

Abstract
The cacophony (cac) locus in Drosophila encodes a Ca2+ channel α subunit, but little is known about properties of cac-mediated currents and functional consequences of cac mutations in central neurons. We found that, in Drosophila cultured neurons, Ca2+ currents were mediated predominantly by the cac channels. The cac channels contribute to low- and high-threshold, fast- and slow-inactivating types of Ca2+ currents, take part in membrane depolarization, and strongly activate Ca2+-activated K+ current [I K(Ca)]. In cac neurons, unexpectedly, voltage-activated transient K+ current I A is upregulated to a level that matches I K(Ca) reduction, implicating a homeostatic regulation that was mimicked by chronic pharmacological blockade of Ca2+ currents in wild-type neurons. Among K+ channel transcripts, Shaker mRNA levels were preferentially increased in cac flies. However, Ca2+ current expression levels remained unaltered in several K+ channel mutants, illustrating a key role of cac in developmental regulation of Drosophila neuronal excitability.
Introduction
Ca2+ influx through voltage-gated Ca2+ channels regulates neuronal membrane excitability, synaptic transmission, plasticity, and growth. A large family of these channels has been identified molecularly and classified according to their gating kinetics, single-channel conductances, and pharmacological profiles (Catterall, 2000; Ertel et al., 2000; Hille, 2001). Five high-threshold, slow-inactivating subtypes (L, N, P, Q, and R), together with the low-threshold, fast-inactivating T-type, form the major Ca2+ entry pathway in mammalian excitable cells. Clustered Ca2+ channel activities lead to intracellular high-Ca2+ microdomains (Llinas et al., 1981), contribute to membrane depolarization, and activate local Ca2+-activated K+ current [I K(Ca)] (Gola and Crest, 1993; Robitaille et al., 1993; Marrion and Tavalin, 1998; Vergara et al., 1998). Voltage-dependent K+ currents, including inactivating I A and non-inactivating I K (for review, see Coetzee et al., 1999), can also be modulated during increases in internal Ca2+. For example, modifications of I A or I K channels, induced by behavior conditioning or excitability change, are triggered by cyclic nucleotide- and/or protein kinase-related mechanisms (Alkon et al., 1982; Poulain et al., 1994; Enyeart et al., 1996; Yao and Wu, 2001).
Homeostasis highlights the ability of biological systems to adjust the internal physiological milieu in response to external perturbations (Cannon, 1932). It has been documented in a number of preparations that, after experimental manipulations of membrane excitability or synaptic activity, some neurons are capable of maintaining appropriate levels of excitation over time by various compensatory mechanisms, including Ca2+-dependent regulations of ion channel function and synaptic efficacy (Spitzer, 1999; Turrigiano, 1999; Davis and Bezprozvanny, 2001; Marder and Prinz, 2002). The complex long-term homeostatic effects caused by altering Ca2+ influx, as well as the genetic control of the diversity of Ca2+ channels, can be further investigated in established genetic systems.
In Drosophila, neuronal Ca2+ currents are also composed of low-threshold, fast-inactivating and high-threshold, slow-inactivating components (Byerly and Leung, 1988; Saito and Wu, 1991; Schmidt et al., 2000). Two genes, Dmca1D and cacophony (cac, or Dmca1A), are known to encode Ca2+ channel α subunits in Drosophila (Zheng et al., 1995; Smith et al., 1996). They play major but nonredundant roles because null alleles of each cause embryonic lethality (Smith et al., 1996; Eberl et al., 1998). Dmca1D encodes vertebrate L-type-like channels, which are enriched in muscles for mediating dihydropyridine (DHP)-sensitive Ca2+ current (Gielow et al., 1995; Zheng et al., 1995; Ren et al., 1998). The cac locus, identified in a mutant screen for altered courtship song (von Schilcher, 1976), encodes Ca2+ channels homologous to vertebrate N-, P-, and Q-type channels (Smith et al., 1996; Rieckhof et al., 2003). It is thought that cac is expressed in the nervous system, as indicated by the reduced synaptic efficacy and altered motor terminal growth at neuromuscular junctions in several viable cac alleles (Kawasaki et al., 2000, 2002, 2004; Rieckhof et al., 2003; Kuromi et al., 2004). However, how cac channels mediate the diverse neuronal Ca2+ currents and how cac mutations affect other ion currents have not been determined under voltage- and current-clamp conditions.
In this study, we isolated inward Ca2+ and outward K+ currents by performing whole-cell clamp recordings on cultured “giant” neurons differentiated from cytokinesis-arrested embryonic neuroblasts of Drosophila (Wu et al., 1990; Zhao and Wu, 1997; Yao and Wu, 1999). Our results demonstrate that cac mutations greatly suppressed neuronal Ca2+ currents of different biophysical properties. Striking alterations were also observed in different types of K+ currents: a great decrease in I K(Ca) coupled with an increase in I A but with no changes in I K. Additional pharmacological and molecular approaches were adopted to investigate a potential homeostatic mechanism of I A upregulation in cac neurons.
Materials and Methods
Drosophila stocks.
The wild-type (WT) strain was Canton S. A homozygous viable allele cac s (Smith et al., 1996) (from Dr. J. Hell, Brandies University, Waltham, MA) and a cac deficiency line l(1)L13 HC129/In(1) FM7i, P{w +wC = Act GFP} (Kawasaki et al., 2002) (from Dr. R. Ordway, Pennsylvania State University, University Park, PA) were examined. The experiments also included Sh M [Shaker M, a null allele (Zhao et al., 1995)], Shab 3 [Shaker cognate b 3, a null allele (Singh and Singh, 1999)], and slo 1 [slowpoke 1, a hypomorph (Elkins et al., 1986)]. The slo 1 chromosome also carried a visible marker scarlet.
Single-embryo giant neuron culture.
The giant neuron culture system has been described previously (Saito and Wu, 1991; Zhao and Wu, 1997; Yao and Wu, 1999, 2001; Berke and Wu, 2002). Briefly, the interior content of stage 7–8 embryos was sucked out with a glass micropipette and then dispersed in culture medium on an uncoated coverslip. The culture medium contains 80% Drosophila Schneider medium and 20% fetal bovine serum (both from Invitrogen, Carlsbad, CA), with the addition of 200 ng/ml insulin, 50 μg/ml streptomycin, and 50 U/ml penicillin (all from Sigma, St. Louis, MO). To generate giant neurons from neuroblasts, cytochalasin B (CCB) (2 μg/ml; Sigma) was added on the first day to arrest cytokinesis (Wu et al., 1990). Within 1 d after CCB washout, phalloidin staining shows restoration of actin filament structures coupled with profuse growth of filopodia and expanded lamellipodia (Berke et al., 2006). For chronic or acute pharmacological effects, cultures were raised in or treated with media containing the drugs at the final concentrations specified. These drugs included amiloride, NiCl, apamine (all from Sigma), and charybdotoxin (Alomone Labs, Jerusalem, Israel). Before electrophysiological recording, drugs were washed out by standard bath solution (see below).
Electrophysiology.
Whole-cell patch-clamp recording from cultured giant neurons has been described previously (Saito and Wu, 1991; Zhao and Wu, 1997; Yao and Wu, 1999, 2001). Recording electrodes prepared from 75 μl glass micropipettes (VWR Scientific, Chicago, IL) had a tip opening of ∼1 μm and an input resistance of 3–5 MΩ in bath solution. The normal pipette solution contained the following (in mm): 144 KCl, 1 MgCl2, 0.5 CaCl2, and 5 EGTA, buffered at pH 7.1 with 10 HEPES. For inward current measurement, K+-free (replaced by Cs+) pipette solution was used to reduce outward K+ current contamination. The standard bath solution contained the following (in mm): 128 NaCl, 2 KCl, 4 MgCl2, 1.8 CaCl2, and 35.5 sucrose, buffered at pH 7.1 with 5 HEPES. To facilitate measurements of currents through Ca2+ channels, Ba2+ (from 5 to 20 mm, as indicated) was added to serve as a charge carrier, with additional blockers for Na+ channels (10 nm TTX; Sankyo, Tokyo, Japan) and K+ channels [1 mm 4-AP (Sigma), 0.05 mm quinidine (Sigma), and 2.5 mm tetraethylammonium (TEA) (Eastman Kodak, Rochester, NY)]. Recordings were performed on the soma of neurons (diameters ranging from 15 to 18 μm) in 2- to 3-d-old cultures by using a patch-clamp amplifier (Axopatch 1B; Molecular Devices, Palo Alto, CA). The seal resistance was usually >5 GΩ, and junction potentials were nulled before establishing the whole-cell configuration. A personal computer in conjunction with an analog-to-digital converter and pClamp software (version 5.5.1; Molecular Devices) was used to generate the current- and voltage-clamp commands and for data acquisition. All experiments were done at room temperature. Current densities were normalized to membrane capacitance, which was obtained by using a small hyperpolarization step (−4 mV, 25 ms). To determine steady-state inactivating current, the test pulse (+60 mV) was delivered 5 ms after 500 ms preconditioning pulses varying from −100 to +60 mV at 20 mV increments. The computer program for analyzing dV/dt of “Ca2+ spikes” (see Fig. 3 D) was created with C++ Language (Borland, Scotts Valley, CA).
Quantification of mRNA levels of K+ channel genes.
Primers for real-time (RT)-PCR detection of the target genes were designed by using PrimerExpress software of Applied Biosystems (Foster City, CA) and were synthesized by Integrated DNA Technology (Coralville, IA). The primers specifically probe the conserved domains of individual genes reported in current updates in FlyBase (http://flybase.bio.indiana.edu/) and are expected to detect all splice variants for each gene. FlyBase identification numbers are as follows: Sh, FBgn0003380; Shal (Shaker cognate l), FBgn0005564; slo, FBgn0003429; and SK (small conductance calcium-activated potassium channel), FBgn0029761. The primer sequences were as follows (F, forward; R, reverse; 5′ to 3′): glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-F, 5′-AGCGCTGGTGCCGAATAC-3′; GAPDH-R, 5′-AGTGAGTGGATGCCTTGTCGAT-3′; Sh-F, 5′-CGGAT-AATGAGAAACAGAGAAAAGTCT-3′; Sh-R, 5′-TGGCGGCTTGCGAACT-3′; Shal-F, 5′-CCAGAGACAATAGCTGGCAAAA-3′; Shal-R, 5′-GACCAGCACACCGCTAAGC-3′; slo-F, 5′-AAAGACTGGGCTGGAGAGCTT-3′; slo-R, 5′-AACGACCAAAATTCGACCAGTT-3′; SK-F, 5′-GCGAAAGGTTCCACGATGAG-3′; and SK-R, 5′-GCCGTTA-GCCACATGGAGTT-3′.
Flies 1–2 d after eclosion of individual genotypes were frozen in liquid nitrogen, and their heads were detached by a 10 s vortexing. Fifty to 55 heads were grounded in a 2 ml homogenizer, and total RNA was extracted by using the RNeasy Mini kit (Qiagen, Valencia, CA). An on-column DNase digestion was performed during RNA preparation to remove potential genomic DNA contamination. Reverse transcription was performed using the SuperScript III kit (Invitrogen) with random primers. The copies of double-stranded (ds) DNAs containing the conserved domain corresponding to the individual K+ channel probes were amplified in PCR in proportion to the initial abundance. Using Power SYBR Green PCR Master Mixer (Qiagen), the fluorescent reaction products were quantified optically with an ABI Prism 7000 machine (Applied Biosystems). For each reaction, a single PCR product was indicated by a characteristic dsDNA dissociation temperature. Triplicates of PCR runs were performed for each gene on each of the three independent RNA preparations. The abundance of transcripts for individual K channel genes was determined by normalizing their optical measurements to that of GAPDH from the same genotypes (Guan et al., 2005).
Results
Currents mediated by Ca2+ channels in cultured Drosophila giant neurons
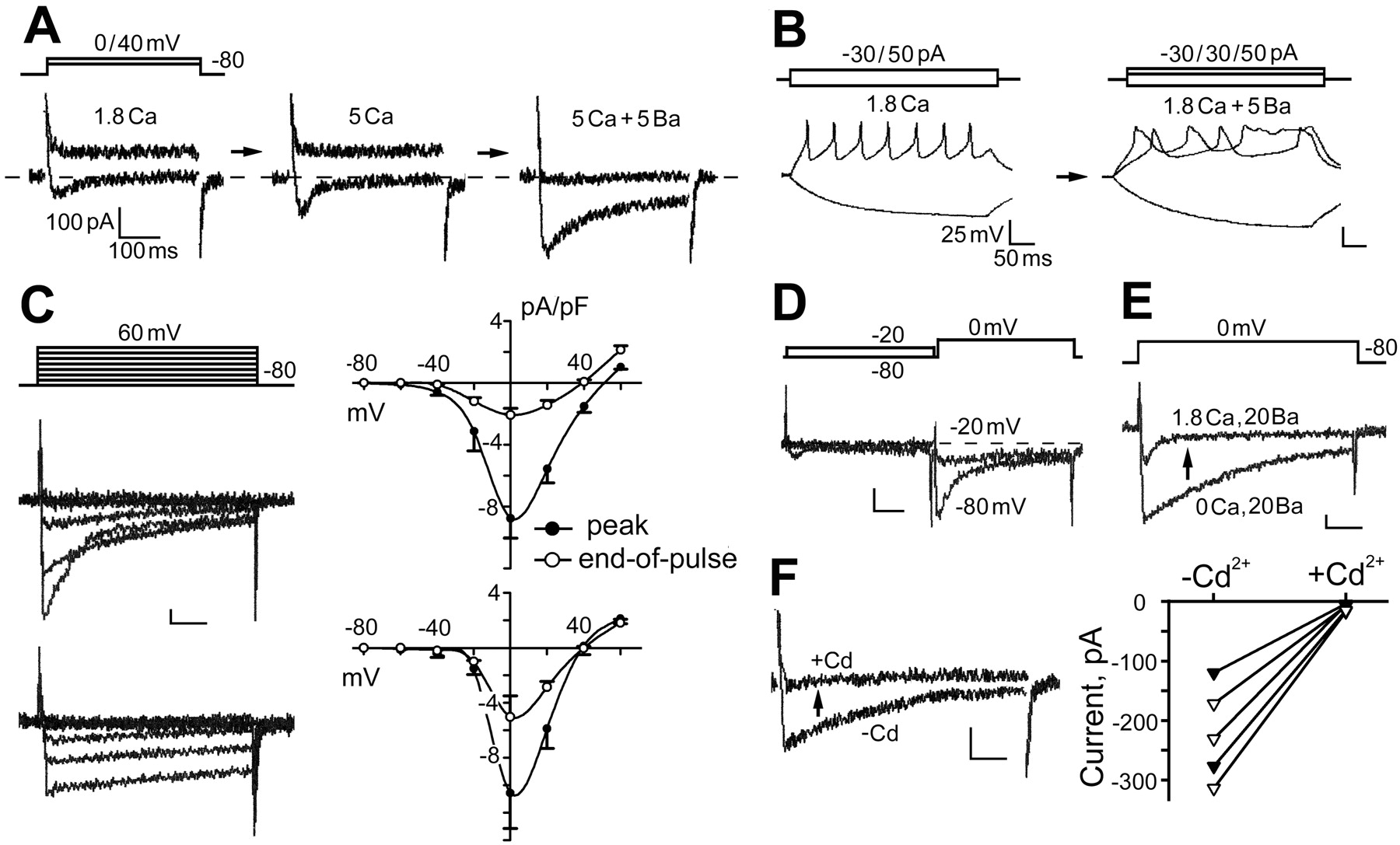
Whole-cell voltage-clamp recordings were performed on Drosophila giant neuron cultures to investigate the currents flowing through Ca2+ channels. Ca2+ currents (I Ca) were indicated by the remaining inward current measured in TTX-containing (10 nm) standard bath solution and K+-free (replaced by Cs+) pipette solution (Fig. 1 A). During a depolarization step from −80 to 0 mV, a small inward current could be detected in WT neurons. An elevated external Ca2+ (from 1.8 to 5.0 mm) increased the amplitude of this inward current. The response turned outward beyond an extreme positive voltage of above +40 mV (Fig. 1 A), presumably reflecting some remaining Ca2+-activated K+ currents [similar to the observations in other Drosophila preparations (Byerly and Leung, 1988)]. Ba2+ is known to be a more effective charge carrier through Ca2+ channels (Hille, 2001). Adding Ba2+ in the bath solution further enhanced the inward current on the same cell and suppressed the outward currents at +40 mV (Fig. 1 A). In addition, adding Ba2+ also affected action potential shape and firing pattern (Fig. 1 B). We regularly observed broadened spikes, sometimes followed by plateau potentials, reflecting the balancing acts of the modified depolarization and repolarization forces after Ba2+ treatments.

Properties of Ca2+ channels in cultured Drosophila giant neurons. A , Whole-cell voltage clamping of Ca2+ currents in standard bath solution containing TTX to block Na+ current, with K+ currents suppressed by using K+-free, Cs+ pipette solution. Superimposed current traces elicited by voltage-clamp steps from −80 to 0 and +40 mV are shown. The amplitude of inward currents (I Ca) at 0 mV increased with the elevation of extracellular Ca2+ (from 1.8 to 5 mm). An additional 5 mm Ba2+ further enhanced the inward current [I Ba(Ca)]. Note the suppression of outward currents at +40 mV by Ba2+ application. B , Spike trains initiated during current clamping in standard bath solution. Note broadening of the action potentials after 5 mm Ba2+ application. C , Kinetics and voltage dependence of I Ba(Ca). To isolate inward currents, standard bath solution contained 20 mm Ba2+, 1.8 mm Ca2+, TTX, 4-AP, TEA, and quinidine (see Materials and Methods). As shown in the two sets of superimposed current traces, two distinct kinetic components of I Ba(Ca) were evident, which displayed fast versus slow inactivation (τ of <100 vs >300 ms at 0 mV). The ensemble I–V curves present data from neurons with a dominant fast-inactivating component (n = 49; top) and from neurons with only slow-inactivating components (n = 35; bottom). Note that the fast-inactivating component had a lower threshold. D , Physiological separation of fast- and slow-inactivating components. The superimposed traces show consecutive current records for the effect of a 0.5 s, −20 mV prepulse in removing the fast-inactivating component. E , Ca2+-dependent inactivation of I Ba(Ca). Superimposed current traces from consecutive records of the same cell demonstrate that fast inactivation of I Ba(Ca) did not occur in Ca2+-free solution. F , Removal of I Ba(Ca) by 0.2 mm Cd2+ in neurons with different decay kinetics. Neurons displaying fast- or slow-inactivating component were represented by filled or open symbols, respectively. Vertical calibration bars: 100 pA or 25 mV. Horizontal calibration bars: 100 ms for voltage-clamp traces and 50 ms for current-clamp traces. Standard bath solution was composed of the following (in mm): 128 NaCl, 2 KCl, 4 MgCl2, 1.8 CaCl2, and 35.5 sucrose, buffered at pH 7.1 with 5 HEPES.
To improve isolation of currents flowing through Ca2+ channels, Ba2+ (20 mm), in addition to Ca2+ (1.8 mm), was used along with Na+ and K+ channel blockers (10 nm TTX, 1 mm 4-AP, 2.5 mm TEA, and 50 μm quinidine) in standard bath solution, in conjunction with K+-free (replaced by Cs+) pipette solution. After growing 2 d in culture, most WT neurons (∼80%; n = 104) expressed an inward current [termed I Ba(Ca)]. Two types of I Ba(Ca) were classified in the neuronal populations based on their distinct decay kinetics (τ of <100 vs >300 ms for the major decay component) (Fig. 1 C). The current–voltage (I–V) relationships show that both types of inward currents reach their maximum at ∼0 mV. However, the fast-inactivating current was activated at more negative potentials than the slow-inactivating component (approximately −40 vs −20 mV) (Fig. 1 C). Some cells expressed both fast- and slow-inactivating currents, which could be isolated by using a prepulse inactivation protocol (Fig. 1 D). These properties of Ca2+ currents are consistent with previous single-channel and whole-cell studies on other Drosophila neuronal culture systems (Leung et al., 1989; Leung and Byerly, 1991; Schmidt et al., 2000).
Ba2+ is also known to suppress Ca2+-dependent inactivation of Ca2+ channels (Hille, 2001). Consistently, we found that, in Ca2+-free, Ba2+-containing saline (20 mm), neurons no longer displayed fast inactivation (n = 67). Adding a physiological concentration of Ca2+ (1.8 mm) restored inactivation of the inward currents (mean ± SEM, τ0Ca, 267 ± 17 ms vs τ1.8Ca, 86 ± 27 ms; n = 5; p < 0.005, paired t test) (Fig. 1 E). The amplitude of inward current was proportional to the external concentration of Ba2+ (5–20 mm; data not shown). Therefore, throughout the remaining study, the bath solution contained 20 mm Ba2+ and 1.8 mm Ca2+, in addition to Na+ and K+ channel blockers, for characterizing cac channel-mediated currents, with the resultant currents termed I Ba(Ca). Notably, a general Ca2+ channel blocker Cd2+ (0.2 mm) was capable of blocking both types of I Ba(Ca) (Fig. 1 F).
Removal of a major component of neuronal Ca2+ currents by cac mutations
Neuronal currents mediated by cac channels have not been characterized under voltage-clamp conditions. We compared recordings from neurons dissociated from WT and cac mutant alleles. As described above, neurons in WT cultures expressed varying amplitudes and kinetics of I Ba(Ca) (Figs. 1 B, 2 A), and, in some neurons, I Ba(Ca) was not detectable (<1 pA/pF) (Fig. 2 A). The extent of variability is shown in the amplitude histogram (Fig. 2 E). In cultures of cac s, a hypomorph, I Ba(Ca) was drastically reduced (Fig. 2 B). The I–V curves for the peak I Ba(Ca) current (Fig. 2 D) show that, on average, I Ba(Ca) in cac s cultures was less than one-fifth in amplitude of that in WT. Notably, the residual currents in cac neurons still displayed both fast- and slow-inactivating components (Fig. 2 B), but the proportion of neurons with no detectable I Ba(Ca) was greatly increased from 20% in WT to >50% in cac s (Fig. 2 E). To rule out the possibility of undesirable genetic background contributions, we examined another independently isolated allele cac ts2. The results demonstrate that cac ts2 mutation also affects both fast- and slow-inactivating Ca currents (data not shown). On average, I Ba(Ca) in cac ts2 cultures was significantly reduced [mean ± SEM (n), WT, −5.61 ± 0.50 pA/pF (104) vs cac ts2, −1.78 ± 0.46 pA/pF (18); p < 0.005, t test].

Neuronal Ca2+ channels encoded by cac. A , Representative traces from six WT neurons with different I Ba(Ca) density and kinetics. Neurons with no detectable I Ba(Ca) were rare (bottom). B , Strong reduction of I Ba(Ca) in cac s mutant neurons. The remaining I Ba(Ca) still displayed fast- or slow-inactivation kinetics. Neurons without detectable I Ba(Ca) were frequently encountered. C , Near elimination of I Ba(Ca) in neurons of l(1)L13 HC129 deficiency line. Most neurons from this line produced no detectable I Ba(Ca). D , I–V relationships for peak I Ba(Ca) from different genotypes [n = 104, 65, and 25 for WT, cac s, and l(1)L13 HC129]. E , Histograms of I Ba(Ca) density at 0 mV. Note abundance of neurons without detectable I Ba(Ca) in both cac s and l(1)L13 HC129 cultures.
The use of embryonic cultures also allowed studies of lethal mutations. Single-embryo cultures of homozygous deficiency l(1)L13 HC129 could be identified by the absence of green fluorescent protein (GFP) signal, because the stock was maintained over a marked balancer chromosome, FM7, P{w +mC = Act GFP} (Kawasaki et al., 2002). In homozygous cultures, >90% of neurons produced no detectable I Ba(Ca) (Fig. 2 C,E). I Ba(Ca) in this deficiency mutant was much smaller than that found in cac s (Fig. 2 D). Our mutational analysis demonstrates that cac encodes the channels that are responsible for operating major Ca2+ currents of different activation voltages and inactivation kinetics in Drosophila neurons.
Current-clamp experiments effectively revealed the contribution to neuronal depolarization by cac-mediated currents of different amplitudes, kinetics, and voltage dependence. Consecutive voltage- and current-clamp recordings were performed on the same cells to investigate the rising phase and maintenance of membrane depolarization. In the presence of TTX, I Ba(Ca) was the remaining depolarizing force. During depolarizing current injection, neurons with substantial I Ba(Ca) generated all-or-none, regenerative activities (Ca2+ spikes or action potentials) (Fig. 3 A, first two cells). After spike initiation, the membrane potential remained depolarized, outlasting current injection, as a result of Ba2+ inhibition of Ca2+ channel inactivation and reduced repolarization by K+ channel blockers in the saline. In contrast, neurons lacking detectable I Ba(Ca) exhibited typical passive membrane responses (Fig. 3 A, last cell). The threshold levels for Ca2+ spikes, as indicated by the inflection point, differed between neurons that exhibited fast- and slow-inactivating I Ba(Ca) [mean ± SEM (n), −13.8 ± 1.4 mV (12) vs −7.0 ± 1.5 mV (9); p < 0.01]. This also reflects differences in activation voltages of the two currents (compare with Fig. 1 C).

Weakened regenerative potentials in cac neurons. A , Relationships of inward currents and regenerative potentials in WT neurons. In addition to 20 mm Ba2+, TTX and K+ channel blockers (Fig. 1 legend) were present in standard bath solution. Therefore, the inward currents and regenerative potentials sequentially recorded from the same cells reflected Ca2+ channel activity. Note the inflection points at which the regenerative potential was initiated. Depolarization of neurons with I Ba(Ca) reached a plateau level, which is determined by the equilibrium potential of the inward current and is independent of additional increase of current injection. In neurons lacking detectable I Ba(Ca) (bottom), membrane polarization was proportional to the amount of current injected, following the kinetics determined by passive membrane properties. Dash lines indicate 0 mV. B , C , Weakened regenerative potentials associated with decreased I Ba(Ca) in cac mutant neurons. D , Reduced rate of depolarization (dV/dt) during initiation of regenerative potentials in cac mutant neurons. The corresponding rate of membrane potential change of the selected traces in A and B are indicated by circles and diamonds next to the inflection point, around which maximum dV/dt occurs.
In cac cultures, the proportion of neurons unable to generate Ca2+ spikes was drastically increased (for representative traces, see Fig. 3 B,C). Only a few cac s neurons expressed small but detectable I Ba(Ca) (Fig. 2 B,E), and their associated action potentials displayed a slow rise (dV/dt in volts per second) (Fig. 3 D). Note that the maximum rise occurred at the time of inflection point of Ca2+ spikes of the corresponding traces, as indicated by symbols (Fig. 3 A,B,D). Once evoked, the thresholds of Ca2+ spikes in cac neurons [−11.7 ± 1.7 (4) vs −6.2 ± 0.9 (4) for cells with fast- vs slow-inactivating I Ba(Ca)] were not significantly different from those in WT.
Sensitivity of neuronal Ca2+ channels to T- and L-type channel blockers
The sensitivities of vertebrate Ca2+ channels to specific pharmacological agents correspond well to their molecular classifications (Ertel et al., 2000). We characterized the sensitivity of Drosophila neuronal I Ba(Ca) with several blockers commonly used for vertebrate Ca2+ channel identification. Vertebrate T-type channels are characterized by a low threshold of activation and fast inactivation compared with L-type channels. As shown above, Cd2+ that blocks both T- and L-type channels could also remove most of the Drosophila neuronal I Ba(Ca), regardless of the decay kinetics (Fig. 1 F). Unexpectedly, two T-type Ca2+ channel blockers, amiloride (1 mm) and Ni2+ (0.1–0.2 mm), blocked not only the low-threshold, fast-inactivating I Ba(Ca) but also the high-threshold, slow-inactivating I Ba(Ca) (Fig. 4 A,B). In contrast, two L-type channel blockers, nifedipine (10 μm) and diltiazem (0.5 mm), had little effect on either type of I Ba(Ca) (Fig. 4 A,B). In fly muscles, it has been shown that the Ca2+ currents are sensitive to L-type rather than T-type blockers (Gielow et al., 1995). Together, the results indicate distinct pharmacological profiles for the dominant Ca2+ currents in neurons and muscles.
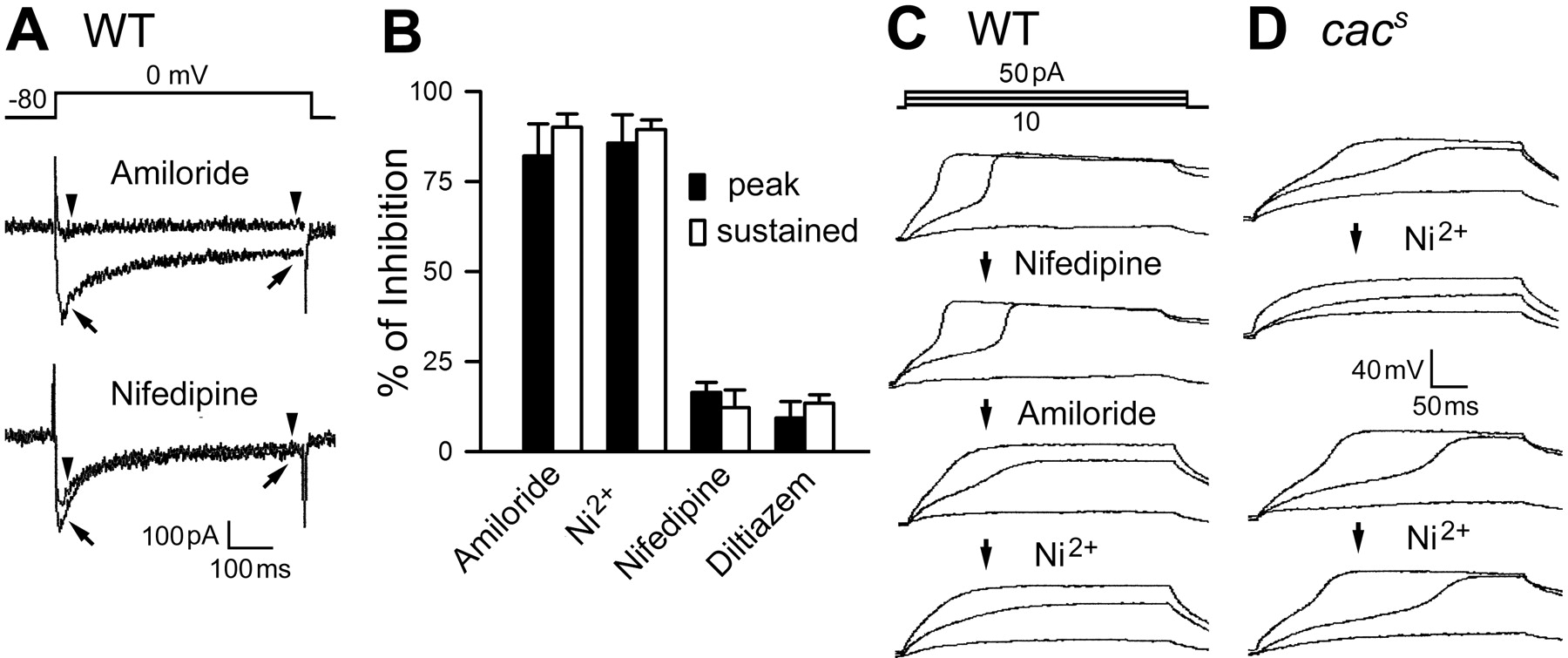
Sensitivity of Drosophila neuronal Ca2+ channels to T-type channel blockers. Two T-type channel blockers, amiloride (1 mm) and Ni2+ (0.1 mm), and two L-type channel blockers, nifedipine (10 μm) and diltiazem (0.5 mm), were examined. A , Superimposed current traces obtained before (arrows) and after (arrowheads) amiloride and nifedipine treatments in WT neurons. B , Percentage of I Ba(Ca) inhibition by different drugs in WT neurons. Reduction in both peak and sustained components (measured at the times indicated by arrows and arrowheads in A ) indicates that both fast- and slow-inactivating components of Ca2+ currents were sensitive to amiloride and Ni2+ (n = 6 and 7, respectively). In contrast, nifedipine and diltiazem removed <20% of the I Ba(Ca) (n = 6 and 5, respectively). C , D , Different drug effects on regenerative membrane potentials in WT and cac neurons. Nifedipine exerted little effect, but amiloride and Ni2+ progressively removed regenerative potentials in WT neurons (an example of sequential recording shown in C ). In contrast, only some cac s neurons examined were sensitive to Ni2+ (4 of 7; top in D ) and amiloride (2 of 4), but the remaining samples were unaffected by either Ni2+ (bottom) or amiloride.
We further tested T- and L-type channel blockers on WT and cac neurons for Ca2+ spike generation. Although the drug effect on the small residual I Ba(Ca) in cac neurons was difficult to quantify with voltage clamping, current-clamp recordings effectively detected the weakening of Ca2+ spikes during drug treatment. In WT cultures, nifedipine treatments had little effects (n = 5) (Fig. 4 C), but either amiloride or Ni2+ application slowed down the rising phase of Ca2+ spikes (n = 4 and 4, respectively) (for sequential applications on the same cell, see Fig. 4 C). These data are consistent with the results from voltage-clamp studies (Fig. 4 A,B). The Ca2+ spikes in cac s cultures were also resistant to nifedipine (n = 4; data not shown). However, unlike the clear blocking effect in WT, amiloride and Ni2+ treatments did not alter the rising phase of Ca2+ spikes in some cac s neurons (Ni2+, 3 of 7; amiloride, 2 of 4) (Fig. 4 D). Therefore, in cac s neurons insensitive to Ni2+ and amiloride, the remaining I Ba(Ca) may be mediated by either cac channels with genetically modified pharmacological sensitivity or non-cac channels.
Reduction of Ca2+-activated K+ currents in cac neurons
The dynamic regulation of intracellular Ca2+ is known to affect a wide range of molecular, cellular, and developmental events. One immediate target is Ca2+-activated I K(Ca) channels, which serve as a feedback mechanism for membrane repolarization and termination of voltage-dependent Ca2+ influx. We investigated how the suppression of Ca2+ influx in cac neurons affects I K(Ca) by using Ba2+-free (1.8 mm Ca2+), TTX-containing bath solution and Cs+-free pipette solution [to compare with standard saline for I Ba(Ca) measurement, see Materials and Methods]. After Cd2+ application, I K(Ca) was extracted by subtracting the remaining outward current from the total current before Cd2+ treatment (Singh and Wu, 1989; Saito and Wu, 1991). In this procedure, contamination from inward I Ca was negligible because of its small size (<50 vs >1000 pA for outward currents) (compare Figs. 1 A, 5 A). The small Ca2+ influx sufficient for activating I K(Ca) is consistent with the high-affinity I K(Ca) channels in Drosophila (submicromolar Ca2+) (Komatsu et al., 1990). The extracted I K(Ca) from WT neurons was variable in current kinetics and size (Fig. 5 A,E). WT neurons could express either or both of the transient and sustained components of I K(Ca) (Fig. 5 A) (cf. Saito and Wu, 1991). The peak current density of I K(Ca) ranged from 1 to 40 pA/pF during depolarization to +60 from −80 mV. Because amiloride was also effective in blocking Ca2+ currents (1 mm) (Figs. 1, 4), amiloride-sensitive K+ currents were also determined as another measure for I K(Ca). As expected, Cd2+- and amiloride-sensitive K+ currents shared similar I–V relationships (Fig. 5 D) and a similarly wide range of current densities (Fig. 5 E). Adding amiloride after Cd2+, or the other way around, did not further reduce the K+ current amplitude (data not shown).

Severe reduction of Ca2+-activated K+ currents [I K(Ca)] in cac neurons. I K(Ca) was extracted from subtracting Cd2+-sensitive currents from total outward current in standard bath saline, in which Cd2+ (0.2 mm) was added to eliminate Ca2+-dependent currents. The difference in current amplitude before and after Cd2+ treatment yields I K(Ca) with a minimal contamination of I Ca (<5%; compare with Fig. 1 A). A , I K(Ca) in WT cultures. Most WT neurons (>60%) produced large I K(Ca) (>6 pA/pF) with a minority displaying little I K(Ca). Application of amiloride to block cac channels produced similar estimates of I K(Ca). B , C , Strong suppression of I K(Ca) in cac s and l(1)L13 HC129 mutant cultures. D , Ensemble I–V curves demonstrating strong suppression of I K(Ca) in cac mutant cultures. Note that estimation of I K(Ca) based on extraction of amiloride-sensitive K+ currents in WT neurons produced results similar to Cd2+-sensitive K+ currents. E , Histograms of I K(Ca) amplitude distribution. Note the similarities between I K(Ca) and I Ba(Ca) density distributions for WT as well as cac mutant alleles (compare with Fig. 2 E). Sample sizes, n = 28, 8, 19, and 10 for WT with Cd2+, WT with amiloride, cac s, and l(1)L13 HC129.
In contrast, most neurons from the two cac alleles carried little detectable I K(Ca) (<2 pA/pF) (Fig. 5 B,C,E). Among the few cac neurons that expressed detectable I K(Ca), the current density was rarely beyond 10 pA/pF (Fig. 5 E). The percentage of cac s neurons carrying I K(Ca) was directly comparable with that of neurons expressing I Ba(Ca) (∼20%) (Figs. 2 E, 5 E). Moreover, I K(Ca) and I Ba(Ca) reduction in the cac mutants was similar in profiles (Figs. 2 D, 5 D). These results suggest that neuronal Ca2+ influx through cac channels provides a major source of Ca2+ for I K(Ca) activation.
Homeostatic upregulation of inactivating IA in cac neurons
Voltage-activated K+ currents are activated in response to membrane depolarization. After removal of inward I Ca and outward I K(Ca) by Cd2+, the remaining outward currents contained two distinct voltage-activated components, inactivating I A and non-inactivating I K, which can be separated by a prepulse inactivation protocol (Zhao and Wu, 1997; Yao and Wu, 1999, 2001; Peng and Wu, 2007). We found in cac s cultures that I A was unexpectedly upregulated, whereas I K was unaltered (Fig. 6 A,B,D).

Homeostatic upregulation of voltage-dependent I A in cac neurons. A–C , Separation of transient I A and sustained I K by a prepulse protocol after removal of Ca2+-dependent currents by Cd2+. Representative traces demonstrate a substantial I A increase in cac s neurons compared with WT neurons. WT neurons pretreated with the cac channel blocker Ni2+ (0.05 mm) for 3 d phenocopied upregulation of I A observed in cac neurons. D , Mean current densities for extracted I A, I K, and I K(Ca). Note upregulation in I A but not I K in cac s neurons and WT neurons with chronic blockade (2–3 d) of cac channels. However, short-term blockade (15–20 min) of cac channels by Ni2+ or amiloride (0.5 mm) did not alter current densities. *p < 0.05 against WT, one-way ANOVA. E , Summation of I A, I K, and I K(Ca) among neurons of different genotypes or with drug treatments. Note the similar total K+ currents, which suggest a homeostatic, compensatory increase of I A for reduction of I K(Ca). F , Properties of upregulated I A in cac neurons compared with WT I A. Steady-state (st-st) inactivation of I A is compared for V 1/2, at which half-inactivation is attained. Solid and dashed lines in the box plots indicate median and mean values. Decay time constant (τ) was determined at +60 mV. Half-time (t 1/2) of I A recovery from inactivation was determined with a twin-pulse (+20 mV) protocol with varying interpulse intervals. Data indicate no significant differences in these biophysical parameters. Error bars represent SEM with sample sizes indicated.
Interestingly, I A increase in cac neurons can be phenocopied by WT neurons after chronic pharmacological blockade of cac Ca2+ channels (Fig. 6 D). Ni2+ (0.05 mm) was applied to WT cultures throughout the culture lifespan (2–3 d). Before recording, Ni2+ was washed out, and I K(Ca), I A, and I K were isolated by application of Cd2+ and the prepulse protocol sequentially. Ni2+ treatment decreased I K(Ca) and, at the same time, increased I A without altering I K (Fig. 6 D). These changes in current density were significant but to a lesser extent compared with the modifications in cac s neurons.
In contrast, short-term Ni2+ treatments (15–20 min) did not cause any detectable alterations in I K(Ca), I A, I K (Fig. 6 C,D), and I Ba(Ca) (data not shown). Short-term treatment of another cac channel blocker, amiloride (1 mm), did not result in detectable changes in K+ currents either, suggesting that the K+ current upregulation requires a relatively slow process. [Long-term incubation with amiloride was not attempted because amiloride is light sensitive (Gielow et al., 1995) and the resultant products appeared to be toxic to cultured neurons.] Remarkably, the size of I A increase in cac neurons as well as in chronically Ni2+-treated WT cultures approximately matched the degree of I K(Ca) reduction (Fig. 6 D), resulting in no appreciable changes in total outward K+ currents (Fig. 6 E).
It is known that posttranslational modifications such as phosphorylation affect channel properties, including voltage dependence and kinetics of channel activation and inactivation (Jonas and Kaczmarek, 1996; Yao and Wu, 2001). To investigate whether upregulated I A in cac neurons displays obvious signs of modified properties indicating channel posttranslational modifications, we examined the parameters including voltage of half steady-state inactivation (V 1/2), kinetics of decay (τ), and recovery from inactivation (t 1/2). We did not detect any significant differences in these properties of I A between WT and cac cultures (Fig. 6 F), despite the striking amplitude increase in cac neurons. These results do not support the possibility of posttranslational modifications of I A channels caused by cac mutations.
mRNA levels of K+ channel genes in cac mutants
In Drosophila, Shaker (Sh) and Shal encode I A channels, whereas slowpoke (slo) and SK code for I K(Ca) channels (Jan et al., 1977; Tanouye et al., 1981; Wei et al., 1990; Atkinson et al., 1991; Adelman et al., 1992). To determine whether transcriptional modifications of these identified genes could be responsible for the altered K+ current expression observed in cac mutants, we quantified their mRNA levels in adult heads by performing RT-PCR. The amplification rate during RT-PCR cycles gave an estimate of the abundance of individual K+ channel transcripts (Fig. 7).

Determination of mRNA abundance for K+ channels in WT and cac neurons by RT-PCR. A , Amplification rates of GAPDH and Sh transcripts from WT and cac fly heads based on the accumulation of fluorescence intensity throughout PCR cycles. Inset, Enlargement for the segment between cycles 16 and 20. The rate for the Sh fragment in cac mutants was faster than that in WT, but the rate for the control GAPDH fragment was nearly identical for the two genotypes. B , Differences in transcript abundance of Sh and Shal I A channels and of slo and SK I K(Ca) channels between WT and cac. The mRNA levels for individual K+ channel genes are estimated by the fluorescence change normalized to GAPDH control. The positive value in the bar graph indicates an increase in RNA abundance in cac over WT. Data from three independent RNA purifications. Error bars indicate SD. *p < 0.05, two-way ANOVA for genotype and channel subtype comparisons.
Our RT-PCR results from adult heads showed only slight but statistically insignificant decrease in slo and SK mRNA in cac mutants (Fig. 7 B), although electrophysiological results from embryonic neuronal cultures demonstrate a drastic I K(Ca) reduction in cac cultures (Fig. 5). These results support the idea of a normal density of I K(Ca) channels with deprived activity attributable to insufficient I Ca in cac neurons. Unlike slo and SK mRNAs, we detected a significant increase of Sh mRNA levels in cac mutants (42%) (Fig. 7 A,B). The data suggest that a modification at the mRNA level may be a contributing factor for the increase of I A in cac cultures. Apparently, Sh was preferentially upregulated because Shal mRNA levels were not increased.
Unaltered Ca2+ current density in K+ channel mutants
The above results demonstrate an important role of cac Ca2+ channels in the long- and short-term regulations of neuronal K+ currents (Figs. 5, 6). It would be of interest to determine whether K+ channel mutations could in turn affect Ca2+ current expression. We measured I Ba(Ca) in neurons dissociated from Sh, Shab (Singh and Singh, 1999), and slo mutants, which lack voltage-gated inactivating, non-inactivating, and Ca2+-gated K+ channels, respectively. Among the null or nearly null mutant alleles examined (Sh M, Shab 3, and slo 1) (Jan et al., 1977; Tanouye et al., 1981; Elkins et al., 1986; Wei et al., 1990; Singh and Singh, 1999), we did not detect any significant alterations in I Ba(Ca) amplitude (Fig. 8). Furthermore, prolonged incubation (2 d) of WT neurons with two I K(Ca) blockers, apamine (5 μm) and charybdotoxin (50 nm), did not affect the densities of I Ba(Ca). These results indicate a central role of cac Ca2+ channels in neuronal homeostatic regulation; manipulations of Ca2+ influx through cac channels strongly affect K+ current expression, whereas elimination of individual K+ channels is not sufficient to cause homeostatic changes in Ca2+ channels.

Lack of modifications in Ca2+ channel-mediated currents in K+ channel mutants. Mean I Ba(Ca) densities among neurons in cac and Ni2+-treated WT cultures are compared with cultures of K+ channel mutants. Sh M, with altered I A, Shab 3, with defective to I K, slo 1, with defective to I K(Ca). No modifications of I Ba(Ca) densities were found in K+ channel mutant neurons and in WT neurons with chronic blockade (2–3 d) of Ca2+-activated K+ channels by 5 μm apamine and 50 nm charybdotoxin (Apa + ChTx). Error bars represent SEM with sample sizes indicated. **p < 0.005 against WT control, one-way ANOVA.
Discussion
Short- versus long-term regulations of K+ currents by cac channel-mediated Ca2+ currents
Intracellular Ca2+ is critical for regulation of neuronal membrane excitability and neurotransmission. In excitable cells, intracellular Ca2+ triggers Ca2+-activated I K(Ca), which contributes to repolarization of action potential and the subsequent hyperpolarizing after potential (for review, see McManus, 1991; Vergara et al., 1998). In the giant neuron culture of Drosophila, we confirmed the tight functional coupling between cac-mediated Ca2+ current and I K(Ca), as indicated by a same proportion of cac s neurons that expressed I Ba(Ca) and I K(Ca) (both ∼20%) and by a similar extent of reduction in the two currents (Figs. 2, 5). The result reflects a unidirectional functional dependence, because the Ca2+ current amplitude is not affected by the I K(Ca) mutation slo or by chronic treatment with I K(Ca) blockers (Fig. 8).
A number of potential mechanisms could be responsible for the I A upregulation observed in cac mutant neurons (Fig. 6), including transcriptional, translational, and posttranslational modifications. Posttranslational modifications by Ca2+-dependent protein kinase phosphorylation and dephosphorylation can substantially change K+ channel activities and properties within tens of seconds to minutes (Jonas and Kaczmarek, 1996; Wicher et al., 2001). Such posttranslational modulation of K+ currents is known to underlie activity-dependent plasticity of synaptic transmission in molluscan species (Byrne and Kandel, 1996; Alkon, 1999) and is altered in Drosophila cAMP pathway mutants defective in memory processes (Zhong and Wu, 1993; Wright and Zhong, 1995). Drosophila Sh channels are modified by cAMP-dependent modulation in a heterologous expression system (Drain et al., 1994) and in larval muscles (Zhong and Wu, 1993) as well as in embryonic and larval cultured neurons (Wright and Zhong, 1995; Yao and Wu, 2001). We examined properties of the upregulated I A in cac or pharmacologically treated WT cultures (Fig. 6) for indications of I A modulation. However, our results did not provide any compelling evidence for channel modulation. Furthermore, the homeostatic regulation observed requires long-term blockade of cac channels (days instead of minutes) (Fig. 6), beyond the ordinary timescale of modulation by protein kinases.
In contrast to the above short-term regulations, there are also long-term Ca2+-dependent effects on neuronal differentiation, growth, and plasticity, in which protein synthesis or gene expression is required. For example, when neuronal spike activity is manipulated, a homeostatic regulation is initiated, in which the activity-dependent intracellular Ca2+ accumulation induces compensatory excitability changes by adjusting the relative abundance of ion channels (Spitzer, 1999; Turrigiano, 1999). In Xenopus embryonic cultures, changing the timing of Na+ channel expression or pharmacologically suppressing Na+ or Ca2+ channel function alters the subsequent developmental time course of K+ current (O'Dowd et al., 1988; Linsdell and Moody, 1994, 1995). In addition, overexpression of Shal I A in lobster neurons triggers a compensatory increase of hyperpolarization-activated inward I h (MacLean et al., 2003). Our study presents a case of homeostatic regulation of voltage-dependent K+ currents by genetic manipulation of cac channel-mediated Ca2+ currents in Drosophila. In cac neuronal cultures, a significant increase was observed in voltage-activated I A but not I K, an upregulation that could be mimicked by chronic blockade of cac channels in WT cultures (Fig. 6). The lack of I K(Ca) in cac neurons appeared to be compensated by an increase in I A, and consequently the total outward K+ currents remained essentially unaltered (Figs. 5, 6).
It has been demonstrated in identified neurons that transcript numbers of several types of K+ channels correspond to their current sizes in crustacean species (Baro et al., 1997; Schulz et al., 2006). In microarray experiments, Drosophila mutations affecting neuronal excitability and nerve terminal development can lead to upregulation or downregulation of ion channel and receptor RNA levels (Guan et al., 2005). Increase in synaptic input levels by genetic manipulations could decrease transcript numbers of the para (paralytic) Na+ channels in identified Drosophila motor neurons (Mee et al., 2004). Furthermore, the mRNA level of slo BK (large-conductance calcium-activated) channels is sensitive to behavior conditioning, strongly upregulated after alcohol sedation (Cowmeadow et al., 2006). In Drosophila giant neuron cultures, Shab mutant neurons, defective in slow-inactivating K+ currents, show an upregulation of fast-inactivating K+ currents (Peng and Wu, 2007). In the current study, an upregulation of I A was also observed in cac mutant neurons (Fig. 6). Our quantitative RT-PCR studies on adult brain tissue indicate a 40% increase in Sh mRNA levels in cac mutants (Fig. 7). Such results are consistent with the idea that a mechanism at the RNA level contributes to the observed homeostatic upregulation of I A in embryonic neuronal cultures (Fig. 6) and suggest the possibility that I A is upregulated in adult cac neurons as well. It is worth noting that Shal mRNA level was not altered in cac neurons, although Shal, rather than Sh, channels are considered to be the dominant contributor to I A in the neuronal soma (Baker and Salkoff, 1990; Baro et al., 1997; Gasque et al., 2005; Peng and Wu, 2007). These data suggest an interesting possibility that Sh plays a role in neuronal plasticity for upregulation and downregulation of neuronal excitability during environmental challenges or behavioral conditioning. It has been shown that Sh mutations affect learning in a courtship conditioning paradigm and a non-associative habituation paradigm (Cowan and Siegel, 1984; Engel and Wu, 1998).
Major Ca2+ currents in Drosophila neurons encoded by the cac gene
In vertebrate nervous systems, low-threshold, fast-inactivating T type Ca2+ channels coexist with high-threshold, slow-inactivating L-, N-, P-, and Q-type Ca2+ channels (Hille, 2001). Molecular characterizations have revealed that these channels are encoded by separate genes, and their differential expression leads to different patterns of intracellular Ca2+ dynamics. In Drosophila, studies of dissociated embryonic neurons demonstrated two distinct components of Ca2+ currents, which parallel the vertebrate low-threshold, fast-inactivating and high-threshold, slow-inactivating Ca2+ currents (Figs. 1, 2) (Byerly and Leung, 1988; Saito and Wu, 1991; Schmidt et al., 2000). The two components can also be distinguished pharmacologically (Leung et al., 1991). Nevertheless, our study suggests that the cac channels mediate a major Ca2+ currents in Drosophila embryonic neurons, because cac deficiency, l(1)L13 HC129, nearly eliminates both fast- and slow-inactivating Ca2+ currents in our soma recording (Fig. 2). This conclusion is also supported by the observation that both components in WT were inhibited by vertebrate T-type channel blockers (Fig. 4). However, a minor component of Ca2+ current still remained in the cac deficiency mutant, implying a role of Dmca1D or other unidentified genes in neuronal Ca2+-dependent functions. It remains to be investigated, in neurons at the larval, pupal, and adult stages, to what extent Ca2+ currents are reduced and whether both fast- and slow-inactivating Ca2+ currents are affected by cac mutations.
Deletion of either Dmca1D or cac, two known Ca2+ channel genes in Drosophila, causes lethality, suggesting that their roles in the organism are not redundant (Smith et al., 1996; Eberl et al., 1998). Genetic, electrophysiological, and pharmacological experiments have demonstrated that Drosophila muscle Ca2+ currents consist of a major, DHP-sensitive and a minor, amiloride-sensitive component (D- and A-currents, respectively) (Gielow et al., 1995; Ren et al., 1998). In the present study, the dramatic reduction in Ca2+ currents observed in the soma of cac neurons implies an important role of cac channels in other neuronal compartments as well. Previous neurite recording and Ca2+ imaging on cultured giant neurons have indicated a high Ca2+ current density along the neurites and in the terminals (Saito and Wu, 1991; Berke et al., 2006). In some cell bodies that do not support full-blown action potentials, spike activities in the distal compartments could still be detected as electrotonically degraded non-overshooting oscillations (Saito and Wu, 1991; Yao and Wu, 2001). However, in cac neurons, we did not detect any electrotonic spread of such spike activities from distal neurites, even under conditions that promote prolonged Ca2+ spikes (Fig. 3). Consistently, in vivo studies have demonstrated a role of cac channels in the function and development of axonal terminals of motor neurons. Mutations of cac result in greatly reduced synaptic transmission and restricted nerve terminal projection (Kawasaki et al., 2000, 2002, 2004; Rieckhof et al., 2003). Therefore, in the Drosophila nervous system, a single gene, cac, appears to play a major role to exert many short- and long-term Ca2+-dependent regulations in neuronal development, function, and plasticity.
In vertebrates, different splice isoforms of Ca2+ channel α subunits display distinct physiological properties, drug sensitivities, phosphorylation, and subcellular distributions (Hell et al., 1994; Moreno Davila, 1999; Lipscombe et al., 2002). For instance, N-type channels, with high sequence similarity with cac (Rieckhof et al., 2003), have different isoforms with distinct voltage-dependent activation and decay kinetics (Stea et al., 1999). Therefore, the reported RNA splicing and editing of the cac transcripts (Smith et al., 1996, 1998) might account for the variation in voltage dependence and kinetics of Ca2+ currents mediated by cac channels. Additional investigations into additional cac alleles or heterologous expression of the identified splice variants may provide additional clues to the mechanisms underlying the remarkable functional diversity of Ca2+ currents generated by the cac gene.
Footnotes
-
This work was supported by National Institutes of Health Grants HD18577 and NS26528. We thank Dr. J. Miller and Dr. J. Lin for the advice and support on real time-PCR experiments. We also thank J. Lee for comments on this manuscript.
- Correspondence should be addressed to Chun-Fang Wu, Department of Biological Sciences, University of Iowa, Iowa City, IA 52242. chun-fang-wu{at}uiowa.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}