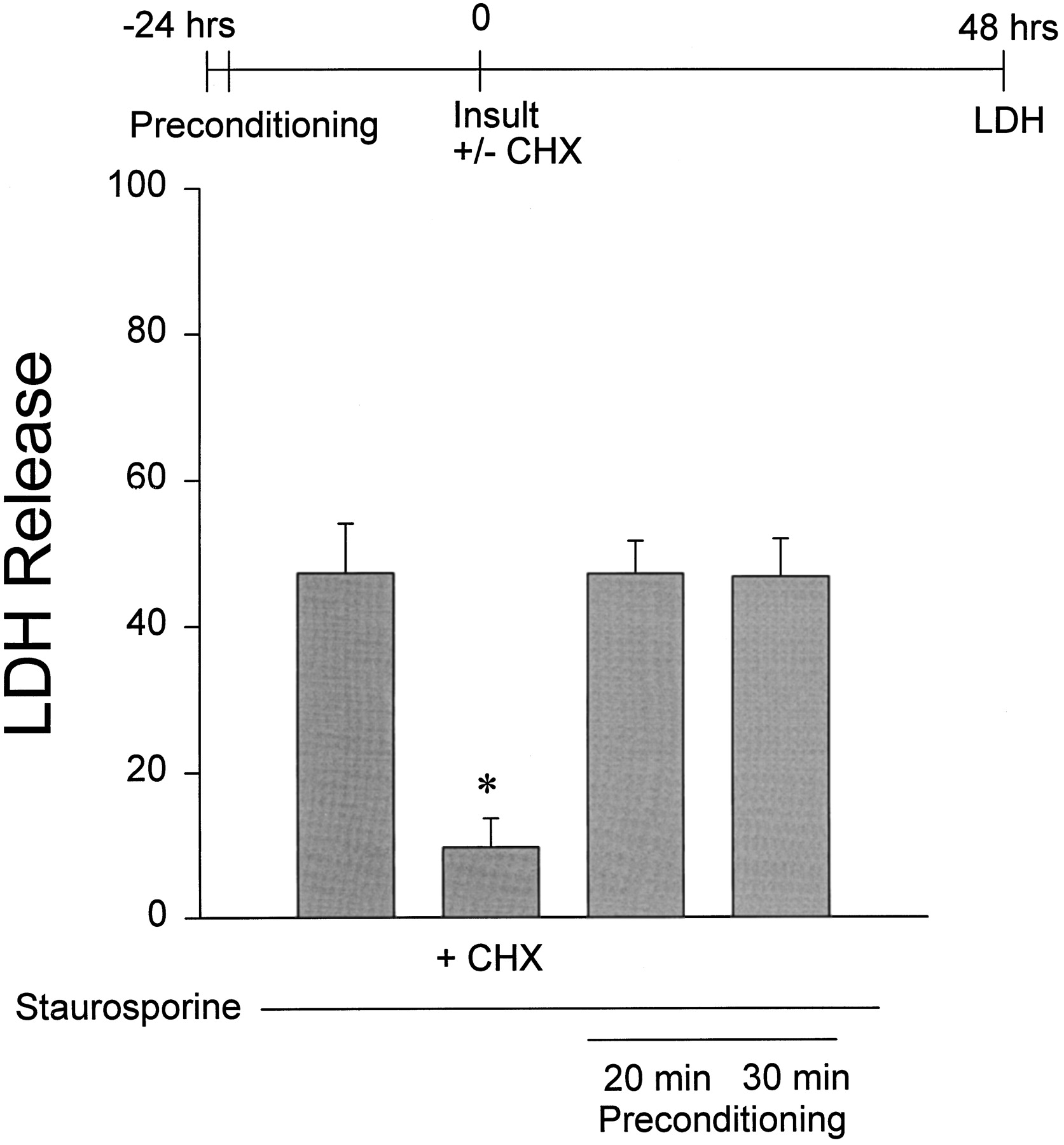
Preconditioning does not protect neurons from staurosporine-induced apoptosis. Cultures were preconditioned with 20 or 30 min oxygen–glucose deprivation and then 24 hr later were exposed to 150 nm staurosporine for 48 hr. Neuronal death was assessed at the end of the staurosporine exposure by LDH release to the bathing medium (mean + SE; n = 8 cultures per condition pooled from 2 experiments). *p < 0.05 versus control (no preconditioning) by one-way ANOVA followed by Student–Newman–Keuls multiple comparisons test. Concurrent addition of 0.5 μg/ml cycloheximide (CHX), but not preconditioning, attenuated staurosporine-induced apoptosis.
Although combined oxygen–glucose deprivation produces multiple perturbations that could account for the observed preconditioning effect, we tested the hypothesis that the associated excitotoxic stress bore primary responsibility. Therefore, we tried to induce tolerance by exposing cultures for 30 min to maximal sublethal concentrations of NMDA (5–10 μm) or kainate (10–45 μm), as well as 100–200 μmtrans-ACPD (a nontoxic agonist), 24 hr before oxygen–glucose deprivation (Table2). Application of NMDA did induce a mild protective effect in the cortical cultures; however,trans-ACPD or kainate did not. In fact, kainate pretreatment actually enhanced the death induced by subsequent oxygen–glucose deprivation. Glutamate application for 30 min (10–20 μm) required a change in the standard preconditioning protocol, because glutamate levels dropped to 50% of the original levels in the culture media after 10 min of 30 μm glutamate exposure as assessed by HPLC analysis (M. C. Grabb, D. Lobner, and D. W. Choi, unpublished observations), presumably reflecting cellular uptake. Therefore, glutamate was added again at 10 and 20 min after the initial exposure onset, to a final media concentration of 10 μm. Both an initial concentration of 10 μm and an initial concentration of 20 μm glutamate produced a mild neuroprotective effect against subsequent oxygen–glucose deprivation challenge, although the latter higher concentration of glutamate did itself produce some neuronal cell death (Table 2).
Glutamate receptor agonists induce preconditioning
Preconditioning-induced neuroprotection could also be induced by the application of 45 mm K+ for 15 min, an exposure that was just below a toxic level in most cultures (Fig.5). This concentration of KCl was selected on the basis of dose-finding experiments. Application of 60 mm KCl itself produced >10% neuronal death; 30 mm KCl did not induce tolerance (data not shown).

An NMDA antagonist blocks preconditioningin vitro. Cells were preconditioned with 30 min of oxygen–glucose deprivation (OGD) or 15 min of 45 mm KCl, either alone or with 100 μmd-CPP in the bathing media; any added drug was washed out at the end of the preconditioning period. Twenty-four hours later, cells were exposed to test challenge (oxygen–glucose deprivation for 45–55 min). d-CPP alone (added for 30 min and then washed out 24 hr before test challenge) did not reduce neuronal death. Neuronal death was assessed 24 hr later by LDH release to the bathing medium (mean + SE; n = 16 cultures per condition pooled from 4 experiments). *p < 0.05 versus control (no preconditioning) by one-way ANOVA followed by Student–Newman–Keuls multiple comparisons test.
Application of the NMDA receptor antagonist d-CPP (100 μm) during the preconditioning exposure to high K+ or sublethal oxygen–glucose deprivation completely blocked the subsequent development of resistance to oxygen–glucose deprivation-induced death (Fig. 5). Application ofd-CPP alone without high K+ or sublethal oxygen–glucose deprivation did not itself induce a preconditioning effect (Fig. 5). In addition, the competitive NMDA receptor antagonist d-(−)-2-amino-5-phosphonopentanoic acid also blocked the preconditioning effect at a concentration of 100 μm (data not shown).
To test the possibility that this NMDA antagonist effect was explained by a nonspecific reduction in preconditioning injury, we examined whether increasing the duration of preconditioning exposure could overcome the ability of d-CPP to block preconditioning-induced neuroprotection. We found that a preconditioning exposure duration of 50 min was the longest that could be tolerated without itself producing lethal injury, even if the concentration of d-CPP were increased to 300 μm to ensure that NMDA receptors were fully blocked (thed-CPP concentration–response relationship for neuroprotection against oxygen–glucose deprivation in our system reaches maximum at 100 μm drug; data not shown). However, even this just sublethal preconditioning stress, minus NMDA receptor activation, failed to produce any protective effect against a subsequent lethal oxygen–glucose deprivation insult (data not shown).
DISCUSSION
We describe here a cell culture model of ischemic tolerance, in which mouse cortical neurons preconditioned by a sublethal exposure to oxygen–glucose deprivation were rendered resistant to injury induced by a subsequent longer exposure to oxygen–glucose deprivation. Demonstration of such preconditioning-induced tolerance in vitro supports the view that ischemic tolerance in vivomay be predominantly explained by alterations in brain parenchymal rather than by alterations in blood flow or systemic response to ischemia and fits with observations that preconditioning does not alter regional cerebral blood flow associated with a subsequent lethal ischemic insult (Matsushima and Hakim, 1995; Chen et al., 1996).
Using the experimental leverage gained in such a reductionist model system, we demonstrated here a critical role for NMDA receptor activation in mediating preconditioning-induced tolerance. Besides oxygen–glucose deprivation, several other insults capable of increasing NMDA receptor activation—increasing extracellular K+ or adding exogenous glutamate or NMDA itself—induced tolerance, whereas activating AMPA/kainate or metabotropic glutamate receptors failed to induce tolerance. Furthermore, we found that the ability of an NMDA antagonist to block oxygen–glucose deprivation-induced preconditioning could not be overcome by increasing insult duration, even to a maximal sublethal level, arguing that the NMDA antagonist effect was not mediated by a nonspecific reduction in cellular stress and injury as Kato et al. (1992) had postulated (see above). Finally, we showed evidence that oxygen–glucose deprivation-induced tolerance was specific for a subsequent lethal oxygen–glucose deprivation insult. Tolerance did not extend to staurosporine-induced apoptosis and, unexpectedly, also did not extend to glutamate, NMDA, or kainate neurotoxicity. The present data are consistent with previous reports of preconditioning-induced tolerance in vivo or in vitro but make several novel points: (1) the specificity of NMDA receptor activation versus AMPA/kainate or metabotropic glutamate receptor activation in inducing tolerance; (2) the inability of overall stress and injury reduction to explain NMDA antagonist reduction of tolerance; and (3) the specificity of tolerance with regard to subsequent oxygen–glucose deprivation-induced insults versus other apoptosis or excitotoxicity paradigms.
Besides the demonstrations by Kato et al. (1992) that MK801 blocked ischemic tolerance in gerbils, the finding that tolerance could be induced by the application of high K+ concentrations (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996; Taga et al., 1997) fits with a postulated key role for NMDA receptor activation. Application of K+ would be expected to depolarize neurons and increase neurotransmitter glutamate release, leading to enhanced NMDA receptor activation. In these studies of high K+-induced ischemic tolerance, the application of K+-triggered spreading depression is also dependent on NMDA receptor activation (McLachlan, 1992; Nellgard and Wieloch, 1992). In fact, Taga et al. (1997) used an experiment comparable with that of Kato et al. (1992) when they blocked both the spreading depression and subsequent tolerance to ischemia using the NMDA antagonist ketamine.
As in the initial gerbil studies by Kitagawa et al. (1990) and Kato et al. (1991), we observed that tolerance developed slowly over many hours after the preconditioning episode, suggesting that changes in gene expression may be involved. We would have liked to have determined whether inhibitors of protein synthesis would block the development of tolerance but did not do this experiment, because the inhibitors themselves attenuate neuronal death after oxygen–glucose deprivation (Lobner and Choi, 1996).
The observation that oxygen–glucose deprivation-induced tolerance was not associated with a reduction in neuronal vulnerability to NMDA, kainate, or glutamate toxicity argues against the possibility that this tolerance was explained by postsynaptic changes in glutamate receptor structure. Previous studies have observed alterations in hippocampal neuronal glutamate receptor expression consequent to hypoxic and ischemic insults, including selective downregulation of NMDA receptor subunits NR2A and 2B (Zhang et al., 1997), selective downregulation of the Ca2+ gatekeeper AMPA receptor subunit GluR2/GluRB (Pellegrini-Giampietro et al., 1992; Gorter et al., 1997), and selective upregulation of AMPA receptor subunit GluR4/GluRD (Ying et al., 1997). The current failure to observe altered vulnerability to excitotoxicity after conditioning oxygen–glucose deprivation may reflect differences in the regulation of glutamate receptor expression in cortical versus hippocampal neurons.
At face value, the present observation that preconditioning oxygen–glucose deprivation did not protect cortical neurons against subsequent staurosporine-induced apoptosis is at odds with the prominence of DNA fragmentation associated with the hypoxic neuronal death in vitro studied by Bruer et al. (1997), an injury paradigm that was successfully attenuated by hypoxic preconditioning. However, those investigators used younger cortical neuronal cultures (rat DIV 8–10) than used here (mouse DIV 13–15), a feature that likely enhances the propensity to undergo programmed cell death (see above). It is therefore possible that their experiments, and our experiments, induced similar shifts in the mechanisms responsible for hypoxic neuronal death but that the ultimate phenotype of this death, necrosis versus apoptosis, was determined by age. Although further experiments will be needed to delineate the basis of oxygen–glucose deprivation-induced tolerance in the present model system, the observed absence of associated changes in neuronal vulnerability to excitotoxicity or staurosporine-induced apoptosis favors the possibility of changes in glutamate release or uptake.
Footnotes
This work was supported by National Institutes of Health Grant NS 30337 (D.W.C.) and National Institutes of Health Fellowship NS 10291 (M.C.G.). We thank Min Tian for technical assistance and Doug Lobner and Debra Babcock for helpful discussions.
Correspondence should be addressed to Dennis W. Choi, Washington University School of Medicine, Department of Neurology, Box 8111, 660 South Euclid, St. Louis, MO 63110.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}