

Abstract
Members of a subclass of hairy/Enhancer of split [E(spl)] homologs, calledhesr genes, are structurally related to another subclass of hairy/E(spl) homologs,Hes genes, which play an important role in neural development. To characterize the roles of hesr genes in neural development, we used the retina as a model system. In situ hybridization analysis indicated that allhesr genes are expressed in the developing retina, but only hesr2 expression is associated spatially with gliogenesis. Each member was then misexpressed with retrovirus in the retinal explant cultures prepared from mouse embryos or neonates, which well mimic in vivo retinal development. Interestingly,hesr2 but not hesr1 orhesr3 promoted gliogenesis while inhibiting rod genesis without affecting cell proliferation or death, suggesting that the cells that normally differentiate into rods adopted the glial fate by misexpression of hesr2. The gliogenic activity ofhesr2 was more profound when it was misexpressed postnatally than prenatally. In addition, double mutation of the neuronal determination genes Mash1 andMath3, which increases Müller glia at the expense of bipolar cells, upregulated hesr2 expression. These results indicate that, among structurally related hesrgenes, only hesr2 promotes glial versus neuronal cell fate specification in the retina and that antagonistic regulation between hesr2 and Mash1–Math3 may determine the ratios of neurons and glia.
Neurons and glial cells differentiate from common precursors. In Drosophila, the gene glial cells missing (gcm) determines the glial versus neuronal cell fate (Hosoya et al., 1995; Jones et al., 1995; Vincent et al., 1996), whereas in mammals gcm homologs are not involved in gliogenesis but regulate trophoblast cell development in the placenta (Kim et al., 1998; Anson-Cartwright et al., 2000; Schreiber et al., 2000), suggesting that the mechanism of gliogenesis has diverged during evolution. We and others have recently found that in mammals Notch1 and its effectors,Hes1 and Hes5 (Kageyama and Nakanishi, 1997), promote gliogenesis at the expense of the neuronal fate (Furukawa et al., 2000; Gaiano et al., 2000; Hojo et al., 2000; Morrison et al., 2000). Hes1 and Hes5, mammalian homologs ofDrosophila hairy and Enhancer of split[E(spl)], encode a transcriptional repressor with a basic helix-loop-helix (bHLH) domain and negatively regulate neuronal bHLH genes such as Mash1 (Akazawa et al., 1992; Sasai et al., 1992; Ishibashi et al., 1995; Tomita et al., 1996a; Chen et al., 1997). It is likely that this anti-neuronal activity may contribute to promotion of gliogenesis. However,Hes1(−/−)–Hes5(−/−) neural precursor cells (Ohtsuka et al., 1999; Cau et al., 2000) can still differentiate into glia (our unpublished data), and therefore it is likely that there may be other bHLH genes that promote gliogenesis.
For the analysis of such factors that regulate neural development, the retina is an ideal model system. It has only six types of neurons and one type of glia (Müller glia), which all differentiate from common precursors (Cepko, 1999). These retinal cells constitute three cellular layers: the outer nuclear layer (ONL), which contains rod and cone photoreceptors; the inner nuclear layer (INL), which contains bipolar, horizontal and amacrine interneurons, and Müller glia; and the ganglion cell layer, which contains ganglion cells. In addition to the simple structure, the majority of retinal neurons and glial cells differentiate postnatally, and therefore it is easy to examine the process of neuronal and glial fate determination.
To identify new gliogenic genes, we focused on the recently characterized bHLH genes, hesr family (Hey,HRT, gridlock, CHF), which belong to a related but different subclass from Hes genes (Kokubo et al., 1999; Leimeister et al., 1999; Nakagawa et al., 1999;Chin et al., 2000; Zhong et al., 2000). There are three members inhesr family, and each member contains three Hes-related domains: a bHLH domain, an orange domain, which confers the specificity of protein interaction (Dawson et al., 1995), and a domain related to the WRPW sequence, which is known to interact with corepressor (Paroush et al., 1994; Fisher et al., 1996; Grabavec and Stifani, 1996). Here, we found that hesr genes are expressed in the developing retina and that misexpression of hesr2 with retrovirus promotes gliogenesis at the expense of rod genesis, whereas that ofhesr1 and hesr3 does not. Thus, among the three structurally related members, only hesr2 functions as a gliogenic gene and may substitute for Hes1 andHes5 in the retina.
MATERIALS AND METHODS
cDNA library screening. To obtain Hes-related cDNAs, poly(A)+ RNA was prepared from mouse retina of postnatal day (P) 0, P5, and P10 and subjected to reverse transcription (RT) using oligo(dT) as a primer. The primers for hesr1 used in PCR were 5′-ATCAGTGTGCACGCACCTCC-3′ and 5′-TCCAAGGGCACTGGGTACCAG-3′ for upper and lower primers, respectively. The PCR product was used as a probe to screen ∼200,000 clones of cDNA library produced from mouse embryos at day 9.5 (E9.5). One hesr1 clone was obtained. To clone other Hes-related cDNAs, RT-PCR was performed again, and the fully degenerated primers corresponding to the following sequences were synthesized and used: RG(I/L/V)(M/L/V)EK and KLE(K/N)A(D/E) for the upper and lower primers, respectively. The PCR products were used as probes to screen ∼1,000,000 clones of a mouse embryo (E9.5) cDNA library. Four clones for hesr2 and three clones for hesr3 were obtained. The protein coding region of each hesr was subcloned into the EcoRI site of the retroviral expression vector pCLIG (Hojo et al., 2000). Three copies of the Myc sequence (MEQKLISEEDLNE) were tagged in frame at the amino-terminal site of each hesr.
Northern blot analysis and in situ hybridization.Total RNA (15 μg) from mouse retinas were electrophoresed on a formaldehyde/1.2% agarose gel and transferred to a nylon membrane (NEN). The full-length of each hesr cDNA was used as a probe, and hybridization was performed as described previously (Sasai et al., 1992). In situ hybridization analysis was performed as described previously (Shimizu et al., 1995; Takebayashi et al., 1997). Digoxigenin-labeled antisense RNA probes corresponding toEcoRI-SalI (1.8 kb),EcoRI-HindIII (1.6 kb), andEcoRI-HindIII (1.35 kb) fragments of hesr1, -2, and -3, respectively, were synthesized in vitro. These probes were hybridized to 16 μm cryostat sections of embryonal and postnatal retinas.
Retinal explant culture and retrovirus infection. Retroviral DNAs were transfected with LipofectAMINE (Life Technologies, Gaithersburg, MD) into ψ2mp34, an ecotropic packaging cell line (Yoshimatsu et al., 1998). The supernatant was collected 2 d later and concentrated with Centriprep 100 (Amicon), as described previously (Ishibashi et al., 1994; Tsuda et al., 1998). The retinal explant culture and retroviral infection were performed as described previously (Tomita et al., 1996a; Hojo et al., 2000). Briefly, eyes were isolated from E17.5 mouse embryos or P1 neonates and transferred to PBS solution. The neural retina without pigment epithelium was placed on a Millicell chamber filter (Millipore: diameter 30 mm, pore size 0.4 μm) with the ganglion cell layer upward. The chamber was transferred to a six-well culture plate. Each well contained 1 ml of culture medium (50% MEM with HEPES, 25% Hank's solution, 25% heat-inactivated horse serum, 200 μml-glutamine, and 5.75 mg/ml glucose). Explants were cultured at 34°C in 5% CO2, and the medium was changed every other day. Two weeks after infection, retinas were fixed with 4% paraformaldehyde, dehydrated in 25% sucrose, and embedded in OCT compound (Miles), and cryosections (16 μm) were made.
Immunostaining and terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling assay. For immunohistochemistry, sections on slides were preincubated in PBS containing 5% goat serum and 0.1% Triton X-100 for 1 hr and then incubated in 1% goat serum and 0.1% Triton X-100 with the following antibodies: rabbit anti-green fluorescent protein (GFP) (diluted 1:500; Medical and Biological Laboratories), mouse anti-Myc (1:400; Invitrogen, San Diego, CA), mouse anti-vimentin (1:1; Histofine), and mouse anti-glutamine synthetase (GS) (1:200; Chemicon, Temecula, CA). To detect these antibodies, biotinylated anti-rabbit antibody (1:200; Vector Laboratories, Burlingame, CA), fluorescein isothiocyanate avidinD (1:1000; Vector), and Fluorolink Cy3-labeled goat anti-mouse antibody (1:400; Amersham Pharmacia Biotech) were used. Retinal cell types were determined by their morphology, their locations, and the following antibodies: anti-HPC1 (amacrine cells), anti-PKC (bipolar cells), anti-calbindin (horizontal and amacrine cells), anti-glutamine synthetase (Müller glia), anti-vimentin (Müller glia), and anti-rhodopsin (rods). For Ki-67 staining, anti-human Ki-67 antibody (1:100; PharMingen, San Diego, CA) was used. Terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) assay was performed with a detection kit (Boehringer Mannheim, Indianapolis, IN). All pictures were taken by a confocal microscope (Carl Zeiss, Thornwood, NY).
Mash1–Math3 mutant mice. Mash1–Math3double-mutant mice were obtained by crossingMash1(+/−)–Math3(−/−) male andMash1(+/−)–Math3(+/−) female mice (Tomita et al., 2000). Because double-mutant embryos survived until E15.5 but died by E17.5, embryos were harvested at E15.5.
RESULTS
Expression of hesr genes in the developing retina
Expression of hesr genes in the retina was first determined by Northern blot analysis. At P0, when there are many retinal precursors, all hesr genes were expressed at comparable levels (Fig.1A). However, after P3 the expression patterns were different from each other.hesr1 expression was maintained at a relatively constant level until adulthood (Fig. 1A). In contrast, bothhesr2 and hesr3 expression was downregulated at P5 but the former was maintained afterward, whereas the latter became undetectable at P7 (Fig. 1A). Thus, hesr1and hesr2 are expressed by both undifferentiated and differentiated cells, whereas hesr3 is expressed transiently by undifferentiated cells.

Expression of hesr genes in the developing retina. A, hesr1–3 expression is examined by Northern blot analysis. All hesr genes are expressed at comparable levels at P0. Although the level ofhesr1 expression is relatively constant until adulthood,hesr2 and hesr3 expression is decreased at P5; hesr3 expression disappears at P7. Glyceraldehyde-3 phosphate dehydrogenase (G3PDH) cDNA was used as a control. B–J, hesr1–3 expression is examined by in situ hybridization. At E17, allhesr genes are expressed in the ventricular zone (V) (B, E,H). hesr1 and hesr3are also expressed in the ganglion cell layer (GCL) (B,H). At P5, hesr1 expression is observed mainly in the outer and inner regions of the INL, which contain horizontal and amacrine cells, respectively, as well as in the GCL (C). In contrast, hesr2 is mainly expressed in the middle region of the INL, which contains bipolar and Müller glial cells (F). Thus,hesr1 and hesr2 display complementary expression patterns. hesr3 is only weakly expressed in the INL (I). At P10, hesr1is again expressed in the outer and inner regions of the INL (D), whereas hesr2 is expressed in the middle region of the INL (G). In contrast,hesr3 expression is not detectable at P10 (J). Scale bar, 25 μm.
The expression was further examined by in situhybridization. At E17, all hesr genes were expressed in the ventricular zone, which contains common precursors for neurons and glia (Fig. 1B,E,H).hesr1 and hesr3 were also expressed in the ganglion cell layer, which contains projection neurons (Fig.1B,H). At P5, when the ventricular cells were differentiating into neurons and glial cells, which form the INL and ONL, hesr expression was shifted to the INL (Fig.1C,F,I).hesr1 expression was observed mainly in the outer and inner regions of the INL, which contain horizontal and amacrine cells, respectively (Fig. 1C). In contrast, hesr2 was mainly expressed in the middle region of the INL, which contains bipolar and Müller glial cells (Fig. 1F). Thus,hesr1 and hesr2 displayed complementary expression patterns in the INL. At P10, when the majority of retinal cells finished differentiation, hesr1 was again expressed mainly in the outer and inner regions of the INL (Fig.1D), whereas hesr2 was expressed in the middle region of the INL (Fig. 1G). In contrast,hesr3 expression was not detectable at P10 (Fig.1J). Thus, the three hesr genes exhibited different expression patterns in the developing retina.
Misexpression of hesr genes in the developing retina at E17.5
To examine the functions of hesr genes, each gene was misexpressed with retrovirus in the developing retina. We used a replication-incompetent retrovirus, CLIG, which directs GFP expression as a marker from the upstream long terminal repeat (LTR) promoter (Fig.2A) (Hojo et al., 2000). Each hesr gene was inserted at the upstream of the internal ribosomal entry site (IRES) so that hesr and GFP genes can be expressed bicistronically (Fig. 2A). Retinal explants were prepared from mouse embryos at E17.5, and virus was applied on the same day. Two weeks later, by which time most retinal cells finished differentiation, the fate of the virus-infected cells was examined by monitoring GFP+cells.

Misexpression of hesr genes in the retinal explants starting at E17.5. A, Schematic structure of the retroviral vector CLIG. Each hesr cDNA fused with three repeats of the Myc epitope is inserted in the upstream of IRES (arrow). EGFP, Enhanced green fluorescent protein; IRES, internal ribosomal entry site; LTR, long terminal repeat. B–K, Retinal explants were prepared from E17.5 mouse embryos and infected with CLIG-hesr1 (B–D), CLIG-hesr2 (E–G), CLIG-hesr3 (H–J), and CLIG (K). After 2 weeks of culture, the explants were subjected to immunohistochemistry using anti-GFP and anti-Myc antibodies. When infected with CLIG-hesr, almost all virus-infected cells expressed both GFP and Myc. Rod genesis is decreased and gliogenesis is increased (E–G) only when CLIG-hesr2 is applied. Scale bar, 25 μm.
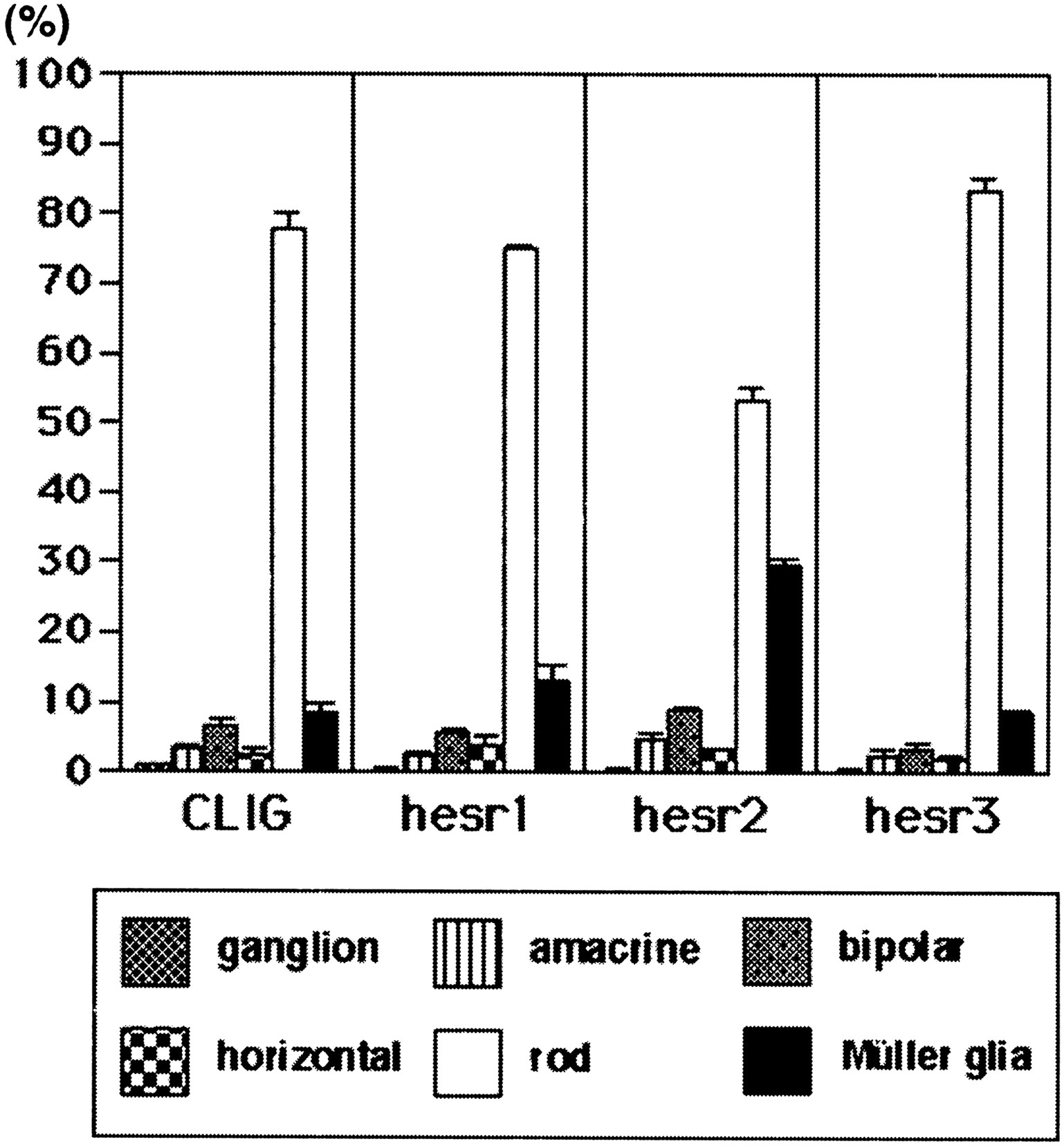
When the control virus CLIG was applied to the retinal explants, ∼80% of the virus-infected cells differentiated into rods in the ONL, whereas the others differentiated mostly into bipolar and Müller glial cells in the INL (Figs. 2K,3), as described previously (Turner and Cepko, 1987). Similarly, when CLIG-hesr1 was applied, ∼80% of the virus-infected cells differentiated into rods in the ONL, and the others differentiated mostly into bipolar and Müller glial cells in the INL (Figs. 2B, 3). Thus, no significant effects by misexpression of hesr1 were detectable. When CLIG-hesr3 was applied, slightly more cells differentiated into rods (Figs. 2H, 3), although this increase was not statistically significant. In contrast, when CLIG-hesr2 virus was applied, >30% of the virus-infected cells differentiated into Müller glia, whereas <60% differentiated into rods (Figs. 2E, 3). Thus, hesr2exhibited an approximately threefold increase of Müller glial cell genesis with concomitant decrease of rod genesis. Thesehesr2-induced Müller glial cells exhibited a typical morphology: a cell body in the INL and long processes to both the ganglion cell layer and the ONL (Fig. 2E). To verify the coexpression of hesr genes with GFP, three repeats of the Myc epitope were fused to the amino terminus of each hesr, and we confirmed double staining of GFP and Myc in almost all virus-infected cells (Fig. 2B–J). In separate experiments, we also misexpressed each hesr without the Myc tag and obtained the same results, which suggests that the Myc tag did not affect the activity of hesr proteins (data not shown). These data indicate that among the three structurally related members, onlyhesr2 may have a gliogenic activity in the retina.
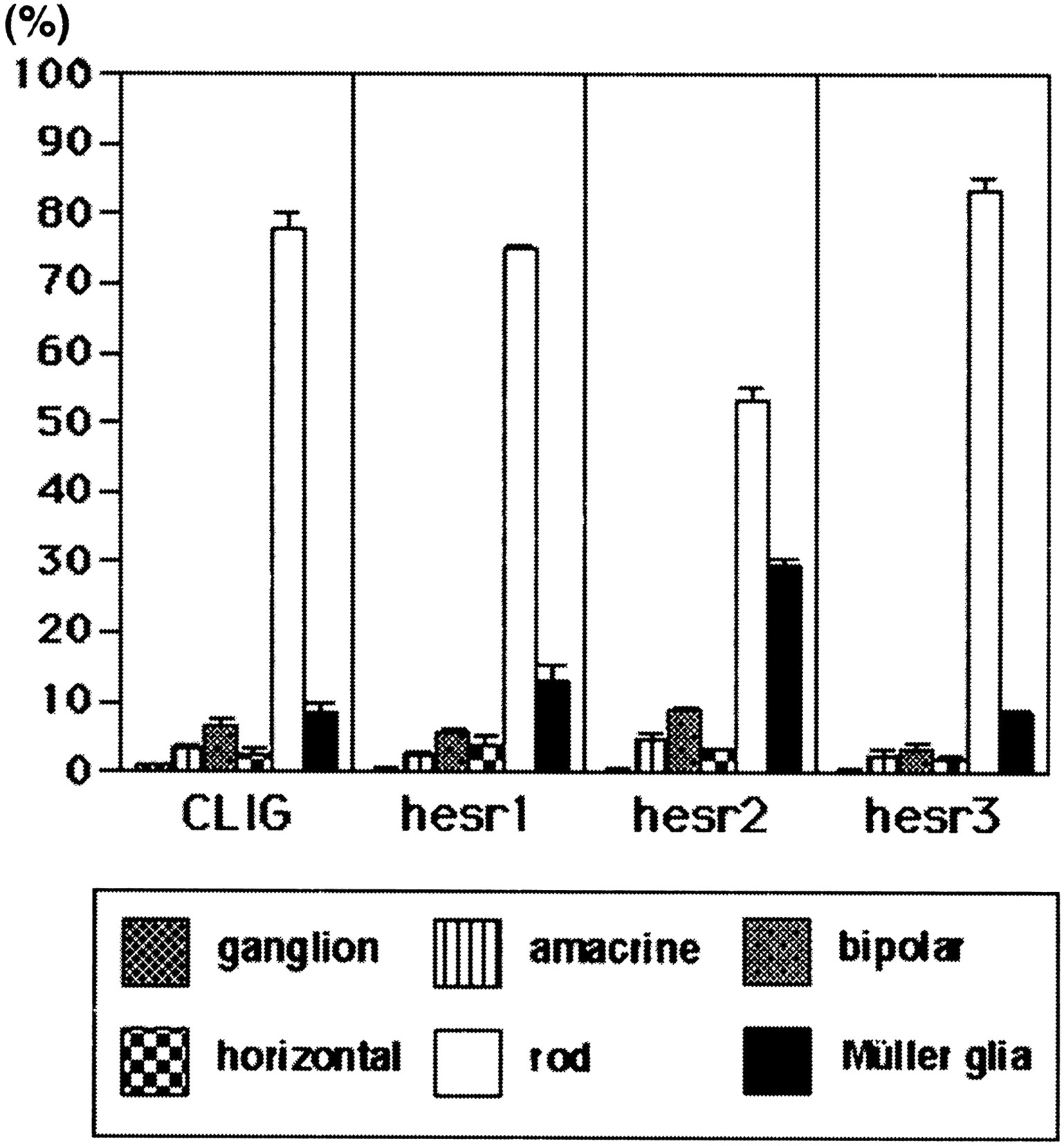
Ratios of the virus-infected cells in the explants starting at E17.5. Ratios of retinal cell types infected with CLIG, CLIG-hesr1, CLIG-hesr2, and CLIG-hesr3 at E17.5 are shown. When infected with CLIG or CLIG-hesr1, ∼80% of the virus-infected cells differentiated into rods, whereas 10% differentiated into Müller glia, indicating that hesr1 does not affect the ratios of retinal cell types. When infected with CLIG-hesr3, slightly more cells differentiated into rods, although this increase is not statistically significant. In contrast, when infected with CLIG-hesr2, ∼30% of the virus-infected cells differentiated into Müller glia, and <60% differentiated into rods. Thus, misexpression ofhesr2 displayed an approximately threefold increase of gliogenesis. Ratios with a SE are the average of at least three independent experiments.
We next examined whether Müller glia-like cells induced byhesr2 expressed glia-specific markers, vimentin and GS. Many of the hesr2+ cells expressed the Müller glial markers vimentin (Fig.4A–C) and GS (Fig. 4D–F). These results indicated that hesr2 promotes gliogenesis but inhibits neurogenesis, as do Hes1 and Hes5.
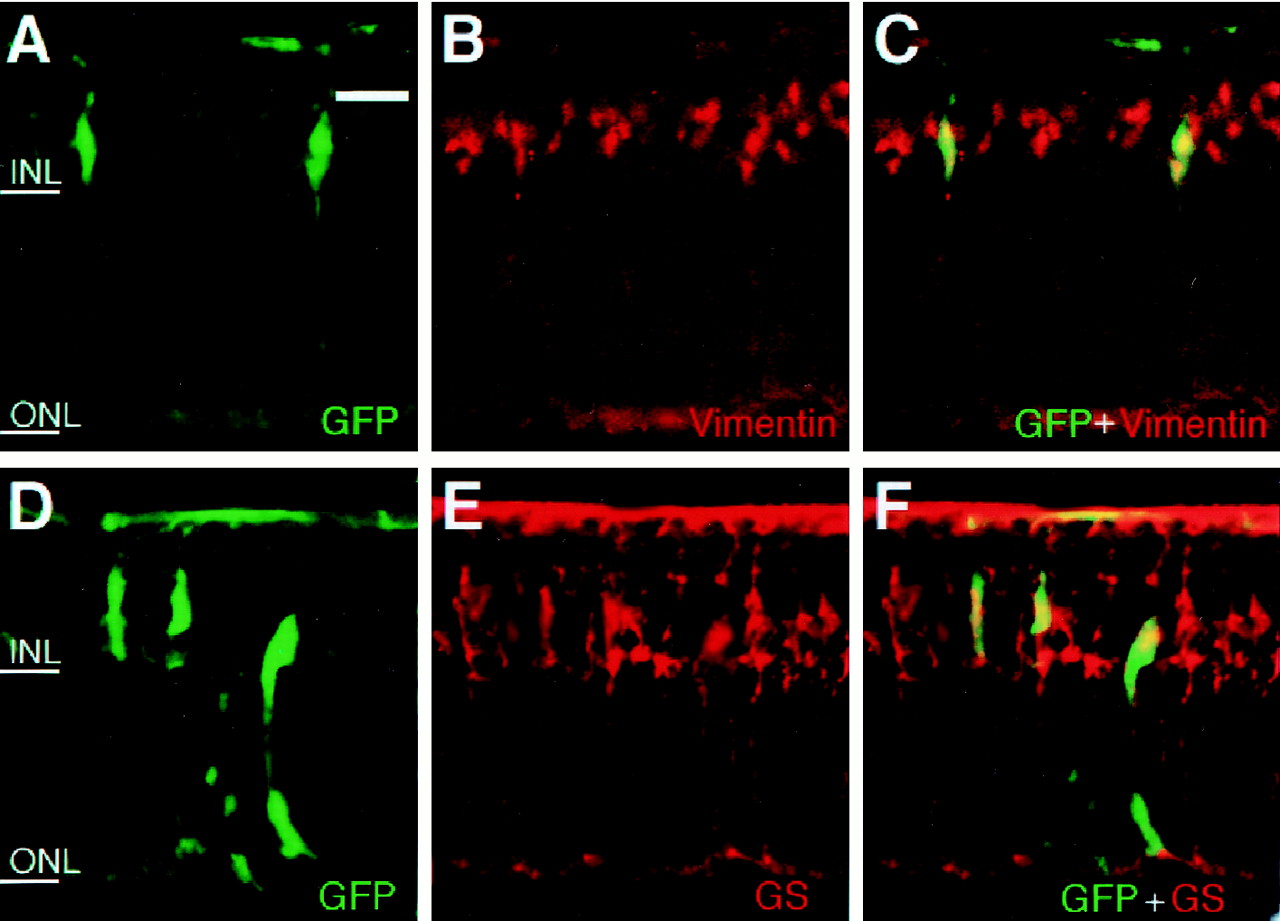
Misexpression of hesr2 promotes Müller glial development. Retinal explants were prepared from E17.5 mouse embryos and infected with CLIG-hesr2. After 2 weeks of culture, the explants were subjected to immunohistochemistry using anti-GFP, anti-vimentin, and anti-GS antibodies. Many of the virus-infected cells expressed the Müller glial markers, vimentin and GS. Scale bar, 25 μm.
Misexpression of hesr genes in the developing retina at P1
Because most Müller glial cells, one of the last-born cell types, differentiate postnatally, we next examined the effects ofhesr genes at a later stage of retinal development. Retinal explants were thus prepared at P1, and virus was applied on the same day. When the control virus CLIG was applied, the ratios of the last-born cell types, bipolar and Müller glial cells, were slightly increased, but still, most of the virus-infected cells differentiated into rods (Figs.5A,6). Similarly, when CLIG-hesr1 was applied, ∼80% of the virus-infected cells differentiated into rods, indicating that hesr1 did not affect the choice between the neuronal and glial fates (Figs. 5B, 6). When CLIG-hesr3 was applied, ∼90% of the virus-infected cells differentiated into rods and only 5% differentiated into Müller glial cells, thus suggesting that hesr3 promotes rod genesis and inhibits gliogenesis (Figs. 5D, 6). In contrast, when CLIG-hesr2 was applied, ∼75% of the virus-infected cells differentiated into Müller glia, whereas only <20% became rods (Figs.5C, 6). In addition, many of hesr2+cells expressed the Müller glial markers, vimentin (Fig.5E–G) and GS (Fig.5H–J). Thus, hesr2 exhibited a profound effect on gliogenesis when misexpressed in the postnatal retina.

Misexpression of hesr genes in the retinal explants starting at P1. Retinal explants were prepared from P1 mouse neonates and infected with CLIG (A), CLIG-hesr1 (B), CLIG-hesr2 (C,E–J), and CLIG-hesr3 (D). After 2 weeks of culture, the explants were subjected to immunohistochemistry using anti-GFP, anti-vimentin, and anti-GS antibodies. The majority of CLIG-, CLIG-hesr1-, and CLIG-hesr3-infected cells differentiated into rods in the ONL (A, B, D). In contrast, most CLIG-hesr2-infected cells became Müller glia (vimentin+, GS+) (C, E–J). Scale bar, 25 μm.

Ratios of the virus-infected cells in the explants starting at P1. Ratios of retinal cell types infected with CLIG, CLIG-hesr1, CLIG-hesr2, and CLIG-hesr3 at P1. When infected with CLIG or CLIG-hesr1, ∼80% of the virus-infected cells differentiated into rods. When infected with CLIG-hesr3, ∼90% of the virus-infected cells differentiated into rods, indicating that hesr3promotes rod genesis. This change is statistically significant (t test; p < 0.0001). When infected with CLIG-hesr2, ∼75% of the virus-infected cells differentiated into Müller glia. Thus, hesr2 significantly promoted gliogenesis when misexpressed postnatally. Ratios with a SE are the average of at least three independent experiments.
Cell proliferation and death are not affected by hesr2
The increase of Müller glia by hesr2 could be the result of proliferation of glial cells and apoptosis of neurons including rods, rather than conversion of precursors to the Müller glial cell fate at the expense of neurons. To distinguish between these possibilities, proliferation and death of virus-infected cells were analyzed. Cell proliferation was examined by Ki67, a nuclear antigen expressed by proliferating cells. Four or seven days after viral infection, most of the cells infected with CLIG or CLIG-hesr2 were negative for Ki67 (Fig.7A,B, and data not shown), indicating that hesr2 did not promote cell proliferation. To determine the extent of cell death, the retinal explants were subjected to TUNEL assay 4 or 7 d after viral infection. Most of the virus-infected cells were negative for the TUNEL assay at both time points (Fig. 7C,D, and data not shown). These results suggested that the Müller glial cell genesis induced by hesr2 was not the result of glial proliferation or neuronal apoptosis but most likely of conversion of precursors toward the Müller glial cell fate at the expense of neurons.

Proliferation and death of the virus-infected cells. A–D, Retinal explants infected with CLIG (A, C) and CLIG-hesr2 (B,D) were subjected to immunohistochemistry with anti-Ki67 antibody (A, B) and to TUNEL assay (C, D) after 1 week of culture. Most of the virus-infected cells were negative for Ki67 expression and TUNEL assay. Scale bar, 25 μm. E, F, Comparison of the clonal sizes of the cells infected at E17.5 (E) and P1 (F). The clonal sizes of CLIG- and CLIG-hesr-infected cells are very similar in all infections. These results indicate that misexpression ofhesr did not affect cell proliferation or survival. Ratios with a SE are the average of at least three independent experiments.
To further determine the extent of cell proliferation and survival, the clonal size of the virus-infected cells was examined. The sizes of clones infected with CLIG-hesr at both E17.5 (Fig.7E) and P1 (Fig. 7F) were mostly one or two cells, and they were very close to the size of CLIG-infected clones (Figs. 7E,F). These results indicated that hesr2 did not affect cell proliferation or death, in agreement with the above data of Ki67 staining and TUNEL assay.
hesr2 expression in the retina double-mutant forMash1 and Math3
We have recently found that in the retina double-mutant forMash1 and Math3, the cells that normally differentiate into bipolar cells are blocked from neuronal differentiation and instead adopt the Müller glial fate (Tomita et al., 2000). To determine whether hesr2 is involved in this increase of Müller glia, we examined hesr2expression in the double-mutant retina. Because all double-mutant embryos die by E17.5, retinal explants were prepared from E15.5 embryos and cultured to examine hesr2 expression at a later stage.
At E15.5, hesr2 expression was not affected in the double-mutant retina (Fig.8A,D). However, at day 7 of retinal explant culture, when more Müller glial cells (vimemtin+) were differentiating in the double-mutant than in the wild type (Fig.8C,F), hesr2 expression was upregulated in the INL of the double-mutant retina (Fig.8B,E). These results suggest that upregulation of hesr2 expression may contribute to the increase of the Müller glial cell number in the double-mutant retina. In contrast, expression of Hes1 and Hes5was not altered in the double-mutant retina both at E15.5 and at day 7 of explant cultures (data not shown).

hesr2 expression in the retina double-mutant for Mash1 and Math3. The wild-type (A–C) and Mash1-Math3 double-mutant retinas (D–F) were examined at E15.5 (A, D) and at day 7 of explant culture (B, C, E,F). A, B,hesr2 is initially expressed in the ventricular zone (V) and later in the INL.C, Müller glial cells (vimentin+) are present in the INL.D, E, hesr2 expression is not altered in the double-mutant retina at E15.5 but significantly upregulated at day 7 of explant culture, compared with the wild type.F, Müller glial cells (vimentin+) are increased in the double-mutant INL. Scale bar, 25 μm.
DISCUSSION
hesr2 promotes the glial versus neuronal fate choice in the retina
Previous studies revealed that twohairy/E(spl) homologs, Hes1 andHes5, play an important role in gliogenesis (Furukawa et al., 2000; Hojo et al., 2000). Here, we found that a related bHLH gene,hesr2, but not hesr1 or hesr3, promotes gliogenesis and inhibits rod genesis in the retina. Analysis of Ki67 and TUNEL staining as well as of clonal sizes demonstrated that glial proliferation and neuronal apoptosis cannot account forhesr2-induced gliogenesis. Therefore, it is most likely that the cells that normally differentiate into rods adopt the glial fate by misexpression of hesr2, indicating that hesr2regulates the glial versus neuronal fate choice in the retina. This activity is very similar to that of Hes1 andHes5, and thus hesr2 may substitute for the twoHes genes in gliogenesis.
The gliogenic activity of hesr2 correlates very well with its expression pattern. hesr2 is initially expressed by common precursors of neurons and glia in the ventricular zone, but during the postnatal period, hesr2 expression is shifted to the middle region of the INL, where Müller glial cells are differentiating. Thus, the expression pattern agrees well with the function of hesr2, which directs precursors to adopt the glial fate. Because hesr2 continues to be expressed until adulthood, it could also be involved in maintenance of mature glial cells in addition to glial fate determination.
Roles of hesr2 in gliogenesis
The mechanism by which hesr2 as well asHes1 and Hes5 promote gliogenesis remains to be determined. One mechanism would be that these genes may downregulate neuronal bHLH genes such as Mash1 and NeuroD, which promote neurogenesis at the expense of gliogenesis (Tomita et al., 1996b; Morrow et al., 1999). We have recently found that in mice double-mutant for the neuronal determination genes Mash1 andMath3, the cells that normally differentiate into neurons are blocked from neuronal commitment and instead adopt the Müller glial fate (Tomita et al., 2000). Thus, downregulation ofMash1 and Math3 is sufficient to initiate the gliogenic program. Because Hes1 and Hes5 are transcriptional repressors that inhibit the activity and expression of neuronal bHLH factors (Akazawa et al., 1992; Sasai et al., 1992; Ishibashi et al., 1995; Chen et al., 1997), it is likely that Hes1 and Hes5may suppress Mash1 and Math3, thereby promoting gliogenesis. In addition, it was recently reported that Id1, which dominant-negatively regulates positive bHLH genes (Benezra et al., 1990), also promotes gliogenesis (Cai et al., 2000). Thus,Hes1, Hes5, and Id1 may specify the glial fate by inducing the same effects as the double mutation ofMash1 and Math3, although it remains to be determined whether this is the only mechanism forHes1/Hes5/Id1-induced gliogenesis. It is not known whetherhesr2 has such an inhibitory activity on Mash1and Math3, but the structural conservation suggests that it does: hesr2 and Hes1/Hes5 share a high homology in two important domains. Hes1 and Hes5 have a conserved bHLH domain, which is important for the DNA binding and dimer formation, and the C-terminal WRPW domain, which interacts with the corepressor Groucho/TLE (Paroush et al., 1994; Fisher et al., 1996; Grabavec and Stifani, 1996). These two domains are essential for transcriptional repression activity of Hes1 and Hes5. Interestingly, hesr2 has a conserved bHLH domain in the amino-terminal region and a WRPW-related sequence, YQPW, in the C-terminal region (Leimeister et al., 1999; Nakagawa et al., 1999; Chin et al., 2000; Zhong et al., 2000), suggesting that hesr2 functions as a transcriptional repressor like Hes1 and Hes5. Supporting this idea, it has been shown recently that hesr2 (CHF1) represses transcription induced by ARNT/EPAS1 (Chin et al., 2000). Interestingly, the increase of Müller glia in the Mash1-Math3double-mutant retina is associated with upregulation ofhesr2, suggesting that Mash1 and Math3normally inhibit gliogenesis by repressing hesr2. Thus, antagonistic regulation between hesr2 andMash1/Math3 may determine the ratios of neurons and glia.
Is gliogenesis a default pathway after downregulation of neuronal bHLH genes? Recent studies demonstrated that oligodendrocyte development is regulated by two related bHLH genes, Olig1 andOlig2 (Lu et al., 2000; Zhou et al., 2000), suggesting that glial development may not be a simple default pathway but require glia-specific transcription factors. However, although Olig1and Olig2 can upregulate some glial-specific gene expression, they alone are not sufficient for oligodendrocyte development (Lu et al., 2000; Zhou et al., 2000). Thus, it is possible that for glial development these glia-specific bHLH genes may depend on negatively acting genes such as hesr2, Hes1,Hes5, and Id1, which suppress neurogenesis and switch on the gliogenic program.
Interestingly, the gliogenic activity of hesr2 seems to be stage-dependent, and when it is misexpressed at a later stage (P1), it displays a more profound effect: ∼75% of hesr2+ cells differentiate into Müller glia, whereas normally ∼10% become glia at this stage. Because some neuronal bHLH genes such as Mash1 are downregulated postnatally (Tomita et al., 1996b), it is possible thathesr2 could more effectively antagonize the neuronal bHLH genes at later stages.
Outside of the retina, hesr genes are also expressed in the developing nervous system. Although hesr1 is expressed in the ventricular zone, which contains neural precursors,hesr2 is expressed in both the ventricular zone and cortical plate (Leimeister et al., 1999), suggesting that these twohesr genes have distinct functions. It remains to be determined whether hesr2 has a gliogenic activity in the cortical plate.
Other functions of hesr genes
hesr genes are also expressed outside of the nervous system. Although hesr1 is expressed in the cardiac atrium,hesr2 is in the ventricles, suggesting that the twohesr genes have again distinct functions in heart development (Kokubo et al., 1999; Leimeister et al., 1999; Nakagawa et al., 1999; Chin et al., 2000; Zhong et al., 2000). They are also expressed in the blood vessels. Interestingly, in the zebrafish mutant for hesr2 (gridlock), assembly of the dorsal aortas is affected and blood flow is blocked (Zhong et al., 2000), indicating that hesr2 plays an important role in development of aorta. However, other defects such as neural defects have not been noted. Because gridlock mutation is hypomorphic rather than null, it is possible that defects are observed only in the most susceptible regions. Null mutation analysis is required to characterize the hesr2 functions in other regions.
It is interesting that hesr3 promotes rod differentiation in the retina, although less efficiently, suggesting that hesr3may have an opposite activity to hesr2. In this regard, the function of hesr3 is similar to Hes6, which also promotes rod genesis in the retina (Bae et al., 2000). Hes6 is structurally related to but functionally antagonizes Hes1 by direct physical interaction. When Hes1 and the positive regulator Mash1 are coexpressed, Hes1 represses Mash1-induced transcription (Sasai et al., 1992). However, when Hes6 is additionally coexpressed, Hes6 suppresses Hes1 and thereby supports the activity of Mash1, which promotes neuronal specification (Bae et al., 2000). Thus, misexpression ofHes6 promotes neuronal differentiation. It is possible that hesr3 may also have a Hes6-like activity and inhibit hesr2 by physical interaction, thereby inducing neurogenesis, although the precise mechanism remains to be determined.
In our present study, the three hesr genes have distinct expression patterns and functions in neural development, although they are structurally related. Further study of hesr genes would help determine the mechanism for the binary cell fate decision between neurons and glial cells.
Footnotes
This work was supported by Special Coordination Funds for Promoting Science and Technology and research grants from the Ministry of Education, Science, Sports, and Culture of Japan and the Japan Society for the Promotion of Science.
Correspondence should be addressed to Ryoichiro Kageyama, Institute for Virus Research, Kyoto University, Shogoin-Kawahara, Sakyo-ku, Kyoto 606-8507, Japan. E-mail:rkageyam{at}virus.kyoto-u.ac.jp.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}