

Abstract
Slow periodic EEG discharges are common in CNS disorders. The pathophysiology of this aberrant rhythmic activity is poorly understood. We used a computational model of a neocortical network with a dynamic homeostatic scaling rule to show that loss of input (partial deafferentation) can trigger network reorganization that results in pathological periodic discharges. The decrease in average firing rate in the network by deafferentation was compensated by homeostatic synaptic scaling of recurrent excitation among pyramidal cells. Synaptic scaling succeeded in recovering the network target firing rate for all degrees of deafferentation (fraction of deafferented cells), but there was a critical degree of deafferentation for pathological network reorganization. For deafferentation degrees below this value, homeostatic upregulation of recurrent excitation had minimal effect on the macroscopic network dynamics. For deafferentation above this threshold, however, a slow periodic oscillation appeared, patterns of activity were less sparse, and bursting occurred in individual neurons. Also, comparison of spike-triggered afferent and recurrent excitatory conductances revealed that information transmission was strongly impaired. These results suggest that homeostatic plasticity can lead to secondary functional impairment in case of cortical disorders associated with cell loss.
- cortex
- EEG
- encephalopathy
- encephalitis
- cerebral damage
- deafferentation
- computational model
- homeostatic plasticity
- periodic complexes
- Alzheimer's disease
- Creutzfeldt-Jakob disease
Introduction
Repetitive low-frequency discharges is the most common electroencephalographic (EEG) abnormality in a broad spectrum of diseases with CNS manifestation (e.g., encephalitis, tumors) (Bauer and Pieber, 1974; Niedermeyer, 2005). These periodic EEG patterns at low frequencies occur over prolonged epochs, in contrast to epileptic seizures that are usually transient and evolving (Kuroiwa and Celesia, 1980). Although periodic complexes are a typical clinical finding in encephalopathies associated with structural cerebral damage (Fisch, 1999), the underlying pathophysiology remains unknown (Brenner and Schaul, 1990; Niedermeyer, 2005). Understanding the cellular and network basis of this aberrant EEG state may facilitate the development of clinical intervention approaches to reduce disruption of neuronal function during cortical disease.
The cortex is endowed with feedback control mechanisms (i.e., homeostatic plasticity) that maintain activity levels of neurons and neural circuits (Davis, 2006). In particular, fast excitatory synaptic transmission is enhanced by synaptic scaling in response to prolonged activity blockage (Desai, 2003; Rich and Wenner, 2007; Turrigiano, 2007). Here, we used computational modeling to investigate how homeostatic plasticity reorganizes network dynamics in response to decrease in afferent input caused by transient or irreversible loss of presynaptic neurons. In our model, global homeostatic plasticity upregulates the recurrent excitatory synaptic conductances to compensate for deafferentation of a random subset of neurons (partial deafferentation). We hypothesized (1) that the average firing rate of the network is recovered and (2) that the distribution of firing rates across cells and the fine temporal structure of the spike patterns in individual cells differ from before deafferentation. According to our hypothesis, these changes in network dynamics are attributable to the shift in balance between afferent and recurrent excitatory inputs caused by partial deafferentation (reduced afferent excitation) and synaptic scaling (increased recurrent excitation) (Houweling et al., 2004). Of particular interest is the interaction between deafferented and intact cells during this homeostatic reorganization process. Our hypothesis is motivated (1) by recent studies of homeostatic plasticity in vivo (Desai et al., 2002; Goel and Lee, 2007; Mrsic-Flogel et al., 2007) and (2) by observation of gradual recovery of activity levels over weeks with altered EEG patterns in response to partial cortical deafferentation (Topolnik et al., 2003a,b; Nita et al., 2006, 2007).
In our simulations, we found a critical degree of partial deafferentation (fraction of neurons deafferented) that determined the final outcome of homeostatic network reorganization. For deafferentation degrees below this threshold, network dynamics recovered without any qualitative reorganization that would predict macroscopic EEG aberrations. At the critical deafferentation degree, however, a relative abrupt change in network dynamic reorganization occurred with the following key characteristics that were absent in the intact and the moderately deafferented network: (1) prominent periodic network activation, (2) decreased sparseness, (3) bursts of action potentials in individual cells, and (4) strongly nonlinear recovery time course of average firing rate. On the basis of these results, we suggest that the clinically ubiquitous periodic complexes in CNS disorders with cortical manifestation may be caused by a paradoxical inability of homeostatic plasticity to maintain sparse, asynchronous network activity in case of change in input structure by partial deafferentation.
Materials and Methods
Neuron models.
We used two-compartment conductance-based neuron models (Mainen and Sejnowski, 1996) as described previously in detail (Frohlich and Bazhenov, 2006; Frohlich et al., 2006) Briefly, neurons consisted of two electrically coupled compartments, the dendritic and axosomatic compartment. The coupling strength of these two compartments determined the firing pattern of the cells in response to a simulated depolarizing current injection [regular spiking for pyramidal cells (PYs) and fast-spiking for inhibitory interneurons (INs)]. Each compartment was endowed with a set of ionic conductances to model a specific complement of ion channels. The axosomatic compartment contained transient voltage-gated sodium and delayed-rectifying potassium channels for spike generation (GNa = 3000 mS/cm2, GKv = 200 mS/cm2), persistent sodium channels (GNaP = 4.0 mS/cm2), and voltage-independent potassium leak channels (GKl = 0.1 mS/cm2). The dendritic compartment included high-threshold calcium, calcium-activated potassium, slowly activating potassium, persistent sodium, hyperpolarization-activated depolarizing mixed cationic, potassium leak, and mixed cationic leak ion channels (GHVA = 0.016 mS/cm2, GKCa = 3.5 mS/cm2, GKm = 0.01 mS/cm2, GNaP = 4.0 mS/cm2, Gh = 0.05 mS/cm2, GKl = 0.01 mS/cm2, GL = 0.033 mS/cm2). INs had a similar ion channel complement (different values for GNa = 2500 mS/cm2, GNaP = 0.0 mS/cm2 in both compartments; dendritic leak conductance, GKl = 0.005 mS/cm2, GHVA = 0.01 mS/cm2, GKCa = 0.3 mS/cm2, GKm = 0.0 mS/cm2). Equations and parameters for these ion channels and intracellular calcium dynamics were described previously in detail (Frohlich and Bazhenov, 2006). Network heterogeneity was introduced by drawing random values for the potassium leak conductance from a normal distribution (mean ± SD, 0.01 ± 0.001 mS/cm2 for PYs; 0.005 ± 0.0005 mS/cm2 for INs).
Network model.
The cortical network model consisted of 80 PYs and 20 INs. Similar to previously studied network configurations (Bazhenov et al., 2002; Frohlich et al., 2006), synaptic connectivity in the model was local such that each PY projected to five neighboring PYs on each side (both AMPA and NMDA receptor channels). Also, each PY targeted three neighboring INs (both AMPA and NMDA receptor channels) that in turn backprojected to 11 neighboring PYs (GABAA receptor channels). In addition, all PYs and INs received independent afferent input that was modeled by a 100 Hz Poisson process. Postsynaptic receptor channels were modeled with a simplified first-order kinetic scheme of binding and unbinding of neurotransmitter described by instantaneous rise and exponential decay of synaptic conductances (Destexhe et al., 1994). To increase computational efficiency, each cell had a single synaptic conductance (for each receptor channel type) that was updated by the according change in conductance each time a spike in one of the presynaptic cells occurred. Maximal total synaptic conductances per cell were chosen such that (1) both afferent input and network input contributed to the firing before deafferentation and (2) average cross-correlogram of action potential firings in PY cells was flat before deafferentation. Synaptic conductances for connections within the network were gAMPA(PY–PY) = 3.2 nS, gNMDA(PY–PY) = 0.32 nS, gAMPA(PY–IN) = 3.0 nS, gNMDA(PY–IN) = 0.30 nS, and gGABA(IN–PY) = 4.0 nS. These maximal synaptic conductances were divided by the number of synapses targeting a given cell to determine the unitary conductances. AMPAergic synapses between PYs included short-term depression with use constant U = 7% per action potential and exponential recovery with time constant τ = 700 ms (Tsodyks and Markram, 1997). Afferent AMPAergic input conductances were set to gPY = 0.5 nS and gIN = 1.0 nS for PYs and INs, respectively.
Deafferentation and homeostatic plasticity.
We modeled neuronal dysfunction with partial deafferentation. In case of deafferentation, the frequency of the afferent excitatory Poisson input was reduced from 100 to 50 Hz. Afferent input was simultaneously reduced for all deafferented cells. We simulated networks with different fractions of neurons subject to deafferentation (deafferentation degree D with values between 10 and 100%). For example, a deafferentation degree D = 40% indicated that a random 40% of both PYs and INs were subject to reduced (50 Hz) afferent excitatory synaptic input. The subset of deafferented cells was random. We assumed (1) that different diseases accompanied by varying degrees of pathological changes are modeled by different values of deafferentation degree D and (2) that disease progression corresponds to increasing values of D. Although a direct mapping from values of D to morphometric neuropathology data are not feasible, we used data on cortical cell loss in brains from patients with Creutzfeldt-Jakob disease (CJD) to determine a rough estimate of the upper limit of deafferentation to be considered [up to 50% cell loss as a function of duration of dementing phase, in our model corresponding to 50% loss of inputs (input reduction from 100 to 50 Hz) in all cells, D = 100% (Masters and Richardson, 1978)]. We chose CJD data for this estimate because EEGs from patients with sporadic Creutzfeldt-Jakob disease exhibit periodic activation that ranges from frontal rhythmic delta activity to periodic sharp wave complexes as function of disease stage (Wieser et al., 2006).
We simulated four different random deafferentation patterns for each deafferentation degree. Firing rate analysis was based on the average of these four simulations. In all simulations, the firing rate of the network was computed every 4 s by averaging over all PY spikes in the preceding 4 s interval. AMPA conductances G between PYs were then updated at these checkpoints by adding ΔG = ε (f* − f)G, where ε is the scaling rate (here ε = 0.05), f* is the target firing rate (5 Hz), and f is the current average firing rate. In a subset of simulations (see Fig. 4), we applied the same homeostatic scaling rule but with opposite sign to the inhibitory conductances onto PYs (homeostatic downregulation of synaptic inhibition). Our homeostatic scaling rule ensured multiplicative scaling of the synapses. In the limit in which the activity mismatch f* − f is small, our homeostatic plasticity rule represents a discrete-time implementation of a first-order exponential recovery scheme. If not stated otherwise, time points in the figures correspond to the checkpoints in which the firing rate was computed and the synaptic conductances were updated based on this homeostatic scaling rule. The choice of the nature of this updating rule was motivated by (1) the computational impossibility to implement the biological timescale for homeostatic scaling in such a model (hours to days) and (2) the fact that homeostatic regulation of synaptic conductances is sufficiently slower than the effect of a change in conductance on the firing behavior. We therefore separated the two timescales and approximated the slow synaptic regulation with a discrete-time update scheme. Intervals between update time points can therefore be considered of arbitrary length in time and are only meaningful compared with other simulations with the same update rule (e.g., different deafferentation degrees).
Data analysis.
Simulated local field potential (lfp) was computed by averaging PY spike trains smoothed with a Gaussian kernel (SD of 20 ms). For presentation purposes, the lfp trace was filtered with a fifth-order Butterworth bandpass filter (0.5 and 400 Hz). The spectrogram of the lfp trace was determined by the Matlab (MathWorks, Natick, MA) function specgram with 5 s window size and 2 s overlap between windows. The spectrogram was smoothed by 10-fold oversampling and two-dimensional linear interpolation. Averaged cross-correlograms were calculated by computing the cross-correlation between all possible pairs of 40 randomly selected PY smoothed spike trains. Mean cross-correlograms are shown in black, and the corresponding SEM are shown in gray. Burst index quantifies the relative fraction of interspike intervals (ISIs) shorter than 50 ms in all PYs for a given 4 s interval. This definition of burst index differs from more commonly definitions that require intraburst interspike intervals to be shorter than 10 ms. Our choice is motivated by the longer duration of intraburst interspike intervals in our simulations. In all cases, we checked for bimodality of the interspike interval distribution by visual inspection to ensure that nonzero burst index indeed reported bursting and not high-frequency tonic firing. The spike-triggered average afferent and recurrent excitatory conductances were determined from the corresponding conductance time courses in all PYs of the network model for a 10 s interval. For each spike in every PY, a 500 ms window before spike occurrence was extracted from the corresponding conductance time courses. The average spike-triggered conductance was then determined by averaging these waveforms. The spike-triggered total excitatory conductance was determined by adding the spike-triggered afferent and recurrent spike-triggered synaptic conductances. If not noted otherwise, the figures show mean values (error bars indicate SEM) that were determined by pooling the four simulations with different random deafferentation patterns for a given degree of deafferentation. All analysis was performed with custom-written Matlab (MathWorks) routines.
Results
Homeostatic plasticity of excitatory synapses may control overall activity levels in neural circuits. Decrease in activity level caused by partial loss of excitatory input can be compensated by synaptic scaling of the remaining excitatory synapses. We studied how a dynamic homeostatic scaling rule for recurrent excitatory synapses reorganizes network dynamics in case of input loss by partial deafferentation. In our computational model of a cortical circuit, homeostatic synaptic scaling succeeded in recovering target average frequency of pyramidal cells for all deafferentation degrees tested (D = 10 to 100%). We found, however, substantial reorganization of fine temporal structure of spike trains, network synchronization, and firing rate distributions as a function of deafferentation degree. The interplay between cells with intact input and deafferented cells shaped the dynamics of the recovery from deafferentation. Although in reality homeostatic plasticity occurs on the timescale of many hours to days, we applied the homeostatic scaling at regularly spaced time points (4 s intervals) because of technical limitations in simulation length. The underlying approximation of this separation of timescales is well justified by the fact that the effect of changes in synaptic conductances on firing rates is immediate, whereas homeostatic scaling triggered by changes in activity levels occurs on a much slower timescale (see Materials and Methods). Time as denoted on the abscissa in the following figures represents the time points at which the homeostatic scaling rule was applied. Because of the compression of the homeostatic plasticity timescale, these “time” values have no absolute meaning but rather serve as a reference for comparison of different recovery time courses.
Random partial deafferentation can cause periodic network activation
Severe deafferentation (D = 90%) of a cortical network model composed of PYs and INs caused an initial drop in activity level (Fig. 1, arrowhead) with subsequent recovery of activity by upregulation of recurrent synaptic excitation (membrane voltages of PYs and INs color coded in Fig. 1A; lfp in B; smoothed time-dependent frequency decomposition in C). Before deafferentation, the network exhibited asynchronous firing (low-amplitude lfp, mean ± SD firing rate of PYs 5.03 ± 3.77 Hz) in response to independent Poisson input to all PYs and INs (frequency, 100 Hz). After deafferentation, as recovery of activity level by synaptic scaling progressed, slow periodic activation of the network occurred (oscillations in lfp in Fig. 1B; red band around 1 Hz in spectrogram of lfp in C). Eventually, average PY firing rate recovered to 5.02 ± 2.83 Hz (mean ± SD). In contrast to before deafferentation, individual PYs fired bursts of action potentials (Fig. 2A, zoom in of network activity after scaling from Fig. 1A; sample membrane voltage trace in B; dominant peak for small interspike intervals in D “after scaling”). The different temporal fine structure of the network activity before deafferentation and after synaptic scaling is reflected in the averaged cross-correlograms (Fig. 2C, average in black, gray lines delimit SEM). Before deafferentation, network activity was asynchronous (flat trace). After synaptic scaling, the cross-correlogram revealed periodic modulation of firing activity with an approximate period of 950 ms (measured from central peak to first sideband peak, oscillatory cross-correlogram). Synaptic scaling changed not only the spiking patterns but also the distribution of firing rates across PYs. Before deafferentation, the distribution of resting potentials defined a decaying distribution of firing rates with a peak for cells with firing rate below 1 Hz (Fig. 2E, left). After scaling, the peak in distribution of firing rates was shifted to higher rates (Fig. 2E, right).

Network reorganization after partial deafferentation (deafferentation degree D = 90%). A, Activity map of all 80 PYs (top) and 20 INs (bottom). Cool and hot colors indicate hyperpolarization and depolarization, respectively. Deafferentation with consecutive drop in network activity (arrowhead). Recovery of target firing rate by homeostatic scaling resulted in prominent periodic network activation. B, Simulated lfp. Same timescale as in A. High-frequency activity with low amplitude before deafferentation. Drop in lfp level after deafferentation (arrowhead). Recovery of activity level is characterized by slow high-amplitude lfp oscillations. C, Spectrogram of lfp. Same timescale as in A. Cool and hot colors indicate low and high power on arbitrary logarithmic scale, respectively. Drop in power after deafferentation (arrowhead). Prominent peak in power at approximately 1 Hz after homeostatic scaling (dark red band).

Network activity after deafferentation (D = 90%) and homeostatic synaptic scaling (zoom of Fig. 1A, after synaptic scaling). Average firing frequency across network matched target rate f* = 5 Hz. A, Strong periodic activation of entire PY population (bands of lighter color). B, Sample membrane voltage trace exhibits low-frequency bursting. Some action potentials are truncated as a result of finite sampling of membrane voltage. Same timescale in A and B. C, Average cross-correlogram for PY activity before deafferentation (flat line) and after homeostatic scaling triggered by deafferentation (oscillatory trace). D, Histogram of ISIs before deafferentation (left) and after homeostatic scaling in response to deafferentation (right). Dominant peak for short ISI after scaling indicates bursting in individual cells. E, Firing rate distribution across the network. Peak for very low firing rate before deafferentation (left) is shifted to higher frequencies and less prominent after synaptic scaling (right).
We expected the steady-state network dynamics after synaptic scaling to depend on the deafferentation degree. Therefore, we simulated the same network for different deafferentation degrees (Fig. 3A, rastergrams; B, average cross-correlograms; network dynamics after synaptic scaling). For weak deafferentation (D = 20%, top row), we found no oscillatory firing (flat average cross-correlogram). Very mild oscillatory modulation of instantaneous firing rates occurred for moderate deafferentation (D = 40 and 60%, middle rows). For strong deafferentation (D = 80 and 100%, bottom rows), however, we observed pronounced synchronized periodic network activation. In summary, cross-correlograms progressively showed more oscillatory structure with growing period for increasing deafferentation degree (Fig. 3B, from top to bottom). Thus, the more severe the deafferentation, the slower and the more pronounced the periodic activation patterns of the network became. Interestingly, the oscillatory structure of the cross-correlograms did not increase linearly with deafferentation degree. Instead, we observed a rather abrupt transition between D = 60% (low-amplitude cross-correlogram) and D = 80% (high-amplitude cross-correlogram).

A, Rastergrams for PYs after homeostatic scaling in response to different deafferentation degrees (D = 20 to 100% from top to bottom). B, Average cross-correlograms show very little oscillatory activity up to D = 60%, but for D = 80 and 100%, strong pronounced oscillations occurred. Oscillation frequency decreased for increasing degree of deafferentation.
In principle, the oscillatory network activity that we found in deafferented networks after homeostatic scaling may not occur if homeostatic scaling of synaptic inhibition is included in the model. Specifically, homeostatic downregulation of inhibitory synapses may reduce the required scaling of AMPAergic PY–PY synapses and therefore prevent the occurrence of network-wide bursts. However, simulations in which we included homeostatic downregulation of inhibition revealed only a very small reduction in PY–PY scaling (<3%) and no change in macroscopic network dynamics (data not shown). To exclude the possibility that this absence of a substantial effect is the result of the relatively weak synaptic coupling between PYs and INs in our model, we performed additional simulations of a network with matched firing behavior before deafferentation but stronger coupling between PYs and INs (gAMPA(PY–PY) = 6.0 nS, gNMDA(PY–PY) = 0.6 nS, gAMPA(PY–IN) = 5.0 nS, gNMDA(PY–IN) = 0.50 nS, gGABA(IN–PY) = 35.0 nS, and gPY = 0.7 nS and gIN = 0.9 nS for PYs and INs) (Fig. 4). A comparison between networks with and without inhibitory homeostatic scaling revealed a modest difference in scaling of PY–PY synapses (Fig. 4A, differences significant for all deafferentation degrees, p < 0.05). Nevertheless, the overall macroscopic reorganization into a network with pathological network-wide bursts remained the same in both cases (Fig. 4B,C, for D = 100%). This limited effect is caused by the reduced firing rate of the INs after deafferentation attributable to loss of afferent excitatory input. Whereas the PYs recovered their firing rate by upregulation of the PY–PY connection strength, the INs only partially recovered their firing rate. Given this limited role of homeostatic scaling of inhibition in the mechanisms studied here, the remainder of this study was performed with networks that did not include homeostatic scaling of inhibition.

A, Homeostatic scaling factor of PY–PY AMPAergic synapses as a function of deafferentation degree in networks without (gray bars) and with (black bars) homeostatic downregulation of synaptic inhibition. B, PY network rastergram after homeostatic scaling of excitation. C, PY network rastergram after homeostatic scaling of both excitation and inhibition.
Roles for deafferented and intact cells in recovery of target activity level
So far, we have discussed the “steady-state” network dynamics after recovery of the target firing rate by synaptic scaling. Now, we consider the temporal evolution of activity during synaptic scaling (Fig. 5). The following key observations become evident. (1) Firing rates as a function of time for increasing degrees of deafferentation (color coded in Fig. 5A, D = 20 to 100%, panels from left to right) show that recovery of overall activity levels by synaptic scaling is shaped by cells with high firing rate before deafferentation (continuous horizontal bands of hot colors in Fig. 5A). After synaptic scaling, nonzero firing rates occurred preferentially in cells in close proximity to these highly excitable cells. (2) For strong degrees of deafferentation (D = 80 and 100%), network-wide activation occurred and cells that were silent before deafferentation became active (decreased sparseness compared with before deafferentation). The observed reorganization of global network dynamics in response to partial deafferentation by homeostatic synaptic scaling is a result of interplay between the excitability of deafferented and intact PYs cells. Therefore, we below compare the initial firing rates α before deafferentation (measure of excitability because all cells received the same amount of afferent input), firing rates β immediately after deafferentation, and firing rates γ after recovery of target activity level by synaptic scaling (time points indicated in Fig. 5A).

A, Firing rates as a function of time for 80 PYs (panels correspond to increasing degrees of deafferentation from left to right). Hot colors indicate high firing rates; cool colors indicate low firing rates. For severe deafferentation, network-wide activation occurred (D = 80 and 100%). Arrowheads indicate three time points considered in B and C: firing rates α before deafferentation, firing rates β shortly after deafferentation, and firing rates γ after homeostatic scaling in response to deafferentation. B, Scatter plots with firing rates β immediately after deafferentation as a function of firing rates α before deafferentation. Red squares represent firing rates of deafferented cells, and blue triangles indicate intact cells (not subject to deafferentation). Black lines denote identity lines (no change in firing rate caused by deafferentation). Intact cells showed growing decreases in firing rate for progressively more severe degrees of deafferentation (from left to right) as indicated by blue triangles falling below identity line. Deafferented cells fell silent if they exhibited low firing rate before deafferentation (red squares on abscissa). C, Same representation as in B but with firing rates γ after synaptic scaling on ordinate. Intact cells show recovery and overcompensation (blue triangles above unit line). Deafferented cells partially recovered their previous firing rate α before deafferentation (red squares closer to unity line than in B). For severe degrees of deafferentation, however, less excitable cells with low firing rates before deafferentation showed overcompensation (red squares above unity line for D = 80 and 100%).
We next established the relative contributions of deafferented and intact cells to the recovery of the target firing rate as function of their excitability. In theory, it is conceivable that reorganization is mediated by (1) intact cells increasing their firing rate, (2) deafferented cells recovering their firing rate, or (3) a combination of the latter two mechanisms in any temporal order. To test which hypothesis is correct, we separated intact (blue triangles) from deafferented (red squares) cells and plotted firing rate β (immediately after deafferentation) and firing rate γ (after synaptic scaling), respectively, as a function of initial firing rate α (before deafferentation) (from left to right in Fig. 5B,C, from 20 to 100% deafferentation; β as a function of α in B; γ as a function of α in C). Intact cells (blue) suffered little loss of activity for mild to moderate deafferentation (firing rates β crowded along the diagonal). For stronger deafferentation, however, rates of PYs with intact input fell consistently below the diagonal (Fig. 5B, D = 80%). Quite in contrast, the drop in firing rate for deafferented cells (shown in red) depended on their intrinsic excitability (i.e., firing rate α before deafferentation). Specifically, PYs with low α turned silent in response to deafferentation and thus clustered on the x-axis. Other PYs (i.e., those with relatively high firing rate α before deafferentation) also suffered from a decrease in firing rate but stayed active after deafferentation. After homeostatic scaling (Fig. 5C), PYs with intact afferent input assumed firing rates γ above initial firing rates α for all degrees of deafferentation (blue triangles above diagonal). For deafferented PYs, we found two distinct patterns of behavior depending on the degree of deafferentation. Relatively little increase in firing rates caused by synaptic scaling occurred in case of weak to moderate deafferentation (by comparison of B and C in Fig. 5). In case of strong deafferentation, however, we observed complete recovery of initial firing rates for some fraction of deafferented cells (red squares on and above diagonal for D = 80 and 100%).
Next, we quantified the temporal dynamics of the activity levels of intact and deafferented cells during recovery of target firing level by homeostatic scaling. Specifically, we considered the case of 80% deafferentation in more details (Fig. 6A,B) to determine the time course and relative order of changes in firing frequency in (1) intact cells (blue triangles), (2) deafferented cells with initial firing frequency α below target rate f* (α < f*, red diamonds), and (3) deafferented cells with initial firing frequency α above target firing rate f* (α > f*, red squares). We defined the frequency shift Δf for a given time point t as the arithmetic difference between the firing rate at that time point t and the initial firing rate α before deafferentation. In essence, this measure indicates the change in activity level relative to baseline, i.e., before deafferentation. First, we computed the frequency shift for the time point immediately after deafferentation (Δf = β − α) to quantify the drop in activity by deafferentation (histogram in top row in Fig. 6A). Second, we determined the frequency shift for a time point after homeostatic scaling (Δf = γ − α), which described the change in firing rate induced by deafferentation and subsequent reorganization by homeostatic scaling (bottom row in Fig. 6A). Specifically, values of Δf larger than zero indicate that the neurons were more active than they had been before deafferentation, and values smaller than zero indicate that the neuron did not recover its pre-deafferentation firing rate. As expected, the peaks of these frequency-shift distributions moved toward more positive values as time and therefore homeostatic scaling progressed (by comparison of top to bottom row in A). To gain a more complete picture of the reorganization dynamics, we then extended this analysis by computing the frequency shift Δf for all PYs for each time point at which the homeostatic rule updated the synaptic connections. We fitted the histograms of Δf values at each time point with normal distributions and then determined the respective mean values. These mean values were then plotted as a function of time (D = 80% in Fig. 6B; D = 40% in Fig. 6C). These time courses of mean values determined from frequency-shift histogram fits exhibited sigmoid shapes for all three subpopulations (Fig. 6B,C). These plots show how the three subpopulations contribute to the recovery of the target firing rate at different time points during the recovery process. We here quantify the temporal order of these contributions by comparing the time point at which half of the frequency shift has occurred (half-maximum of sigmoid curves). Specifically, half-maximum frequency shift occurred almost simultaneously for cells with intact input (blue triangles) and deafferented cells with α > f* (red squares, more active cells). Only with a marked delay, however, deafferented cells with α < f* (red diamonds, less active cells) increased their firing rates (Fig. 6B). Thus, the more excitable deafferented cells (α > f*) contributed to the early increase in activity levels after deafferentation and the less excitable cells (α < f*) mostly mediated the later increase in activity levels. In contrast, for D = 40% (Fig. 6C), recovery of target activity level was almost exclusively mediated by intact PYs and deafferented PYs with α > f* (blue triangles and red squares, respectively). In summary, instead of further upregulating the firing rate of the more excitable cells in more deafferented networks (e.g., D = 80%), the scaling process recruited PYs that were silent or displayed only low activity level before deafferentation. Therefore, reorganization of cortical network dynamics by synaptic scaling consists of several distinct processes occurring in a well defined temporal sequence as a function of the severity of partial deafferentation.

Recovery time course of firing rates as a function of their input (intact in blue, deafferented in red) and of their excitability (low excitability with initial firing rate α below target firing rate f*, high excitability with initial firing rate α above target firing rate f*). Frequency shift represents the arithmetic difference between firing rate at a given time point and the firing rate α before deafferentation (negative and positive values indicate undercompensation and overcompensation, respectively). A, Distribution of frequency shifts β − α after deafferentation (top row) and frequency shifts γ − α after scaling (bottom row). Lines indicate Gaussian distribution fits. Both intact (left) and less excitable deafferented (middle) cells showed overcompensation (peak in distribution for positive frequency shifts), whereas more excitable cells exhibited undercompensation (right). B, Same analysis as in A for all time points after deafferentation (time point 6) to steady-state after homeostatic scaling (time point 20). Plots show mean frequency shifts determined by normal distribution fits as in A. Vertical dashed lines indicate 50% of maximal shift determined from logistic functions fits. Both intact (blue triangles) and more excitable (red squares) cells reached 50% recovery at very similar time points. However, less excitable deafferented cells (red diamonds) reached 50% recovery at a later time point (D = 80%). C, In case of D = 40%, however, less excitable cells contributed very little to recovery of the target firing (flat line for red diamonds).
Transition point to pathological network-wide activation
So far, we have established that the recruitment of less excitable deafferented cells constituted a major difference between moderate and severe deafferentation by comparison of the time courses of the respective mean frequency shifts for different deafferentation degrees. We therefore expected to find different firing rate distributions after synaptic scaling as a function of deafferentation degree. Specifically, our previous analysis suggests that strong deafferentation degree will reduce the fraction of silent cells (decreased sparseness). We quantified the time course of firing rates on a cell-by-cell basis by plotting the time-dependent frequency histograms for D = 20 to 100% (Fig. 7A, from left to right, black boxes indicate data replotted in B and C, deafferentation time point marked by arrowhead). We had chosen the initial distribution of PY membrane voltages such that, for intact afferent input, a limited fraction of cells remained silent (hot colored patches in top left corners in Fig. 7A). Deafferentation transiently increased the number of silent cells. Indeed, whereas for mild to moderate deafferentation the fraction of silent cells recovered (in fact, in some cases to values higher than before deafferentation), more severe deafferentation qualitatively changed the firing rate distribution of the network such that all cells were active, resulting in a shift of the peak of the firing rate distribution as a function of time (Fig. 7A, rightmost panel). We next considered the time course of silent cells (split into two subpopulations, cells with intact input and deafferented cells, in blue and red, respectively, in Fig. 7B). In case of mild deafferentation, we observed a quite linear decrease in silent cells during synaptic scaling, mostly mediated by intact cells (blue band becomes narrower and red band assumes constant width as time progresses). This indicates that the network recovered its target activity level in part by activating cells with intact input that were silent right after deafferentation. Yet, for more severe deafferentation, we found a strongly nonlinear time course of fraction of silent cells. In particular, the fraction of silent cells (average firing rate below 1 Hz) sharply increased after deafferentation but then decreased to values lower than before deafferentation (horizontal slice in Fig. 7A; time courses in B). Thus, most of the silent cells, including the deafferented ones, became active during recovery of target activity level. Accordingly, frequency histograms after synaptic scaling exhibited a decrease in fraction of silent cells and a sharpening of the distribution in case of severe deafferentation (vertical slice in Fig. 7A; distributions in C).

A, Distribution of firing rates as a function of time for different deafferentation degrees (from 20 to 100%, from left to right). Black boxes indicate data that is replotted in B and C. Arrowhead indicates time point of deafferentation. B, Fraction of silent cells (firing rate below 1 Hz) as a function of time (intact cells in blue, deafferented cells in red). C, Firing rate histograms after recovery of target firing rate by synaptic scaling.
The average firing rate of the network as a function of time (Fig. 8A) permitted a similar classification into more linear and more nonlinear time courses (shown in gray and black, respectively). Specifically, for deafferentation degrees up to 60% (curves in gray), the network firing rate recovered with approximately even rate with only modest fluctuations in slope. In case of more severe deafferentation (D = 70% and higher), however, an initially relative flat time course was followed by an epoch of high slope recovery (curves in black). In other words, we found linear firing-rate time course for weak to moderate deafferentation and nonlinear firing-rate time course for strong deafferentation. The deafferentation degrees for which this strong nonlinearity occurred corresponded to the values of deafferentation for which we found pronounced network oscillations after synaptic scaling (Fig. 3). These two distinct recovery modes were not a simple consequence of how the PY–PY coupling was scaled by the dynamics of the homeostatic plasticity rule. Time courses of the recurrent AMPA conductance on PYs were well fitted with sigmoids for all degrees of deafferentation (Fig. 8B) independent from whether the firing rate time course was linear or not. Therefore, the scaling rule itself was not the direct source of the nonlinear recovery behavior for severe deafferentation. Rather, the time course of the fraction of silent cells (Fig. 8C, summary plot from data in Fig. 7B, for D = 10 to 100%) exhibited strong similarity to the time courses of the average network firing rate (Fig. 8A). A scatter plot of average network firing rates and fraction of silent cells indeed exhibited tight correlation (Fig. 8D). Thus, the two different network behaviors for mild/moderate and severe deafferentation, respectively, are driven by the differential recruitment of additional cells that resulted in decreased fraction of silent cells (i.e., sparseness) after severe deafferentation.

A, Average PY network firing rate as a function of time (deafferentation at time point 5). Each line corresponds to a deafferentation degree from D = 10 to 100%. The higher D, the stronger the initial drop of activity after deafferentation at time point 5. Linear recovery time course in gray (D = 10… 60%), nonlinear recovery time course in black (D = 70… 100%). B, Time course of normalized PY–PY coupling (crosses, data; lines, logistic function fits) for D = 10 to 100%. Gray and black shading as in A. More severe deafferentation causes more rapid scaling of recurrent excitatory synapses. C, Time course of fraction of silent cells for D = 10 to 100%. Gray and black shading as in A. Similar clustering into linear and nonlinear time course as in A. D, Average network frequency as a function of fraction of silent cells. Scatter plot of all time points in A and C. Gray shading as above. E, Burst index (fraction of ISIs smaller 50 ms) as a function of time for D = 10 to 100%. Sample membrane voltage traces for D = 10% (single spikes) and D = 100% (bursts of spikes), respectively (2 s time window). F, Steady-state value of burst index (dark filled circles) and fraction of silent cells (open squares) as a function of D.
We then determined how this finding related to the observed bursting for strong deafferentation (Fig. 2) by computing the relative fraction of interspike intervals smaller than 50 ms (burst index) as a function of time after deafferentation for D = 10 to 100%. Similar in time course to the fraction of silent cells, burst index strongly increased for D > 60% (Fig. 8E). A comparison of steady-state values of fraction of silent cells and burst index after recovery of target firing rate as a function of deafferentation shows that, as the fraction of silent cells falls below the value before deafferentation, cells in the network fire bursts of action potentials (Fig. 8F). We therefore found a transition point that determines whether the network recovered with limited pathological consequences (no bursting, sparseness maintained, no pronounced oscillation with synchrony) or not. In our case, this transition occurred between D = 60 and 70%.
Effect of homeostatic plasticity on information transfer
We expected pathological reorganization by homeostatic plasticity after severe deafferentation to have a negative impact on the network ability to respond to afferent input. We quantified and compared the impact of afferent and recurrent excitatory input on spiking in PYs by calculating the spike-triggered average afferent and recurrent excitatory conductance. This measure allows for a direct quantitative comparison of the role of afferent and recurrent excitation in driving spiking of postsynaptic cells. For example, if recurrent excitation is sufficiently weak such that the afferent input alone determines when a postsynaptic cell spikes, we would expect, on average, a flat, low-amplitude spike-triggered conductance for recurrent excitation and a pronounced transient increase in spike-triggered afferent conductance. In other words, a comparison of the peak values of the spike-triggered conductances serves as a measure for the relative importance of afferent and recurrent excitation in determining the network dynamics. Here, afferent excitation was dominant before deafferentation (Fig. 9A, left; total excitatory conductance in black, afferent excitatory conductance in red, recurrent excitatory conductance in blue) and after synaptic scaling in response to deafferentation for moderate degrees of deafferentation (Fig. 9A, middle, D = 40%). Recurrent excitation, however, dominated after synaptic scaling in case of more severe deafferentation (Fig. 9A, right, D = 80%). Integrated spike-triggered total excitatory conductances for different degrees of deafferentation were fairly constant (Fig. 9B, black line) The more deafferented the network was, however, the stronger the integrated recurrent excitatory conductance was (Fig. 9B, blue line). A comparison of the peak values further emphasized the critical degree of deafferentation between 60 and 70% (Fig. 9C). At this point, the peak recurrent excitation exceeded the peak afferent excitation. Thus, pathological network reorganization was mediated by a shift between afferent and recurrent excitation that resulted in poor information transmission in case of severe deafferentation because the influence of the afferent input on spiking was very limited (low-amplitude spike-triggered conductance). This shift, therefore, may explain deficiency of normal cortical function as a result of brain disorders associated with cell loss.
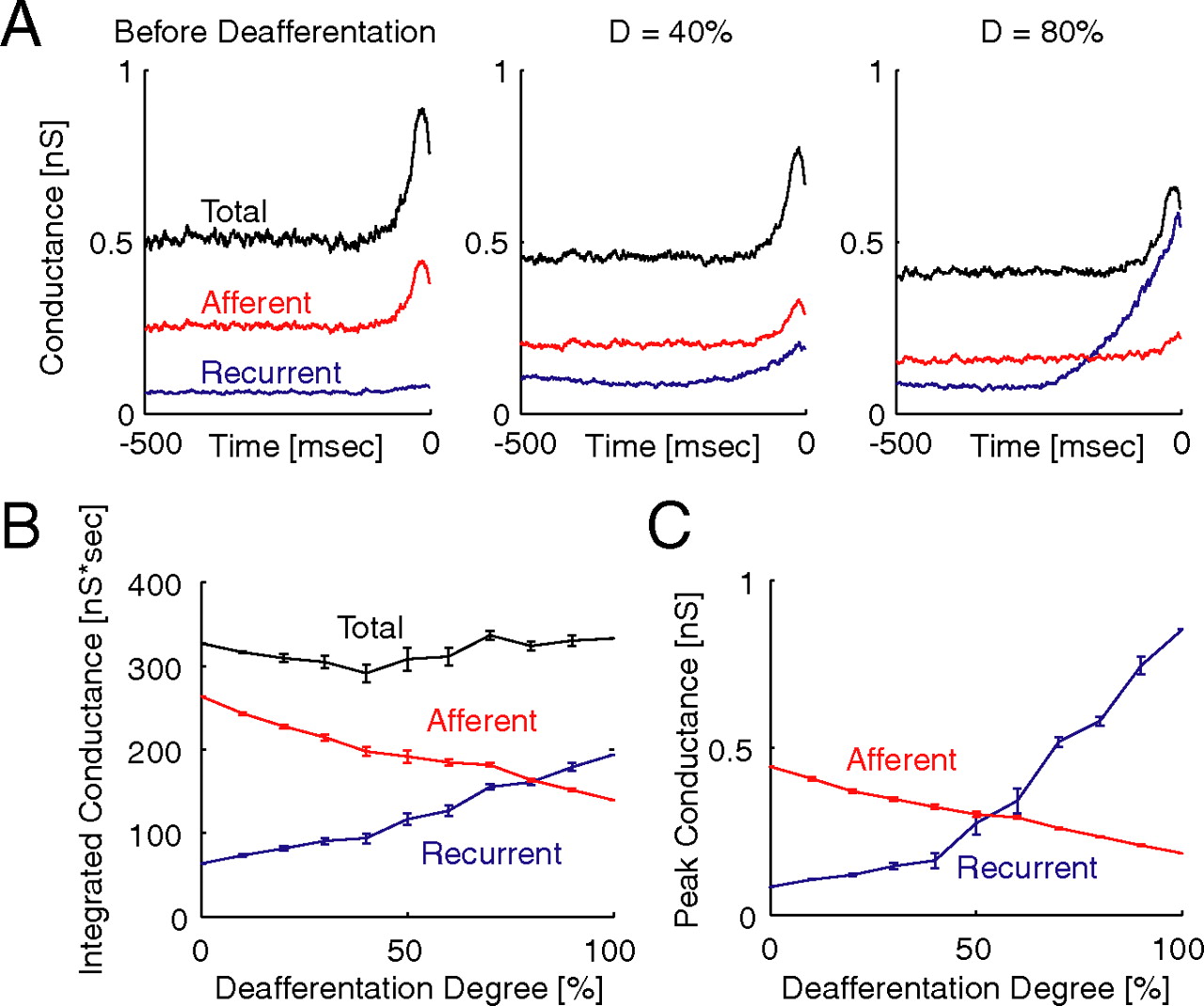
Spike-triggered conductances. A, Afferent excitatory (red), recurrent excitatory (blue), and total (black) spike-triggered synaptic conductance for PYs (left, before deafferentation; middle, D = 40%; right, D = 80%). B, Integrated spike-triggered afferent (red), recurrent (blue), and total (black) excitatory conductance as function of deafferentation degree D. C, Peak values of spike-triggered afferent (red) and recurrent (blue) excitatory conductance as function of deafferentation degree D.
Discussion
Homeostatic plasticity describes the regulation of synapses and intrinsic properties to counteract changes in activity levels and to maintain overall stability of synaptic strength (Turrigiano et al., 1998; Turrigiano and Nelson, 2004; Turrigiano, 2007). Here, we studied homeostatic plasticity as a putative mechanism for cortical network reorganization that occurs during CNS disorders associated with neural dysfunction. We assumed (1) that the loss of inputs that occurs in cortical disorders with structural damage is random, (2) that a homeostatic rule scales the recurrent excitatory synapses on PYs to match the average firing rate of the network before disease onset, and (3) that disease severity can be modeled by varying fractions of deafferented cells. On the basis of these assumptions, we simulated cortical network models subject to different deafferentation degrees and found a critical threshold for pathological network reorganization. For deafferentation more severe than this critical value, we observed periodic network-wide discharges with bursts of action potentials in individual cells after homeostatic scaling of recurrent excitatory synapses. Our findings therefore represent an unexpected and seemingly paradoxical effect of homeostatic plasticity. In fact, the target frequency of the whole network was indeed recovered in our model, but the spike timing was substantially altered such that there were periods of high activity interleaved with epochs of relative quiescence in case of severe deafferentation (periodic EEG complexes). As a result of this pathological network reorganization, representation of afferent input in the spiking of the network was severely diminished because the network dynamics were dominated by recurrent excitation (Fig. 9).
Computational network models of homeostatic plasticity have almost uniquely focused on the role of synaptic scaling in regulating and stabilizing overall synaptic excitability in presence of Hebbian learning (Rabinowitch and Segev, 2006a,b; Lazar et al., 2007; Toyoizumi et al., 2007; Triesch, 2007). Computational models of tinnitus suggested involvement of homeostatic plasticity (Dominguez et al., 2006; Schaette and Kempter, 2006), but there has been little work that examines the role of homeostatic plasticity in more realistic, conductance-based models of cortical networks. In a previous computational modeling study (Houweling et al., 2004), cortical deafferentation was simulated to examine homeostatic plasticity as a potential cause of posttraumatic epilepsy. A recent study with cultured hippocampal slices provided additional support for this hypothesis (Trasande and Ramirez, 2007). In contrast to this previous modeling work, we have now studied the time course of network reorganization after partial deafferentation to understand the specific roles of intact and deafferented cells as a function of time after disease onset. Because we here studied disorders of the cortex that incapacitate a presumably random fraction of neurons, the partial deafferentation scheme is different from the one used previously to investigate posttraumatic epilepsy (Houweling et al., 2004) in which all cells in the network were subject to the same degree of reduction in input.
A priori, the heterogeneity of input levels introduced by partial deafferentation permits two orthogonal strategies of recovering target network activity levels. In the first scenario, intact cells carry the burden of recovering target activity levels by increasing their firing rate above their initial activity level before partial deafferentation. In the second scenario, deafferented cells recover their firing rate. In our simulations, we found the two scenarios to occur in a well defined temporal sequence. In case of mild to moderate deafferentation, we only observed concurrent upregulation of firing rates of both intact cells and more depolarized deafferented cells. Only in case of severe deafferentation, less excitable deafferented cells also became active. Thus, network reorganization in response to partial deafferentation is not a linear process. Rather, we found a nonlinear regimen that essentially mediated a threshold for pathological reorganization in case of more severe deafferentation.
The goal of our work is to provide hypotheses to experimentalists and clinicians about the underlying mechanism of aberrant brain activity in response to CNS disorders. As for any computer simulation, the conclusions drawn are inherently limited by the accurateness and sophistication of our model. We purposefully did not attempt to closely match any specific experimental dataset to keep the model as general as possible. In particular, we tried to limit model complexity by focusing on aspects of real biological circuits that we deemed relevant for the hypotheses we aimed to investigate. As a result, we neglected several important features of real biological networks. In particular, our model is restricted to a single class of inhibitory interneurons (fast-spiking response pattern in response to simulated direct current injection) and therefore omits slower spiking inhibitory interneurons that target the dendritic domain of pyramidal cells (Soltesz, 2006). We consider this simplification of the diversity of interneurons justified by the fact that we determined synaptic inhibition to only play a secondary role in the pathological reorganization behavior shown here (Fig. 4). We chose to use fast-spiking interneurons because of their distinctive role in modulating pyramidal cell spiking by their somatic axonal arborization and presumed role in controlling pyramidal cell activity level (Freund, 2003).
In summary, we used a modified version of a standard cortical network model that consisted of locally connected, conductance-based pyramidal cells and inhibitory interneurons. Nevertheless, two novel aspects of our model deserve closer scrutiny. First, novelty in our model was the choice of homeostatic scaling rule. Most likely, homeostatic plasticity describes a range of different phenomena at different spatial scales and with different expression loci (Turrigiano, 2007). Activity levels could be sensed and regulated at the synapse, cell, or network level. We modeled homeostatic plasticity at the network level (e.g., mediated by a diffusible factor as in the study by Stellwagen and Malenka, 2006) by designing a simple rule that prescribed incremental scaling of synaptic conductances proportional to their size and to the mismatch between target and current average frequency. Exploratory simulations indicated that more local homeostatic rules are likely to result in different patterns of network reorganization after partial deafferentation. In vitro studies of neural cultures exposed to activity blockage showed concurrent upregulation of AMPA synapses and downregulation of GABAA synapses (Turrigiano et al., 1998; Kilman et al., 2002; Mody, 2005). Little is known, however, about homeostatic scaling of inhibition in vivo (Mody, 2005) (but see Echegoyen et al., 2007). Also, we did not find the pathological network reorganization to be qualitatively affected by homeostatic downregulation of synaptic inhibition in our model (Fig. 4). Second, novel design choice was the random pattern of partial deafferentation as a model of the effect of cortical disease on neural firing. This choice is based on the phenomenological approximation that a broad class of CNS disorders cause diffuse random cell loss and thus partial deafferentation of postsynaptic circuits.
Thus, although our model certainly represents a simplification of the underlying processes, it is sufficiently refined and accurate to provide new hypotheses and insights about the pathophysiology of slow periodic discharges in a broad range of CNS disorders affecting cortex. In addition, the mechanisms described here may be applicable to other neurological conditions with primarily subcortical localization. More specific models are required to test whether our findings translate to disorders as chronic pain mediated by deafferentation hypersensitivity (Zimmermann, 2001; Flor et al., 2006; Navarro et al., 2007) and low-frequency oscillations in the subthalamic nucleus–globus pallidus network in patients suffering from Parkinson's disease (Bevan et al., 2002).
There is little in vivo data available on the dynamics of cortical circuits in CNS disease at the cellular and synaptic level. The findings from our modeling work therefore represent predictions that require comprehensive experimental testing. However, there exists ample literature on EEG patterns in patients with CNS disorders (for review, see Niedermeyer, 2005) that we used to qualitatively assess our model simulation results. Our model predicts the following EEG characteristics: (1) slow periodic discharges in severely deafferented networks (Figs. 1, 2), (2) decrease in frequency of periodic discharges during disease progression (Fig. 3), and (3) delayed onset of network reorganization compared with disease progression (Fig. 8). The clinical EEG literature confirms all three predictions. Periodic discharges at low frequencies are a hallmark of many types of CNS disorders associated with transient and permanent cell loss (Niedermeyer, 2005). Delayed onset and recovery of EEG during recovery from CNS disease has been observed for example in case of herpes simplex infections (Upton and Gumpert, 1970; Illis and Taylor, 1972). In additional, albeit indirect, support of our hypothesis, frontal white matter volume and amplitude of resting delta EEG oscillations (1.5–4 Hz) show negative correlation in patients with mild cognitive impairment and Alzheimer's disease (Fernandez et al., 2003; Babiloni et al., 2006).
Little progress has been made in the debate about the nature of periodic discharge types and mechanisms (Chong and Hirsch, 2005; Hirsch et al., 2005; Jirsch and Hirsch, 2007). The variety of structural damages has hindered the development of a unifying theory about the pathophysiology (Yemisci et al., 2003; Gurer et al., 2004; Kalamangalam et al., 2007). We found that homeostatic plasticity is a very likely candidate mechanism underlying pathological reorganization of cortical network dynamics in CNS disease. Hopefully, our findings on reorganization dynamics and critical value of disease progression for occurrence of pathological macroscopic oscillations may serve as a starting point for the development of early intervention approaches to prevent secondary cortical dysfunction caused by homeostatic plasticity.
Footnotes
-
This work was supported by grants from National Institute on Deafness and Other Communication Disorders, National Institutes of Health.
- Correspondence should be addressed to Dr. Maxim Bazhenov, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, CA 92037. bazhenov{at}salk.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}