

Abstract
Dopaminergic projections to the striatum, crucial for the correct functioning of this brain region in adulthood, are known to be established early in development, but their role is currently uncharacterized. We demonstrate here that dopamine, by activating D1- and/or D2-dopamine receptors, decreases the number of functional GABAergic synapses formed between the embryonic precursors of the medium spiny neurons, the principal output neurons of the striatum, with associated changes in spontaneous synaptic activity. Activation of these receptors reduces the size of postsynaptic GABAA receptor clusters and their overall cell-surface expression, without affecting the total number of clusters or the size or number of GABAergic nerve terminals. These changes result from an increased internalization of GABAA receptors, and are mediated by distinct signaling pathways converging at the level of GABAA receptors to cause a transient PP2A/PP1-dependent dephosphorylation. Thus, tonic D1- and D2-receptor activity limits the extent of collateral inhibitory synaptogenesis between medium spiny neurons, revealing a novel role of dopamine in controlling the development of intrinsic striatal microcircuits.
Introduction
The striatum, as the central part of the basal ganglia, integrates excitatory inputs from the cortex and thalamus with dopaminergic inputs from the substantia nigra pars compacta (SNpc), and sends projections to the output nuclei. It is composed primarily of GABAergic medium spiny projection neurons (95–98%), and a small number of GABAergic interneurons and cholinergic neurons (∼5% of all neurons) (Tepper and Bolam, 2004). Inhibition of medium spiny neurons is largely intrinsic to the striatum and occurs primarily via a feedforward mechanism mediated by the interneurons (Tepper and Bolam, 2004; Mallet et al., 2005), and to some extent by a feedback mechanism between projection neurons (Guzmán et al., 2003; Taverna et al., 2008). The essential molecular mediators of both mechanisms are GABAA receptors, members of a diverse family of heteropentameric GABA-gated chloride channels, which can be assembled from seven classes of homologous subunits: α(1–6), β(1–3), γ(1–3), δ, ε, θ, and π (Whiting, 2003). GABAA receptors are highly concentrated at synaptic sites apposed to presynaptic GABA-releasing terminals in the striatum (Fujiyama et al., 2000), and are likely to be dynamically regulated by the lateral migration to extrasynaptic sites (Thomas et al., 2005), internalization and reinsertion (Kittler et al., 2000, 2004), and direct phosphorylation (Brandon et al., 2002; Jovanovic et al., 2004) as reported in other brain regions.
The essential role of dopamine in a wide range of psychomotor functions of the adult striatum has been extensively characterized in both health and disease, and shown to be mediated through the activation of D1- and D2-like dopamine receptors (D1Rs and D2Rs) (Seeman and Van Tol, 1994). Thus, dopamine receptors regulate the activity of striatal neurons (Aosaki et al., 1998; Bracci et al., 2002), GABAergic currents (Yan and Surmeier, 1997; Flores-Hernandez et al., 2000), and glutamatergic synapse numbers (Day et al., 2006). In contrast, the role of dopamine in the developing striatum is unknown despite the evidence that dopaminergic inputs from the SNpc are formed as early as embryonic day 12 (E12) in rats (Voorn et al., 1988; Gates et al., 2006). Nevertheless, a developmental role for dopamine has been suggested by observations following in utero exposure to cocaine, which, via activation of dopamine receptors (Jones et al., 2000; Stanwood and Levitt, 2007), has profound effects on the development of dendrites (Jones et al., 1996; Levitt et al., 1997) and expression of GABAA receptors (Lu et al., 2009). Given that embryonic striatal neurons express both D1Rs and D2Rs (Aizman et al., 2000), we sought to assess whether these receptors regulate GABAergic synaptogenesis in the developing striatum.
We demonstrate that dopamine receptor activity decreases the number of GABAergic synapses formed between striatal neurons in vitro, by causing a reduction in the size of GABAA receptor clusters and their overall cell-surface expression. These changes are mediated by distinct D1R and D2R signaling pathways converging at the level of GABAA receptors to trigger their transient PP2A/PP1-dependent dephosphorylation. Thus, by altering the levels of cell-surface expression of GABAA receptors, dopamine exerts a powerful control of inhibitory synaptogenesis in the developing striatum.
Materials and Methods
Immunohistochemistry.
Whole-brain specimens were isolated from E17 Sprague Dawley rats and fixed overnight in 4% paraformaldehyde, 0.1% glutaraldehyde, and 0.2% saturated picric acid solution in 0.1 m phosphate buffer (PB), pH 7.2, at 4°C. For cryoprotection, brains were immersed in increasing concentrations of sucrose/PBS (10%, 20%, and 30%) until submerged, at 4°C. Brain specimens were frozen, and 20 μm sections were cut through the midbrain–hindbrain region. Sections were washed with PBS and incubated with 0.3 m glycine/PBS to quench PFA. For permeabilization and reduction of nonspecific binding, sections were incubated in 1% BSA/ 0.1% Triton X-100/PBS for 30 min. Sections were incubated with a rabbit anti-tyrosine hydroxylase antibody (1:1000 dilution, Merck Biosciences), or mixture of mouse anti-D1R (1:100 dilution, Abcam) and rabbit anti-D2R (5 μg/ml, Millipore) antibodies in PBS overnight at 4°C. Primary antibodies were visualized after staining with the appropriate goat anti-rabbit and anti-mouse IgGs conjugated to Alexa555 and Alexa488, respectively (3 μg/ml, Millipore Bioscience Research Reagents), in 1% BSA/PBS for 60 min. Sections were washed and coverslips mounted using Vectashield (Vector). Immunoreactivity was visualized using laser scanning confocal microscope (Zeiss LSM 510 Meta) with ×5 objective or ×63 oil-immersion objective.
HPLC analysis of tissue dopamine levels.
Embryonic striata were promptly dissected and weighed before being flash-frozen and stored at −80°C. Individual tissue samples were then placed in ice-cold PBS and homogenized. All homogenates were treated with 0.1 m perchloric acid (1:10, w/v) containing ascorbic acid (0.2 μm) and EDTA (0.2 μm), to precipitate cell debris. These were then centrifuged at 13,000 × g for 15 min at 4°C. The supernatant was passed through a syringe filter (10 μm pore size), and whole-tissue dopamine levels were estimated using HPLC with electrochemical detection (Biggs et al., 1992). Dopamine peak areas were quantified using an external standard method, and dopamine levels expressed as amount of dopamine in nanomoles per milligram of total protein content, with protein levels measured using BCA assay (Thermo Fisher Scientific).
Cell cultures.
Primary striatal neuronal cultures were prepared as described previously (Ventimiglia and Lindsay, 1998) with minor modifications. Briefly, striata were dissected from E16–E17 Sprague Dawley rats, dissociated by trituration in Ca2+- and Mg2+-free HEPES-buffered saline solution (HBSS; Invitrogen), plated at a density of 100,000 cells per cm2 in Neurobasal medium, containing B27 supplement, glutamine (2 mm), penicillin (100 U), streptomycin (100 μg), and glucose (6 mm; all from Invitrogen) on either 0.1 mg/ml poly-d-lysine-coated culture dishes or 0.1 mg/ml poly-l-lysine-coated glass coverslips. Cultures were incubated in a humidified 37°C/5% CO2 incubator for 7 or 14 d before experimentation.
Immunocytochemistry.
Striatal neurons cultured at a density of 100,000 cells/cm2 were treated with vehicle, the D1R agonist SKF-38393 (1 nm), or the D2R agonist quinpirole (100 nm; both from Tocris Bioscience) for 72 h (from 4 to 7 d in vitro, DIV) or 7 d (from 7 to 14 DIV), before fixation with 4% paraformaldehyde/4% sucrose/PBS for 15 min. Cultures were washed and incubated with 1% BSA/0.5% Triton X-100/PBS for 30 min to permeabilize cells and reduce nonspecific binding. Cultures were incubated with mouse anti-D1R (1:100 dilution, Abcam) and rabbit anti-D2R (5 μg/ml, Millipore) antibodies in PBS overnight at 4°C. For analysis of GABAergic synapses, cultures were first incubated with the GABAA receptor β2/3 subunit-specific primary antibody (MAB341, bd17 clone, 10 μg/ml, Millipore Bioscience Research Reagents) in 1% BSA/PBS overnight (14–16 h) at 4°C without permeabilization. Cultures were then washed with PBS and cells permeabilized with 0.5% Triton X-100/PBS for 30 min, followed by incubation with 1% BSA/PBS for 30 min to block nonspecific binding. Cultures were incubated with rabbit anti-vesicular inhibitory amino acid transporter (VIAAT) antibody [1:1000, kindly provided by Dr. A. Dumoulin and Prof. A. Triller, Inserm U497, Ecole Normale Supérieure, Paris, France (Dumoulin et al., 2000)] or glutamic acid decarboxylase (GAD) 65 (1:1000, Millipore Bioscience Research Reagents) in 1% BSA/PBS for 60 min. Primary antibodies were visualized after staining with the appropriate goat anti-mouse and anti-rabbit IgG conjugated to Alexa555 and Alexa488, respectively (3 μg/ml, Millipore Bioscience Research Reagents), in 1% BSA/PBS for 60 min. Cultures were washed and coverslips mounted using Vectashield (Vector). Immunoreactivity was visualized using laser scanning confocal microscope (Zeiss LSM 510 Meta) with a ×63 oil-immersion objective. In each image, laser light levels and detector gain and offset were adjusted to avoid any saturated levels.
Quantification of punctum area, number, and colocalization.
For each treatment, a minimum of 6–10 randomly selected neurons were examined in at least three independent experiments. For these experiments, the number, area, and colocalization of puncta were determined from confocal images using the LSM5 Image program. Immunoreactive puncta were defined as immunoreactivity >0.1 μm2 present along the first 20 μm length of primary processes (Yu et al., 2007). Threshold for each channel in each image was calculated as the mean pixel intensity for the entire image plus two SDs above the mean. Puncta colocalization in two different fluorescence channels was determined by overlaying the images. A punctum in the red channel was considered to colocalize with a punctum in the green channel, resulting in a yellow color, when ∼50% or more of the surface of one punctum overlapped with the other punctum. Colocalization was analyzed by determining the percentage of individual β2/3 subunit-positive puncta along the first 20 μm length of primary processes that were in close apposition to (and thus partially overlap with) VIAAT-1 or FM1-43FX-immunoreactive puncta. Values are expressed as mean ± SEM (number of cells). Statistical analysis was performed using a two-tailed t test (Graphpad Prism 4.0).
FM1-43FX uptake.
To assess the functionality of GABAergic neurons, we determined the uptake of the aldehyde-fixable styryl dye FM1-43FX (Invitrogen) (Sara et al., 2002; Brumback et al., 2004). Uptake of FM1-43FX was performed as previously described with some minor modifications (Ting et al., 2006). Following treatment with dopamine receptor agonists as described in the previous section, cells were briefly washed twice with buffer A (mm: 149 NaCl, 4 KCl, 1.5 CaCl2, 1.5 MgCl2, 10 glucose, and 10 HEPES, pH 7.4). Cultures were then incubated at 37°C with 100 μm bicuculline, 100 μm picrotoxin, 50 μm d-AP5, and 10 μm CNQX (all from Tocris Bioscience) diluted in buffer A. Cultures were further incubated in 60 mm K+ solution (in mm: 69 NaCl, 60 KCl, 1.5 CaCl2, 1.5 MgCl2, 10 glucose, and 10 HEPES, pH 7.4) containing FM1-43FX (10 μm) for 2 min. Excess styryl dye was effectively removed by washing three times in cold buffer A (4°C) for 1 min, followed by washing twice with ADVASEP-7 (500 μm, Sigma-Aldrich) for 2 min (Kay et al., 1999). Cultures were subsequently washed in buffer A with a low concentration of K+ to remove FM1-43FX/ADVASEP-7 at 4°C without causing exocytosis of vesicles loaded with FM1-43FX dye. Cells were fixed in 4% paraformaldehyde/4% sucrose/PBS and processed for immunocytochemistry using anti-β2/3 antibody, followed by goat anti-mouse IgG conjugated to Alexa555 (3 μg/ml) as described above. Immunoreactivity was visualized using a Zeiss LSM 510 Meta laser scanning confocal microscope with a ×63 oil-immersion objective.
Electrophysiology.
Whole-cell recordings were made from striatal neurons in culture, treated with the vehicle, SKF-38393 (1 nm), or quinpirole (100 nm) for up to 30 min, or for 72 h (4–7 DIV), as described above. Neurons were visualized using videomicroscopy under near-infrared differential interference contrast (DIC) illumination. Experiments were conducted at 20–22°C. Patch pipettes (resistance 8–10 MΩ) were pulled from borosilicate glass tubing and filled with an internal solution containing the following (in mm): 144 K-gluconate, 3 MgCl2, 0.2 EGTA, 2 Na2-ATP, 0.2 Na2-GTP, and 10 HEPES, pH 7.2–7.4, 300 mOsm. Spontaneous activity of the neurons was recorded in current-clamp mode (SEC 05L/H, npi electronics) in the presence of TTX (1 μm), d-AP5 (50 μm), and CNQX (20 μm, all from Tocris Bioscience). Synaptic potentials recorded were amplified, low-pass filtered at 2 kHz, and digitized at 5 kHz using a CED 1401 interface and data acquisition program, Signal 4.04 (Cambridge Electronic Design). Electrophysiological recordings were later analyzed offline using Signal. Individual sweeps were observed manually and measurable IPSPs were taken from values >0.05 mV in peak amplitudes. Miniature IPSP amplitudes (measured from the baseline to the peak of the IPSP) and rise times (10–90%) were measured from individual synaptic events. Average amplitudes are given as mean ± SD obtained from 100–250 frames of 1 s duration. Events <0.05 mV were related to the baseline noise without any measurable amplitudes or rise times. The data illustrated in Figure 3 are individual single sweep raw data that have not been smoothed, and illustrate a high signal-to-noise ratio.
Internalization assay.
Cell-surface receptors were tagged in living cultured neurons with the primary antibody against β2/3 subunits as described previously (van Rijnsoever et al., 2005). Briefly, coverslips were incubated with ice-cold buffer A (in mm: 150 NaCl; 3 KCl; 2 MgCl2; 10 HEPES, pH 7.4; and 5 glucose), containing 0.35 m sucrose for 5 min to inhibit receptor internalization, followed by incubation with mouse anti-β2/3 antibody (MAB341, 10 μg/ml, Millipore Bioscience Research Reagents) for 60 min at 4°C in buffer A containing 0.35 m sucrose, 1 mm EGTA, and 1% BSA. Excess antibody was removed by washing in the same medium. Cultures were returned to the incubator (at 37°C) in buffer A containing 1 mm CaCl2, 5 mm glucose, and 5 μg/ml leupeptin in the absence or presence of SKF-38393 (1 nm) or quinpirole (100 nm) for 30 min to allow internalization of the tagged β2/3 subunit-containing GABAA receptors. Cultures were then fixed for 15 min using 4% paraformaldehyde/4% sucrose/PBS, extensively washed with PBS, and incubated in 1% BSA/PBS for 30 min to reduce nonspecific binding. To label GABAA receptors remaining at the cell surface, cultures were incubated with goat anti-mouse IgG conjugated to Alexa555 (3 μg/ml, Millipore Bioscience Research Reagents) overnight (14–16 h) at 4°C. Cultures were extensively washed with PBS, and cells were permeabilized and nonspecific binding blocked for 30 min using 1% BSA/PBS containing 0.5% Triton X-100. To label internalized GABAA receptors, cultures were incubated with goat anti-mouse IgG conjugated to Alexa488 (3 μg/ml, Millipore Bioscience Research Reagents) for 60 min at the room temperature. Cultures were washed extensively and coverslips mounted using Vectashield. Immunoreactivity was visualized as described above using a Zeiss LSM 510 Meta laser scanning confocal microscope.
Determination of GABAA receptor cell-surface levels using ELISA.
Changes in surface and total levels of GABAA receptors were analyzed using a cell-surface ELISA assay as described previously (Noel et al., 1999; Bedford et al., 2001; Jovanovic et al., 2004). Striatal neurons were cultured in 24-well plates at a density of 100,000 cells/cm2. Following both short (up to 2 h) and long (up to 7 d) treatments with vehicle, SKF-38393 (1 nm), or quinpirole (100 nm) as described in the figure legends, cultures were washed with PBS and fixed using 4% paraformaldehyde/4% sucrose/PBS for 15 min. The fixative was removed by extensive washing with PBS followed by HBSS (Invitrogen). Nonspecific cell-surface binding of antibody was reduced by incubating cultures for 30 min in 1% BSA/10% rabbit serum/HBSS. In those cultures where total protein levels were evaluated, cells were permeabilized and nonspecific binding blocked by incubating with 0.5% Triton/1% BSA/10% rabbit serum/HBSS for 30 min. Cultures were incubated with anti-β2/3 antibody (1 μg/ml) overnight at 4°C. After extensive washing and nonspecific binding block as described above, cultures were incubated with anti-mouse IgG conjugated to horseradish peroxidase (HRP, 1:2500, Pierce) for 60 min. Cultures were extensively washed and a color substrate 3,3,5,5-tetramethylbenzidine (TMB; Sigma-Aldrich) reagent added for 20 min until sufficient color reaction had developed. Absorbances were read at λ = 655 nm using a spectrophotometer (Duo 800, Beckman Coulter). Controls were routinely used without using primary antibody to determine background levels of peroxidase and nonspecific binding of secondary antibody. Values are expressed as mean ± SEM (number of experiments, each done in duplicate). Statistical analysis was performed using one-way ANOVA with Dunnett post hoc analysis and two-tailed t test (Graphpad Prism 4.0).
In vitro phosphorylation.
Glutathione S-transferase (GST) fusion proteins encoding the intracellular transmembrane domain 3–4 loops of GABAA receptor β1-3 subunits were purified from Escherichia coli as described previously (McDonald and Moss, 1997; Jovanovic et al., 2004). The catalytic subunit of PKA was purified from bovine heart as described previously (Kaczmarek et al., 1980). Phosphorylation of GST-β1, -β2, and -β3 used the incubation conditions described for the catalytic subunit of PKA (Huttner et al., 1981), in the presence of 150 μm ATP, with trace amounts of [γ-32P]ATP, to yield a final stoichiometry of 0.4, 0.4, and 1.1 molP/mol protein, respectively. Incorporation of 32P was determined using measurement of Cerenkov radiation. The phosphorylated proteins were repurified using NICK columns (GE Healthcare). Dopamine- and cAMP-regulated phosphoprotein (Mr = 32,000; DARPP-32), phosphorylated by PKA to a stoichiometry of 0.3 molP/mol of protein (Girault et al., 1989) and phosphorylase a (Cohen et al., 1988), were phosphorylated and repurified as described.
In vitro dephosphorylation.
Protein phosphatase 1 catalytic subunit (PP1c, Mr = 37,000) and protein phosphatase 2A (PP2A, complex of catalytic and regulatory subunit, Mr = 96,000) were purchased (Millipore), and calcineurin (Mr = 76,000) was purified from rat brain (Nairn et al., 1995). Purified phosphatases were assayed in 50 mm Tris-HCl, pH 7.0, 15 mm 2-mercaptoethanol, and 1 mg/ml BSA at 30°C, as described previously (Desdouits et al., 1998), in the presence of 0.3% Brij-35 and 0.3 mm EGTA in the case of PP1c and PP2A, or 100 μm CaCl2 and 1 μm calmodulin in the case of calcineurin. Reactions were started by the addition of substrate and terminated by the addition of 200 μl of 20% (w/v) trichloroacetic acid. After the further addition of 50 μl of 10 mg/ml bovine serum albumin, samples were centrifuged for 5 min at room temperature at 17,000 × g, and the amount of 32P in the supernatant and the pellet was determined by measurement of Cerenkov radiation. PP1c and PP2A activities were measured using 1 μm [32P] phosphorylase a as substrate (Ingebritsen et al., 1983) under initial rate conditions (the release of phosphate was linear with time and enzyme concentration, and corresponded to ∼25% of the phosphate incorporated into the substrate). For measurements of calcineurin activity, initial rate conditions were determined using 1 μm [32P] phospho-DARPP-32. Under the same conditions, PP1c-, PP2A-, and calcineurin-mediated dephosphorylation of different [32P]-labeled phospho-GST-β1-3 subunits (1 μm) was directly compared to that of standard substrates (phosphorylase a and DARPP-32) (Table 1). Data represent the means of three independent experiments, each done in duplicate. For kinetic analysis, dephosphorylation assays were done using increasing concentrations of phospho-substrates (GST-β1: 0.6–10 μm; GST-β2: 0.4–2 μm; and GST-β3: 0.1–1 μm). The total amount of PP1c and PP2A per reaction was 2.5 ng. The Km and Vmax values were calculated from linear regression analysis of Lineweaver-Burk transformations of data describing the initial rates of dephosphorylation as a function of substrate concentration, and represent the means of three independent experiments, each done in duplicate (Table 2).
GABAA receptor β subunit dephosphorylation by purified protein phosphatases
Kinetic analysis of GABAA receptor β1-3 subunit dephosphorylation by purified protein phosphatases
Treatments with protein kinase and phosphatase inhibitors.
For cell-surface ELISA experiments, E17 embryonic striatal cultures (7 DIV) were treated with SKF-38393 (1 nm), quinpirole (100 nm), or dopamine (1 μm) for 30 min with the exception of time course experiments. Those cultures that received treatment with SCH-23390 (1 μm), sulpiride (1 μm), 8-bromo-cAMP (10 μm), forskolin (10 μm), fostriecin (1 μm), and okadaic acid (100 nm or 1 μm; all from Tocris Bioscience), calphostin C (200 nm), and PD-98059 (50 μm, both from Calbiochem), were treated for 10 min before the addition of dopamine receptor agonists; all others were treated with vehicle during this time. Those cultures treated with EGTA (1 mm, Sigma-Aldrich) or BAPTA-AM (25 μm, Calbiochem) received a 30 min treatment before dopamine receptor agonists; all others received vehicle during this time.
Immunoblotting.
E17 embryonic striatal cultures (7 DIV) were treated with SKF-38393 (1 nm), quinpirole (100 nm), or both for 5 min to 2 h. Those cultures that received treatment with PD-98059 (50 μm), fostriecin (1 μm), or okadaic acid (1 μm) were treated for 10 min before the addition of dopamine receptor agonists for 10 min; all others were treated with vehicle during this time. Samples were lysed with 1% SDS and protein concentration determined using BCA assay. Samples with the same amount of total protein were separated using SDS-PAGE and transferred onto a nitrocellulose membrane. Nonspecific binding was blocked by incubation of membranes with TBS/Tween buffer containing 2 mg/ml BSA. Membranes were incubated with rabbit anti-phospho-β subunit antibody purified against a phospho-Ser409-peptide column (Jovanovic et al., 2004) or rabbit total β3 antibody (0.5 μg/ml, Phosphosolutions, Aurora, CO), for 90 min, followed by washing with TBS/Tween. Membranes incubated with the total β3 antibody were blocked using TBS/Tween buffer containing 1.5% (w/v) powdered milk. Membranes were incubated with [125I]-anti-rabbit polyclonal antibody (GE Healthcare) for 60 min, and washed as before using TBS/Tween buffer. Detection of immunoreactivity was performed using a phosphoimager (Molecular Devices).
Results
Dopaminergic projections and dopamine receptors are present in the embryonic E17 striatum
In the ontogeny of the rat, dopaminergic fibers positive for tyrosine hydroxylase (TH) arrive in the striatal enlarge on E14 (Specht et al., 1981; Voorn et al., 1988). To establish the presence of dopaminergic system in the embryonic rat (E17) striatum in vivo, whole-brain sections cut through the midbrain–hindbrain region were stained using antibodies specific for TH, D1R, and D2R. Prominent TH fibers and terminal-like staining were detected at this stage of development (Fig. 1A). The expression of dopamine D1R and D2R, and a high degree of colocalization between these receptors, was also observed throughout the embryonic striatum (Fig. 1B). The levels of tissue dopamine were measured in acutely prepared homogenates using HPLC, revealing 11.4 ± 0.5 nmol of dopamine per milligram of total protein in E17 and 14.4 ± 0.6 nmol of dopamine per milligram of total protein in E18 striatal tissue. In contrast, levels of tissue noradrenaline and 5-HT were below detection in these experiments.

Dopaminergic projections and dopamine receptors are present in the embryonic striatum. A, Dopaminergic projections positive for TH (×5, scale bar corresponds to 200 μm) form a prominent network of fibers in the ventral part of the striatal enlargement, while terminal-like staining (×63, scale bars correspond to 20 μm) is abundant in the dorsal part of the embryonic (E17) striatum. B, Dopamine D1 receptors and D2 receptors are expressed throughout the whole striatal enlargement and show a high-degree of colocalization (×63, scale bars correspond to 20 μm).
Dopamine receptor activation decreases the number of GABAergic synapses and the size of GABAA receptor clusters
Embryonic striatal neurons in vitro form a highly homogenous population of precursors of GABAergic medium spiny neurons that display a number of properties that are similar to their in vivo counterparts; they form functional GABAergic synapses, express D1Rs and D2Rs, and display similar electrophysiological properties (Bockaert et al., 1986; Aizman et al., 2000; Falk et al., 2006). The coexpression of D1R and D2R demonstrated in vivo (Fig. 1B) was also detected in vitro, where all the neurons imaged showed a strong immunoreactivity for both receptors at 7 DIV (Fig. 2A) and 14 DIV (Fig. 2B). To examine whether dopamine regulates the extent of synaptic connectivity between these neurons, we used confocal microscopy to analyze the number of GABAergic synapses upon the application of SKF-38393, a D1R-specific agonist, or quinpirole, a D2R-specific agonist. Localization of VIAAT-1, a presynaptic marker of GABAergic and glycinergic synapses (Dumoulin et al., 1999) was found to completely overlap with that of GAD-65, a presynaptic marker specific for GABAergic synapses (data not shown). As these presynaptic markers colocalize with surface-expressed GABAA receptor β2/3 subunit clusters, reflecting the close apposition between the presynaptic and postsynaptic components of GABAergic synapses (Chih et al., 2005; Dong et al., 2007; Yu et al., 2007), we estimated the number of these synapses along defined length of primary neuronal processes.

Dopamine receptor activation decreases the colocalization of postsynaptic GABAA receptor β2/3 subunit puncta with presynaptic VIAAT-1 puncta. Embryonic striatal neurons express both D1Rs and D2Rs when cultured for 7 d (7 DIV) (A), and 14 d (14 DIV) (B). Scale bars correspond to 20 μm. Striatal cultures were treated for 72 h (from 4 to 7 DIV) (C) or 7 d (from 7 to 14 DIV) (D) with vehicle, SKF-38393, or quinpirole, and stained for GABAAR β2/3 subunit (in red) and VIAAT-1 (in green). Scale bars correspond to 10 μm. The percentage of β2/3 subunit-positive puncta that colocalized with VIAAT-1 puncta was significantly decreased in cultures treated with SKF-38393 or quinpirole for 72 h (E) or 7 d (F). G, H, Relative cumulative frequency plots of β2/3-immunoreactive puncta and VIAAT-1-positive puncta (in square micrometers) measured along a defined length of primary processes (20 μm) following the treatment with vehicle (white), SKF-38393 (black), or quinpirole (dark gray) for 72 h (4–7 DIV, left) (G) or 7 d (7–14 DIV, right) (H). I, SKF-38393 (black) and quinpirole (gray) significantly decreased the average β2/3 subunit punctum area in comparison with controls (vehicle treated, white) cells, following treatments for 72 h or 7 d. J, SKF-38393 (black) and quinpirole (gray) had no effect on the average VIAAT-1 punctum area in comparison with controls (vehicle treated, white), following treatments for 72 h or 7 d. K, The total number of β2/3-positive puncta remained unchanged following the treatment with vehicle (white), SKF-38393 (black), or quinpirole (gray) for 72 h or 7 d. L, The total number of VIAAT-1-positive puncta remained unchanged following the treatment with vehicle (white), SKF-38393 (black), or quinpirole (gray) for 72 h or 7 d. Bars represent mean ± SEM. Statistical analysis was performed using paired t test: ***p < 0.001.
Upon treatment with SKF-38393 (1 nm) or quinpirole (100 nm) for 72 h or 7 d, we observed a significant decrease in the colocalization between GABAA receptor β2/3 subunit clusters expressed at the cell-surface and VIAAT-1-positive presynaptic terminals: 79.6 ± 3.0% of surface β2/3 subunit clusters, which were colocalized with VIAAT-1 puncta in control cultures (7 DIV), was significantly decreased to 50.1 ± 3.7% (p < 0.01, paired t test, n = 26) by 72 h treatment with SKF-38393, or to 49.8 ± 3.6% (p < 0.01, paired t test, n = 21) by 72 h treatment with quinpirole (Fig. 2C,E). Similarly, a prolonged 7 d treatment of cells (from 7 to 14 DIV) with either of the agonists resulted in a significant decrease in colocalization between β2/3 subunit clusters and VIAAT-1-positive presynaptic terminals: 90.6 ± 1.2% colocalization observed in control vehicle-treated cells was significantly reduced to 68.0 ± 2.4% (p < 0.01, paired t test, n = 24) by SKF-38393, and to 70.0 ± 3.0% (p < 0.01, paired t test, n = 21) by quinpirole (Fig. 2D,F). Thus, both D1Rs and D2Rs decrease the number of GABAergic synapses formed between embryonic precursors of medium spiny neurons in vitro.
The observed reduction in the number of GABAergic synapses may occur due to a reduction in the number of presynaptic GABAergic terminals or postsynaptic GABAA receptor clusters, or inhibition of the formation and maintenance of synaptic contacts. Alternatively, these changes may result from changes in the structure of presynaptic or postsynaptic elements. To evaluate these possibilities, we performed detailed quantitative analysis of the area and the number of β2/3 puncta and VIAAT-1 puncta along the initial 20 μm of primary neuronal processes following dopamine receptor activation.
Our analysis revealed a significant decrease in the area of surface β2/3-positive puncta following dopamine receptor activation. In control cells cultured for 7 DIV, the area of β2/3 subunit puncta was 0.45 ± 0.04 μm2, which was significantly decreased to 0.29 ± 0.02 μm2 (p < 0.001, paired t test, n = 26), or to 0.31 ± 0.03 μm2 (p < 0.001, paired t test, n = 21) by 72 h treatment with SKF-38393 or quinpirole, respectively (Fig. 2G,I, 72 h). Dopamine receptor activation produced similar effects in cells cultured for 14 DIV: in control cells, the area of β2/3 subunit puncta was 0.67 ± 0.04 μm2 compared to 0.46 ± 0.03 μm2 after SKF-38393 treatment (p < 0.001, paired t test, n = 24) for 7 d and 0.48 ± 0.03 μm2 after 7 d treatment with quinpirole (p < 0.001, paired t test, n = 21) (Fig. 2H,I, 7 d). However, we observed no significant change in the number of β2/3 puncta at the cell surface following activation of either D1Rs or D2Rs for 72 h or 7 d. In control cells cultured for 7 DIV, there were 5.31 ± 0.31 β2/3 subunit puncta compared to 5.59 ± 0.29 in those treated with SKF-38393 (p > 0.05, paired t test, n = 26) and 5.36 ± 0.26 in those treated with quinpirole for 72 h (p > 0.05, paired t test, n = 21) (Fig. 2K, 72 h). The number of β2/3 subunit puncta in neurons cultured for 14 DIV was 7.25 ± 0.28 in control cells and 7.43 ± 0.30 in those treated with SKF-38393 for 7 d (p > 0.05, paired t test, n = 24) and 7.83 ± 0.41 in those treated with quinpirole for 7 d (p > 0.05, paired t test, n = 21) (Fig. 2K, 7 d).
To assess changes in presynaptic GABAergic terminals, we analyzed the area and the number of VIAAT-1-positive puncta. There was no significant change in the area of VIAAT-1 puncta following 72 h treatment with SKF-38393 or quinpirole: the average area of puncta in vehicle-treated cells was 0.49 ± 0.03 μm2 in comparison with 0.45 ± 0.06 μm2 in SKF-38393-treated (p > 0.05, paired t test, n = 26) or 0.49 ± 0.05 μm2 in quinpirole-treated (p > 0.05, paired t test, n = 21) cultures (Fig. 2G,J, 72 h). Similar results were obtained in cultures treated for 7 d with SKF-38393 or quinpirole: the average area of puncta in vehicle-treated cells was 0.56 ± 0.03 μm2 in comparison with 0.55 ± 0.03 μm2 in SKF-38393-treated (p > 0.05, paired t test, n = 24) or 0.57 ± 0.04 μm2 in quinpirole-treated (p > 0.05, paired t test, n = 24) cultures (Fig. 2H,J, 7 d). The number of VIAAT-positive puncta also remained unchanged during 72 h treatment with SKF-38393 or quinpirole: vehicle-treated cells had 5.33 ± 0.30 puncta compared to 5.20 ± 0.30 in SKF-treated (p > 0.05, paired t test, n = 24) and 5.31 ± 0.29 in quinpirole-treated (p > 0.05, paired t test, n = 21) cultures (Fig. 2L, 72 h). Similarly, the number of VIAAT-positive puncta in controls (6.47 ± 0.19, p > 0.05, paired t test, n = 24) remained unchanged during 7 d treatment with SKF-38393 (6.57 ± 0.30, p > 0.05, paired t test, n = 24) or quinpirole (6.49 ± 0.30, p > 0.05, paired t test, n = 24) (Fig. 2L, 7 d).
To assess whether the observed dopamine receptor-mediated changes in morphology correlated with changes in the number of functional GABAergic synapses, we used confocal microscopy to analyze the degree of colocalization between GABAergic terminals labeled by activity-dependent uptake of aldehyde-fixable styryl dye FM1-43FX (Sara et al., 2002), and GABAA receptor β2/3 subunit clusters. We demonstrate that colocalization between surface β2/3- and FM1-43FX-positive puncta was significantly decreased from 68.1 ± 2.6% in control conditions to 42.3 ± 3.1% (p < 0.01, paired t test, n = 26) following 72 h treatment with SKF-38393, and to 45.6 ± 3.2% (p < 0.01, paired t test, n = 21) following treatment with quinpirole (Fig. 3A,B). Prolonged 7 d treatment of cells (from 7 to 14 DIV) with either of agonists also resulted in a significant decrease in colocalization between surface β2/3- and FM1-43FX-positive puncta: 75.9 ± 1.8% observed in control conditions was significantly decreased to 36.6 ± 2.4% (p < 0.01, paired t test, n = 24) upon the treatment with SKF-38393, and to 35.8 ± 2.6% (p < 0.01, paired t test, n = 21) upon the treatment with quinpirole (Fig. 3C).

Dopamine receptor activation decreases the number of functional GABAergic synapses. A, Cultures were treated for 72 h (from 4 to 7 DIV) with vehicle, SKF-38393, or quinpirole, followed by activity-dependent labeling with FM1-43FX. Scale bars correspond to 10 μm. SKF-38393 and quinpirole significantly decreased the colocalization of β2/3 puncta (in red) and FM1-43FX puncta (in green) after treatment for 72 h (from 4 to 7 DIV, B), or 7 d (from 7 to 14 DIV, C). Bars represent mean ± SEM. Statistics was performed using t test: ***p < 0.001. D, mIPSPs recorded in striatal neurons after 72 h treatment (from 4 to 7 DIV) with vehicle (control) or SKF-38393 in the presence of TTX (1 μm), d-AP5 (50 mm), and CNQX (20 mm), before and after bath application of GABAA receptor antagonist picrotoxin (50 μm, + picrotoxin). Three representative traces for each condition are shown. Scale refers to all four conditions. E, F, SKF-38393 and quinpirole treatments decreased the amplitude (E) and frequency (F) of mIPSPs. Bars represent mean ± SD. Statistical analysis was performed using two-tailed t test: *p < 0.05.
We performed whole-cell patch-clamp analysis of striatal neurons to measure spontaneous activity at inhibitory synapses (mIPSPs) in response to application of dopamine receptor agonists. We demonstrate that cultured embryonic striatal neurons exhibit mIPSPs with an amplitude of 1.8 ± 0.2 mV and a frequency of 2.3 ± 0.2 Hz (n = 6) (Fig. 3D–F). These spontaneous currents were GABAergic since they were completely eliminated by the addition of picrotoxin (50 μm) to the perfusion bath (Fig. 3D, left, +picrotoxin). Following 72 h treatment with SKF-38393, the amplitudes and frequency of mIPSPs were significantly decreased compared to control: amplitudes of mIPSPs were 0.9 ± 0.3 mV, and frequency was 1.4 ± 0.2 Hz (p < 0.05, two-tailed t test, n = 4) (Fig. 3D–F). The remaining spontaneous currents in SKF-38393-treated cultures were also completely inhibited by picrotoxin (Fig. 3D, right, +picrotoxin). The amplitudes and frequency of mIPSPs were also reduced in cultures treated for 72 h with quinpirole (Fig. 3E,F). However, short treatments (up to 30 min) with SKF also reduced the amplitude and frequency of mIPSPs in these cultures from 2.0 ± 1.0 mV and 3.0 ± 0.8 Hz, respectively, in vehicle-treated cultures, to 0.9 ± 0.7 mV and 0.6 ± 0.4 Hz (p < 0.05, two-tailed t test, n = 4, data not shown). Similar effects were observed following short treatments with quinpirole: amplitudes and frequencies of mIPSPs were reduced from 3.2 ± 0.4 mV and 3.2 ± 0.8 Hz in vehicle-treated cultures to 1.6 ± 0.3 mV and 1.3 ± 0.3 Hz, respectively (p < 0.05, two-tailed t test, n = 3, data not shown). These data provide further evidence that both D1Rs and D2Rs decrease the number of functional GABAergic synapses formed between embryonic precursors of medium spiny neurons.
Dopamine receptors decrease cell-surface expression of GABAA receptors
The reduction in size of GABAA receptor β2/3 subunit-positive clusters at the cell surface observed upon activation of D1Rs and D2Rs may be occurring due to increased lateral diffusion from synaptically clustered receptor pools to more diffuse extrasynaptic pools (Thomas et al., 2005), without an overall decrease in the total surface numbers. Alternatively, it may reflect an overall reduction in their number at the cell surface, without changing a balance between clustered and diffuse receptors. To distinguish between these two mechanisms, we measured the cell-surface levels of GABAA receptor β2/3 subunits using cell-surface ELISA assays (Noel et al., 1999; Bedford et al., 2001; Jovanovic et al., 2004).
We demonstrate that D1R activation by SKF-38393 led to a significant and prolonged decrease in the surface expression of GABAA receptors, which was observed as early as 10 min upon the application of the drug and maintained for up to 7 d in the continuous presence of the drug (one-way ANOVA; with Dunnett post hoc analysis; n = 4) (Fig. 4A, in black): surface β2/3 subunit levels were reduced to 82.4 ± 6.2% of vehicle (p < 0.05) after 10 min, to 81.3 ± 3.5% of vehicle (p < 0.05) after 30 min; to 82.9 ± 5.6% (p < 0.05) of vehicle after 60 min; to 80.4 ± 5.9% (p < 0.05) of vehicle after 120 min; to 82.4 ± 3.1% of vehicle (p < 0.01) after 72 h, and to 80.8 ± 5.2% of vehicle (p < 0.05) after 7-d-long treatment with SKF-38393. D1R activation had no significant effect on the total levels of β2/3 subunits at any of those time points tested (Fig. 4A, in gray). The effects of SKF-38393 (88.1 ± 1.9% of vehicle-treated controls) were blocked by the application of D1R antagonist SCH-23390 (101.3 ± 4.8% of vehicle-treated control, p < 0.05, paired t test, n = 6) (Fig. 4C).

Activation of dopamine receptors decreases the cell-surface expression of GABAA receptors. A, SKF-38393 (1 nm) caused a significant and prolonged decrease in the level of β2/3 subunits (black) expressed at the cell surface, but not in total levels of these subunits (gray). B, Quinpirole (100 nm) caused a significant and prolonged decrease in β2/3 subunit cell-surface expression (black) but not in their total levels (gray). C, D1R antagonist SCH-23390 (1 μm, SCH) abolished the reduction in surface β2/3 levels upon 30 min treatment with SKF-38393 (1 nm, SKF). D, D2R antagonist sulpiride (10 μm, Sulp) significantly attenuated the reduction in surface β2/3 levels upon 30 min treatment with quinpirole (100 nm, Q). E, Dopamine (1 μm, DA)-dependent decrease in the β2/3 subunit surface was significantly attenuated by SCH-23390 (DA + SCH), and sulpiride (DA + Sulp), indicating that both D1Rs and D2Rs mediate the effects of dopamine. F, Activation of both D1Rs and D2Rs by their respective agonists (SKF + Q) caused a reduction in surface β2/3 levels comparable to changes observed upon the treatment with SKF-38393 (SKF) or quinpirole (Q) alone. Bars represent mean ± SEM. Statistical analysis was performed using one-way ANOVA with either a Dunnett post hoc analysis versus control (*p < 0.05; **p < 0.01), or paired t test (#p < 0.05).
We also examined the effects of D2R activation by quinpirole on GABAA receptor surface levels using cell-surface ELISA approach. Quinpirole decreased the surface expression of β2/3 subunits to 94.0 ± 1.1% following 10 min treatment (p > 0.05); to 83.8 ± 2.0% after 30 min treatment (p < 0.01), to 91.6 ± 2.0% after 60 min treatment (p > 0.05); to 84.1 ± 7.2% after 120 min treatment (p < 0.01), to 85.4 ± 3.2% after 72 h treatment (p < 0.05); and to 86.5 ± 3.7% after 7 d treatment (p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 4) (Fig. 4B, in black). D2R activation had no significant effect on the total levels of β2/3 subunits at any of time points tested (Fig. 4B, in gray). The decrease observed after 30 min treatment with quinpirole (74.0 ± 6.3% of control) was prevented by the D2R antagonist sulpiride (93.9 ± 2.9% of control, p < 0.05, paired t test, n = 4) (Fig. 4D).
Furthermore, we demonstrate that the application of dopamine (1 μm) also caused a prominent reduction in the cell-surface expression of GABAA receptors via activation of both D1Rs and D2Rs. Significantly, the decrease in surface β2/3 subunit levels to 75.6 ± 2.3% of vehicle-treated controls was partially attenuated to 86.7 ± 4.3% of vehicle-treated controls in the presence of D1R antagonist SCH-23390 (1 μm), and to 84.7 ± 3.9% of vehicle-treated controls in the presence of D2R antagonist sulpiride (10 μm, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 5) (Fig. 4E).
Although both D1Rs and D2Rs appear to mediate regulation of GABAA receptor cell-surface levels by dopamine, application of specific receptor agonists SKF-38393 (1 nm) and quinpirole (100 nm) at the same time resulted in a reduction in surface β2/3 subunit levels to 71.4 ± 7.7% of vehicle-treated controls, which was slightly but not significantly more pronounced than the application of SKF-38393 (81.3 ± 3.5% of vehicle-treated control) or quinpirole alone (83.8 ± 2.0% of vehicle-treated control, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 4) (Fig. 4F). Thus, both D1R and D2R activation leads to a significant and prolonged decrease in cell-surface levels of GABAA receptors in embryonic striatal neurons.
Dopamine receptor activation enhances GABAA receptor endocytosis
GABAA receptors are continuously internalized by clathrin-dependent endocytosis and replaced by insertion of newly synthesized receptors (Kittler et al., 2000). To examine whether dopamine receptor activation leads to an increase in GABAA receptor endocytosis, we visualized this process using immunocytochemistry (van Rijnsoever et al., 2005). Cultured striatal neurons were incubated with the same anti-β2/3 subunit antibody as in cell-surface ELISA experiments to label GABAA receptors at the cell surface. Cultures were subsequently treated with vehicle, SKF-38393 (1 nm), or quinpirole (100 nm) for 30 min. Following fixation, GABAA receptors remaining at the cell surface were probed with a secondary antibody conjugated to Alexa555 (Fig. 5A,B, in red). Following permeabilization, the internalized receptors were probed with a secondary antibody conjugated to Alexa488 (Fig. 5A,B, in green). The two pools were analyzed using confocal microscopy. We demonstrate that both treatments increased the amount of internalized GABAA receptors with a concomitant reduction in those located at the cell surface relative to vehicle-treated controls (Fig. 5A,B). A small amount of internalized β2/3 subunits observed in neurons treated with vehicle is likely to be due to the constitutive internalization of GABAA receptors (van Rijnsoever et al., 2005).

Dopamine receptor activation decreases the cell-surface expression of GABAA receptors via a dynamin-dependent pathway. A, B, Internalization of GABAA receptors was visualized by staining with β2/3-specific antibody in the presence of vehicle, SKF-38393 (A), or quinpirole (B), as described in Materials and Methods. Binding of β2/3-specific antibody to the surface expressed GABAA receptors was visualized using a secondary anti-mouse IgG coupled to Alexa555 (in red), while binding of the same antibody to internalized receptors was visualized using a secondary anti-mouse IgG coupled to Alexa488 (in green). Scale bars correspond to 10 μm. C, The decrease in surface β2/3 subunit levels caused by SKF-38393 (SKF) was significantly attenuated in the presence of a dynamin inhibitory peptide (SKF/DynP), but not in the presence of its scrambled control (SKF/DynPC). D, The decrease in surface β2/3 subunit levels caused by quinpirole (Q) was also significantly attenuated in the presence of a dynamin inhibitory peptide (Q/DynP), but not by its scrambled control (Q/DynPC). Bars represent mean ± SEM. Statistical analysis was performed using one-way ANOVA with Dunnett post hoc analysis versus control (*p < 0.05; **p < 0.01) or paired t test (#p < 0.05; ##p < 0.01).
To investigate these processes further, we performed cell-surface ELISA assays in the presence of an inhibitory peptide that specifically prevents clathrin-dependent endocytosis by blocking the binding of dynamin to amphiphysin (Gout et al., 1993; Grabs et al., 1997; Lissin et al., 1998; Marks and McMahon, 1998; Nong et al., 2003). This peptide (DynP) significantly attenuated the ability of SKF-38393 to decrease surface β2/3 subunit levels from 86.5 ± 2.2% of control to 96.6 ± 2.1% (p < 0.05, paired t test; n = 4) (Fig. 5C), and quinpirole from 88.1 ± 1.2% of control to 95.6 ± 2.0% of control (p < 0.01, paired t test; n = 4) (Fig. 5D). In contrast, the scrambled control version of this peptide (DynPC) had no significant effect (Fig. 5C,D). SKF-38393 or quinpirole had no effect on total β2/3 levels (102.3 ± 2.7% of control), and this was unaffected by the presence of DynP (97.9 ± 4.6% of control) or DynPC (97.0 ± 5.7% of control; p > 0.05, paired t test; n = 4; data not shown). These results indicate that both D1Rs and D2Rs mediate the reduction in the cell-surface expression of β2/3 subunit-containing GABAA receptors, at least in part, through an increase in dynamin-mediated endocytosis.
D1Rs and D2Rs activate distinct signaling pathways to reduce cell-surface levels of GABAA receptors
D1Rs and D2Rs are coexpressed in the embryonic precursors of medium spiny neurons both in vivo and in vitro (Figs. 1, 2) (Aizman et al., 2000). Furthermore, both receptor types are activated by dopamine to control the formation of inhibitory synapses and regulate cell-surface expression of GABAA receptors. Although evidently impinging on the same functional target, these receptors are likely to activate distinct signaling cascades in these neurons (Greengard, 2001). We used specific activators and inhibitors of a number of intracellular signaling molecules to characterize these signaling cascades.
cAMP/PKA signaling
D1Rs and D2Rs are classified by their positive or negative coupling to Gs or Gi/o G-proteins that regulate adenylyl cyclase (AC); it is this coupling that is responsible for a large number of downstream cellular effects (Missale et al., 1998). To determine whether AC mediates regulation of surface GABAA receptors by D1Rs or D2Rs, we examined the effects of forskolin, an activator of AC (Awad et al., 1983; Laurenza et al., 1989). Neurons were pretreated with vehicle or forskolin (10 μm) for 10 min, after which D1Rs and D2Rs were activated with SKF-38393 (1 nm) or quinpirole (100 nm) in the absence or presence of forskolin. Changes in the surface and total levels of β2/3 subunits were examined using cell-surface ELISA. We demonstrate that forskolin treatment alone caused an apparent increase in β2/3 surface levels to 108.2 ± 3.3% of control, although this was not statistically significant (p > 0.05, one-way ANOVA; n = 4; data not shown). The decrease in surface β2/3 levels after 30 min treatment with SKF-38393 (83.5 ± 3.1% of control, p < 0.05, one-way ANOVA; n = 4) (Fig. 6A, SKF) was significantly attenuated by forskolin (98.5 ± 5.1% of control, p < 0.05; paired t test; n = 4) (Fig. 6A, SKF/F). In contrast, the decrease observed in the presence of quinpirole (85.2 ± 3.9% of control, p < 0.05, one-way ANOVA; n = 5) (Fig. 6A, Q) was unaffected (88.4 ± 4.3% of control, p < 0.05, one-way ANOVA; n = 5) (Fig. 6A, Q/F) by the presence of forskolin. Forskolin had no significant effect on the total levels of β2/3 subunits in the absence (105.8 ± 4.4% of control, p > 0.05, one-way ANOVA; n = 4) or presence of SKF-38393 (113.5 ± 3.2% of control; p > 0.05, one-way ANOVA; n = 4) or quinpirole (105.1 ± 5.1% of control, p > 0.05, one-way ANOVA; n = 5, data not shown). These results suggest that stimulation of AC by forskolin occludes the ability of D1Rs to activate AC and regulate GABAA receptor levels, without affecting the D2R-dependent regulation of these receptors.

D1R-dependent reduction of the GABAA receptor levels expressed at the cell surface is mediated by the activities of PKA, ERK1 and ERK2, and PP2A, whereas D2R-dependent decrease is mediated by the activity of PP1. A, The effect of SKF-38393 (SKF), but not quinpirole (Q), was occluded by a direct activation of adenylyl cyclase (AC) with forskolin (SKF/F, Q/F, respectively). B, The effect of SKF-38393 (SKF), but not quinpirole (Q), was also occluded by a direct activation of protein kinase A (PKA) with 8-bromo-cAMP (SKF/cAMP, Q/cAMP, respectively). C, The effect of SKF-38393 (SKF), but not quinpirole (Q) was attenuated by inhibition of ERK kinases (ERK1 & 2) with PD-98059 (SKF/PD, Q/PD, respectively). D, The inhibition of protein kinase C (PKC) by calphostin C (CC) had no effect on SKF-38393-dependent (SKF vs SKF/CC), or quinpirole-dependent (Q vs Q/CC) decrease in β2/3 surface levels. E, Buffering of extracellular calcium with EGTA (EGTA) had no effect on SKF-38393-dependent (SKF vs SKF/EGTA), or quinpirole-dependent (Q vs Q/EGTA) decrease in β2/3 surface levels. F, Buffering of intracellular calcium with BAPTA-AM (BAPTA) had no effect on SKF-38393-dependent (SKF vs SKF/BAPTA), or quinpirole-dependent (Q vs Q/BAPTA) decrease in β2/3 surface levels. G, The decrease in surface β2/3 levels caused by SKF-38393 (SKF), but not quinpirole (Q) was abolished by a low concentration of okadaic acid (0.1 μm; SKF/OA 0.1, Q/OA 0.1, respectively), while both SKF-38393-dependent (SKF) and quinpirole-dependent (Q) decreases were abolished by a high concentration of okadaic acid (1 μm; SKF/OA 1, Q/OA 1, respectively). H, The effect of SKF-38393 (SKF), but not quinpirole (Q), was significantly attenuated by fostriecin (SKF/Fos, Q/Fos, respectively). Bars represent mean ± SEM. Statistical analysis was performed using one-way ANOVA with either a Dunnett post hoc analysis versus control (*p < 0.05; **p < 0.01) or paired t test (#p < 0.05; ##p < 0.01).
To test whether PKA participates as a downstream component of D1R/AC signaling cascade, we treated striatal neurons with the selective PKA activator 8-bromo-cAMP (10 μm) in the absence or presence of SKF-38393 (1 nm) or quinpirole (100 nm) for 30 min. Direct activation of PKA with 8-bromo-cAMP caused a small but significant increase in the surface expression of β2/3 subunit to 112.8 ± 4.0% of control (p < 0.05, one-way ANOVA with Dunnett post hoc test; n = 5; data not shown), and abolished (101.4 ± 3.1% of control, p < 0.05; paired t test; n = 5) (Fig. 6B, SKF/cAMP) the SKF-38393-dependent decrease in β2/3 levels to 89.0 ± 2.9% of control (p < 0.05; one-way ANOVA with Dunnett post hoc analysis; n = 5) (Fig. 6B, SKF). However, quinpirole-dependent decrease in surface β2/3 levels (81.0 ± 7.3% of control, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 4) (Fig. 6B, Q) was unaffected by 8-bromo-cAMP (82.6 ± 6.3% of control, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 4) (Fig. 6B, Q/cAMP). The total levels of β2/3 subunits in the absence (101.2 ± 8.9% of control) or presence of SKF-38393 (118.2 ± 10.4% of control; p > 0.05, one-way ANOVA; n = 5) or quinpirole (103.5 ± 10.3% of control, p > 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 4) were also unaffected by 8-bromo-cAMP (SKF/8BrcAMP: 107.1 ± 12.4% of control; quinpirole/8BrcAMP: 108.3 ± 7.2% of control, p > 0.05, one-way ANOVA; n = 5 and n = 4, respectively, data not shown).
To further support the role of PKA in D1R/AC signaling pathway, we treated striatal neurons with the PKA inhibitor KT-5720 (5 μm) in the absence or presence of SKF-38393 (1 nm) for 30 min. While PKA inhibition with KT-5720 alone had no significant effect on surface β2/3 levels (96.1 ± 6.1% of control; p > 0.05, one-way ANOVA; n = 3), it significantly enhanced D1R-mediated decrease in β2/3 cell-surface expression from 87.1 ± 4.2% of control to 77.3 ± 5.5% of control (p = 0.024, paired t test; n = 3, data not shown). Total β2/3 levels were unaffected in the presence of KT-5720 (99.5 ± 2.1% of control, p > 0.05, one-way ANOVA; n = 3) or SKF (115.8 ± 6.5% of control) or both (114.7 ± 8.1% of control; p > 0.05, one-way ANOVA; n = 3; data not shown).
MEK/ERK1 and ERK2 signaling
D1Rs, but not D2Rs, are known to stimulate MAP kinases ERK1 and ERK2 (Valjent et al., 2005) via activation of an upstream kinase, MAP kinase/ERK kinase (MEK) (Sefton, 2001). We have observed that SKF-38393 but not quinpirole significantly increased the phosphorylation/activity of ERK1/2 after 30 min treatment (data not shown). Furthermore, we found that inhibition of MAP kinase kinase, MEK, with PD-98059 (50 μm) attenuated the ability of SKF-38393 to decrease GABAA receptor cell-surface levels from 87.0 ± 3.5% (Fig. 6C, SKF) to 98.5 ± 2.5% of control (p < 0.05, paired t test, n = 7) (Fig. 6C, SKF/PD). In contrast, PD-98059 had no significant effect quinpirole-dependent regulation: quinpirole decreased surface β2/3 levels to 86.9 ± 4.2% of control (Fig. 6C, Q) and to 85.9 ± 5.5% of control in the presence of PD-98059 (p > 0.05, paired t test, n = 6) (Fig. 6C, Q/PD). PD-98059 did not significantly affect cell-surface β2/3 levels when added alone (data not shown).
PKC signaling
PKC is known to regulate surface levels of GABAA receptors in cultured hippocampal and cortical neurons by inhibiting the recycling of internalized receptors to the plasma membrane (Connolly et al., 1999). To determine whether PKC is involved in modulating surface levels of GABAA receptors in cultured striatal neurons, we used calphostin C (0.2 μm), a PKC inhibitor that inhibits both typical and atypical isoforms of this enzyme (Kobayashi et al., 1989). In the presence of calphostin C alone, surface β2/3 levels were unaffected (97.5 ± 10.1% of control; p > 0.05, one-way ANOVA; n = 5; data not shown). A significant reduction in surface β2/3 levels by SKF-38393 to 80.7 ± 4.7% of control (p < 0.05, one-way ANOVA; n = 5) (Fig. 6D, SKF), or by quinpirole to 85.2 ± 3.9% of control (p < 0.05, one-way ANOVA; n = 5) (Fig. 6D, Q), was unaffected by calphostin C [72.6 ± 5.3% of control, p > 0.05, paired t test; n = 5 (Fig. 6D, SKF/CC); and 84.8 ± 5.1% of control, p > 0.05, paired t test; n = 5 (Fig. 6D, Q/CC)]. Total β2/3 levels remained unchanged following the treatment with calphostin C alone, or in combination with SKF-38393 or quinpirole (data not shown). Thus, PKC activity is not required for either D1R- or D2R-dependent regulation of surface β2/3 levels in the embryonic striatal neurons.
Ca2+-dependent signaling
Ca2+ plays a fundamental role as an intracellular signal by directly regulating the activity of a large number of signaling molecules including protein kinases and phosphatases (Walaas and Greengard, 1991), which have been previously implicated in both D1R (Surmeier et al., 1995; Tang and Bezprozvanny, 2004; Iwakura et al., 2008) and D2R (Surmeier et al., 1992; Aizman et al., 2000; Tang and Bezprozvanny, 2004) signaling in striatal neurons. To examine a possible requirement for Ca2+ and Ca2+-dependent signaling pathways in the regulation of GABAA receptor surface levels, we selectively buffered extracellular or intracellular Ca2+ before the activation of dopamine receptors. To examine the contribution of extracellular Ca2+, we applied a Ca2+-chelating agent EGTA (1 mm). We demonstrate that EGTA had no effect on the surface levels of β2/3 subunits (p > 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 5; data not shown). Importantly, SKF-38393-dependent (88.5 ± 1.2% of control, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 5) (Fig. 6E, SKF) and quinpirole-dependent (85.5 ± 3.4% of control, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 4) (Fig. 6E, Q) decreases in surface β2/3 levels were unaffected by the presence of EGTA (82.5 ± 5.0% of control, SKF/EGTA, and 85.8 ± 2.6% of control, Q/EGTA, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 5 and n = 4, respectively) (Fig. 6E). We also demonstrate that changes in total β2/3 levels were not statistically significant following the treatment with EGTA alone or in combination with SKF-38393 or quinpirole (data not shown).
To buffer intracellular Ca2+, we applied a high-affinity, membrane-permeable Ca2+ chelator, BAPTA-AM (25 μm). BAPTA-AM treatment alone had no effect on surface β2/3 levels (p > 0.05, one-way ANOVA; n = 3; data not shown). Neither the SKF-38393-dependent (89.5 ± 1.0% of control, p < 0.01, one-way ANOVA; n = 3) (Fig. 6F, SKF), or quinpirole-dependent (72.3 ± 4.5% of control, p < 0.05, one-way ANOVA with Dunnett post hoc analysis; n = 5) (Fig. 6F, Q) decrease in surface β2/3 levels was affected by BAPTA-AM [89.5 ± 0.7% of control, p < 0.01, one-way ANOVA; n = 3 (Fig. 6F, SKF/BAPTA); and 78.9 ± 4.9% of control p < 0.05, one-way ANOVA; n = 5 (Fig. 6F, Q/BAPTA)]. Total β2/3 levels were not significantly affected following treatment with BAPTA alone or in combination with SKF-38393, or quinpirole (data not shown). Together, these data suggest that changes in [Ca2+]i and Ca2+-dependent signaling proteins are not involved in the regulation of surface GABAA receptors by D1Rs or D2Rs.
Protein phosphatase signaling
Protein phosphatases PP1 and PP2A have been shown previously to associate with GABAA receptors (Jovanovic et al., 2004; Terunuma et al., 2004). To investigate whether their activity is required for D1R- or D2R-dependent regulation of surface β2/3 levels, we incubated striatal neurons with 0.1 μm okadaic acid, a concentration sufficient to block PP2A activity, or with 1 μm okadaic acid, a concentration that inhibits both PP1 and PP2A (Nishi et al., 1997). We demonstrate that treatments with both 0.1 and 1 μm okadaic acid significantly attenuated the ability of SKF-38393 to reduce β2/3 surface levels from 82.1 ± 5.9% of control (p < 0.05, one-way ANOVA; n = 5) (Fig. 6G, SKF) to 101.6 ± 5.6% of control, and to 95.3 ± 6.2% of control (p < 0.05, paired t test, n = 3, SKF/OA 0.1, or n = 4, SKF/OA 1, respectively) (Fig. 6G). In contrast, quinpirole-dependent decrease in surface β2/3 levels (87.0 ± 2.7% of control, p < 0.05, one-way ANOVA; n = 5) (Fig. 6G, Q) was unaffected by 0.1 μm okadaic acid (80.6 ± 4.7% of control, p > 0.05, paired t test, n = 5) (Fig. 6G, Q/OA 0.1), but significantly attenuated by 1 μm okadaic acid (105.3 ± 3.4% of control, p < 0.01, paired t test, n = 5) (Fig. 6G, Q/OA 1). Okadaic acid had no significant effect on surface β2/3 levels at either concentration tested (data not shown). Total β2/3 levels were not significantly affected following treatment with okadaic acid alone or in combination with SKF-38393 or quinpirole (data not shown).
The role of PP2A was confirmed using a selective PP2A antagonist fostriecin (100 nm) (Stampwala et al., 1983). The ability of SKF-38393 to decrease β2/3 subunit cell-surface levels (82.0 ± 3.3% of control, one-way ANOVA; n = 4) (Fig. 6H, SKF) was significantly attenuated by fostriecin (97.5 ± 2.7% of control, p < 0.05, paired t test, n = 4) (Fig. 6H, SKF/Fos). In contrast, quinpirole-dependent decrease in surface β2/3 levels (83.8 ± 5.4% of control, p < 0.05, one-way ANOVA; n = 3) (Fig. 6H, Q) was unaffected by fostriecin (77.8 ± 3.0% of control, p > 0.05, paired t test, n = 3) (Fig. 6H). Thus, D1R-dependent regulation of surface GABAA receptors operates via activation of AC/PKA signaling pathway, ERK kinases, and PP2A, while D2R-dependent regulation requires the activity of PP1 in embryonic striatal neurons.
D1R and D2R activation transiently dephosphorylates GABAA receptors
Given that D1R- and D2R-dependent regulation of surface GABAA receptors requires the activity of protein phosphatases PP2A and PP1, we hypothesized that GABAA receptor themselves may be dephosphorylated by these phosphatase to trigger their endocytosis, as already demonstrated in the adult striatal tissue (Kittler and Moss, 2003; Kittler et al., 2005, 2008; Chen et al., 2006). Thus we used a phosphorylation state-specific antibody [anti-Pβ antibody (Jovanovic et al., 2004)], which selectively binds to P-Ser409 in the β1 and β3, and P-Ser410 in the β2 subunit of GABAA receptors, to determine the state of phosphorylation of these receptors in response to D1R or D2R activation. Using quantitative immunoblotting, we demonstrate that SKF caused a rapid dephosphorylation of β subunits (75.9 ± 4.3% of control, p < 0.05, paired t test; n = 3) (Fig. 7A, SKF), which was abolished in the presence of PP2A inhibitor fostriecin (120.8 ± 11.6% of control, p < 0.05, paired t test; n = 3) (Fig. 7A, SKF/Fos). Additionally, fostriecin caused an apparent increase in the basal levels of β phosphorylation to 125.3 ± 21.9% of control (p > 0.05, paired t test; n = 3) (Fig. 7A, Fos). Interestingly, MEK inhibitor PD-98059 (50 μm) also abolished dephosphorylation of β subunits by SKF (124.3 ± 11.0% of control, p < 0.05, paired t test; n = 3) (Fig. 7A, SKF/PD), and produced an apparent increase in β phosphorylation (125.7 ± 14.0% of control, p > 0.05, paired t test; n = 3) (Fig. 7A, PD) on its own. Total levels were not significantly affected by SKF-38393 or any other treatment (data not shown). Activation of D2Rs with quinpirole caused a significant decrease in phosphorylation of β subunit to 79.3 ± 6.2% of control (p < 0.05, paired t test; n = 4) (Fig. 7B, Q). Pretreatment with the PP1 inhibitor okadaic acid (1 μm) increased significantly the basal levels of β subunit phosphorylation to 726.6 ± 76.4% of control (p < 0.05, paired t test; n = 4) (Fig. 7B, OA), and completely abolished dephosphorylation of β subunits caused by quinpirole (664.7 ± 110.4% of control, p > 0.05, paired t test; n = 4) (Fig. 7B, Q/OA). Thus, it appears that PP2A and PP1 dephosphorylate GABAA receptor β subunits in response to D1R and D2R activation, respectively.

D1R and D2R activation leads to a transient dephosphorylation of GABAA receptor β subunits. A, Treatment with SKF-38393 (1 nm, 30 min) led to a decrease in β subunit phosphorylation (SKF), which was abolished by fostriecin (SKF/Fos) or PD-98059 (SKF/PD). Both inhibitors alone caused small but insignificant increases in the basal phosphorylation state of the β subunits (Fos, PD, respectively). A representative immunoblot is shown below the bar graph. B, Treatment with quinpirole (100 nm, 30 min) also led to a decrease in β subunit phosphorylation (Q), which was abolished by okadaic acid (Q/OA). Okadaic acid alone caused a prominent increase in the basal phosphorylation state of the β subunits (OA). A representative immunoblot is shown below the bar graph. C, D, The time course of changes in the phosphorylation state of the β subunits in the presence of SKF-38393 (SKF), quinpirole (Q), or both (SKF/Q). Representative immunoblots (C) and graphs showing percentage values normalized to the appropriate controls treated with the vehicle (100%; D) show a transient decrease in the phosphorylation state of the β subunits at an early time point (5–10 min) and a later time point (60 min) of incubation with either SKF-38393 (SKF, in red), quinpirole (Q, in green), or both (SKF/Q, in blue). Values represent mean ± SEM. Statistical analysis was performed using one-way ANOVA with either a Dunnett post hoc analysis versus control (*p < 0.05) or paired t test (#p < 0.05).
To characterize further PP1- and PP2A-mediated dephosphorylation of GABAA receptors, we performed in vitro phosphatase assays using purified enzymes, catalytic subunit of PP1 (PP1c), PP2A (PP2A), and calcineurin, and purified phosphorylated GST-fusion proteins containing the intracellular TM3-4 loops of the β1-3 subunits (see Materials and Methods). In these experiments, the initial rates of dephosphorylation of [32P]-phospho-GST-β1-3 subunits, and [32P]-phospho-DARPP-32 or [32P]-phosphorylase a used as reference substrates, were assessed by measuring the release of phosphate (Table 1). Compared to the reference substrates, all three β subunits appeared to be good substrates for PP1c and PP2A, but poor substrates for calcineurin. These initial results were extended by a kinetic analysis of [32P]-phospho-GST-β1-3 dephosphorylation with PP1c and PP2A (Table 2). Two kinetic parameters, Km (the apparent affinity for substrate), and Vmax (the rate), were determined demonstrating a high affinity and rapid dephosphorylation of all three β subunits by PP1c and PP2A.
Importantly, activation of dopamine receptors leads to a significant but transient dephosphorylation of GABAA receptor β subunits. We demonstrate that application of SKF-38393 (1 nm), caused a significant decrease in phosphorylation of the β subunits at 10 min to 87.0 ± 3.3% of control (p < 0.05, one-way ANOVA; n = 3), and, at 60 min to 86.5 ± 2.5% of control (p < 0.05, paired t test; n = 5) (Fig. 7C,D, SKF), which was reversed to the basal levels at later time points. The application of quinpirole (100 nm) caused a significant decrease in phosphorylation of the β subunits within 5 min to 82.5 ± 5.2% of control (p < 0.05, one-way ANOVA; n = 4), but only an apparent decrease at 60 min to 88.4 ± 6.0% of control (p > 0.05, one-way ANOVA; n = 4) (Fig. 7C,D, Q). Importantly, when both SKF-38393 and quinpirole were applied at the same time, we observed a similar transient decrease in β subunit phosphorylation to 83.7 ± 4.0% of control at 5 min, and to 79.9 ± 8.0% of control at 60 min, which was reversed to the basal levels at later time points (p < 0.05, one-way ANOVA; n = 4) (Fig. 7C,D, SKF/Q).
Thus, dopamine receptor activity exerts its powerful control of inhibitory synaptogenesis in the developing striatum by regulating the levels of GABAA receptors expressed at the cell surface through a mechanism that involves transient dephosphorylation of these receptors.
Discussion
With its essential role as the major input area to the basal ganglia and its dysfunction in the pathogenesis of diseases such as Parkinson's and Huntington's diseases, the striatum represents one of the primary subjects of scientific interest. While there is increasing research into the function of the adult striatum, very little work has focused on its development. The striatum receives considerable dopaminergic input from the substantia nigra early in development, and the evidence suggests that dopamine may play an important role. For example, in utero exposure to cocaine profoundly affects development of neuron dendrites (Jones et al., 1996; Levitt et al., 1997) through the activation of dopamine receptors (Jones et al., 2000; Stanwood and Levitt, 2007). Here we present evidence for a novel role of dopamine in the formation of GABAergic synapses between embryonic precursors of striatal medium spiny neurons, and uncover the underlying signaling mechanisms.
We first demonstrate the presence of dopaminergic (TH+) fibers and terminals in the embryonic E17 striatum in vivo, and estimate the in vivo levels of tissue dopamine using HPLC. Moreover, we reveal that dopamine receptors (both D1Rs and D2Rs) are coexpressed in the large population of neurons within the embryonic striatum. Our experiments further demonstrate that activation of these receptors produces a robust decrease in the number of GABAergic synaptic contacts as indicated by a reduction in colocalization between GABAA receptor-positive clusters and VIAAT-1/FM1-43FX-positive presynaptic nerve terminals. Similar type of analysis has been used extensively to study formation of GABAergic synapses (Chih et al., 2005; Dong et al., 2007; Yu et al., 2007). Furthermore, our functional experiments demonstrate that the observed mislocalization of synaptic elements leads to a reduction in the frequency of spontaneous GABAA receptor-mediated synaptic potentials (mIPSPs), as well as a reduction in their amplitudes. The latter also correlates with a reduction in the number of GABAA receptors present at the postsynaptic membrane (Nusser et al., 1997, 1998). Accordingly, immunocytochemical analysis of GABAA receptor-positive clusters, as well as biochemical measurements of cell-surface levels of GABAA receptors using ELISA, clearly demonstrates that this is indeed the case. However, it is important to note that despite the reduction in size, the overall number of postsynaptic GABAA receptor clusters, as well as the size and number of presynaptic VIAAT-1-positive terminals, remains unaltered. This suggests that formation and/or stability of functional GABAergic synapses may depend on the size of GABAA receptor clusters present at the postsynaptic membrane, in addition to the type of GABAA receptors that form these clusters. In support of this hypothesis is the observation that, in many brain regions, extrasynaptic clusters of GABAA receptors are significantly smaller than their synaptic counterparts (Christie et al., 2002a,b). Overall, the ability of striatal neurons to form functional synaptic contacts with each other appears to be restricted by dopamine during development. Dopamine is known to decrease the number of glutamatergic synapses present on D2R-containing medium spiny neurons in the adult striatum, since the depletion of dopamine in the rat models of Parkinson's disease leads to an increase in the number of synapses (Day et al., 2006). Together with these observations, our experiments suggest that the effects of dopamine on the structure of synaptic contacts may be widespread in the developing striatum as well as in the adult striatum. In addition, given that dopamine receptor activation leads to a reduction in cell-surface levels of GABAA receptors, which is concomitant with a reduction in the number of GABAergic synapses, we hypothesize that GABAA receptors promote development of GABAergic synapses not only as the “building blocks” of GABAergic synapses, but also through their activity. Indeed, GABAA receptor activity has been suggested to play an important role in formation and/or stabilization of synapses in the adult neurons (Li et al., 2005; Chattopadhyaya et al., 2007).
Dopamine-dependent reduction in the levels of GABAA receptors expressed at the cell surface, which we demonstrate is due to increased endocytosis of these receptors, and a decrease in GABAergic currents, appear to be preserved in the striatum until adulthood, when D1Rs activation causes similar effects (Flores-Hernandez et al., 2000; Hernández-Echeagaray et al., 2007). Moreover, in other brain regions, for example, in the nucleus accumbens (Chen et al., 2006) and hippocampus (Graziane et al., 2009), D2Rs cause a decrease in cell-surface expression and GABAA receptor-mediated currents. It is intriguing however, that the functionally opposing signaling pathways coupled to D1Rs and D2Rs, which are clearly segregated to different types of medium spiny neurons in the adult striatum (Surmeier et al., 2007), operate jointly within their embryonic counterparts to produce quantitatively similar changes in GABAA receptor surface levels as demonstrated here.
To elucidate the signaling players underpinning this regulation, we first demonstrate that D1Rs decrease the cell-surface expression of GABAA receptors and their phosphorylation state through an intracellular signaling pathway involving AC/PKA, ERK1/2, and PP2A (Figs. 6⇑–8). The question remains as to how AC, PP2A, and ERK1/2 can act in concert to cause the observed dephosphorylation and endocytosis of GABAA receptors. One possible mechanism is that D1Rs decrease the phosphorylation of GABAA receptors through the activity of PP2A (Fig. 8). Indeed this has been shown to occur in other systems (Jovanovic et al., 2004), and PP2A can be activated by D1Rs through PKA-mediated phosphorylation (Ahn et al., 2007). However, PKA is also known to associate with GABAA receptors and phosphorylate β subunits directly (Brandon et al., 2003). Therefore it seems likely that the balance between these two signaling pathways, possibly mediated by two spatially segregated pools of PKA, will determine the phosphorylation state of GABAA receptors, but is this balance critical for the regulation of GABAA receptor endocytosis? Although we demonstrate a clear correlation between D1R- and D2R-dependent reduction in both GABAA receptor cell-surface expression and phosphorylation state, it must be noted that the former is long lasting, while the latter is only transient. Thus, the receptor dephosphorylation may be required only for the initiation of endocytosis, but not for the subsequent maintenance of the appropriate levels of GABAA receptors at the cell surface. In the adult tissue, dynamin-dependent endocytosis of GABAA receptors has been reported to depend on their phosphorylation state (Kittler et al., 2000, 2005, 2008). Similar phosphorylation-dependent mechanisms have been shown to underlie D3R-induced internalization of GABAA receptors in the adult nucleus accumbens (Chen et al., 2006).

Functional cross talk between D1R and D2R signaling pathways regulating GABAA receptors in the embryonic striatal neurons. Activation of D1R with SKF-38393 (in red) leads to activation of adenylyl cyclase (AC) resulting in activation of conceivably two segregated pools of protein kinase A (PKA). The first pool of PKA is associated with GABAA receptors leading to a rapid phosphorylation of GABAA receptors (i). The second pool of PKA is more distal and unable to associate with GABAA receptors. This pool of PKA directly phosphorylates and activates protein phosphatase 2A (PP2A, ii). Activated PP2A associates with and dephosphorylates GABAA receptors, promoting their internalization from the cell surface (iii). The ability of PP2A to dephosphorylate GABAA receptors is likely to be enhanced due to ERK1/2-mediated phosphorylation and activation of PP2A (iv). ERK1/2 can be activated by two different signaling pathways both involving PKA. First, PKA can directly phosphorylate and thus inactivate striatal enriched phosphatase (STEP, v). In this way, STEP is no longer able to dephosphorylate and deactivate ERK1/2. Second, PKA can lead to the activation of DARPP-32, an inhibitor of protein phosphatase 1 (PP1, vii). Inactivated PP1 cannot dephosphorylate and activate STEP, which is then unable to dephosphorylate ERK1/2 (viii). Both of these pathways lead to an increase in the level of phosphorylated ERK1/2, which acts on PP2A to promote dephosphorylation of GABAA receptors. Activation of D2Rs by quinpirole (in green) leads to a dephosphorylation of GABAA receptors and their internalization by inhibiting AC. Inhibition of AC leads to a reduction in the activation of PKA and consequently to a reduction in PKA-mediated phosphorylation of GABAA receptors (i). At the same time however, inhibition of AC may lead to a decrease in DARPP-32 activity and thus activation of PP1 (vii). Activated PP1 is then capable of associating with GABAA receptors and mediating their dephosphorylation and subsequent internalization (ix).
The role of ERK1 and ERK2 kinases in D1R-dependent regulation of surface GABAA receptors is intriguing. As schematically depicted in the Figure 8, D1Rs may regulate the activity of ERK1 and ERK2 kinases through an inhibition of the striatal-enriched tyrosine phosphatase STEP (Paul et al., 2000; Valjent et al., 2005; Braithwaite et al., 2006b), which was previously shown to dephosphorylate and inactivate ERK kinases (Braithwaite et al., 2006b; Venkitaramani et al., 2009). D1R-triggered increase of ERK activity may also be mediated through DARPP-32-dependent and -independent mechanisms (Gerfen et al., 2008). Namely, D1Rs can lead to an activation of DARPP-32 through a PKA- and/or PP2A-dependent signaling mechanism (Svenningsson et al., 2004) resulting in a decrease in the activity of PP1. This could further lead to an increase in STEP phosphorylation, which is concomitant with an inhibition of its activity and consequent stimulation of ERK kinases. Additionally, DARPP-32 appears to be required for an increase in phosphorylation of MEK, a dual specificity kinase that directly phosphorylates and thus activates ERK kinases (Valjent et al., 2005). A possible DARPP-32-independent mechanism may occur through a PKA-dependent phosphorylation of STEP, which would decrease its activity and consequently stimulate the activity of ERK kinases (Braithwaite et al., 2006b). Although ERK kinases can be activated in a number of ways by D1Rs, it is unclear how this activation impinges on the regulation of surface GABAA receptors. We hypothesize that activated ERK kinases may stimulate PP2A (Belcher et al., 2005) to dephosphorylate GABAA receptors and trigger their internalization. In support of this hypothesis is the observation that inhibition of ERK kinases by PD 98509 not only abolishes dephosphorylation of β subunits by D1Rs, but also increases their phosphorylation on its own. A possible role of STEP in GABAA receptor regulation is interesting since it has been previously shown to regulate endocytosis of both NMDA and AMPA receptors (Braithwaite et al., 2006a; Zhang et al., 2008).
We have demonstrated that D2R-dependent decreases in both phosphorylation and cell-surface expression of GABAA receptor are mediated by PP1 (Figs. 6⇑–8). D2Rs, by virtue of their ability to decrease PKA activity through inhibition of AC, may lead to a dephosphorylation of DARPP-32 (Nishi et al., 1997). At the same time, reduced activity of PKA may lead to a reduction in the activity of PP2A (Ahn et al., 2007) resulting in an increase in phosphorylation of DARPP-32 at the phosphorylation site, which converts this protein into an inhibitor of PKA (Bibb et al., 1999). The overall effect of these changes may be an increase in the activity of PP1 and a corollary inhibition of PKA. This may lead to a decrease in phosphorylation of GABAA receptors by PP1 as shown previously (Kanematsu et al., 2006). Thus, DARPP-32 and STEP may act as signaling integrators of D1R- and D2R-signaling pathways regulating phosphorylation and surface expression of GABAA receptors in the embryonic striatal neurons.
In summary, tonic activity of dopamine receptors regulates formation of GABAergic synapses in the developing striatum, at least in part, through phosphorylation-dependent changes in cell-surface expression of GABAA receptors. The effects of dopamine and consequences of dopamine-depletion on the structure of GABAergic synapses in the adult striatum await future investigations.
Footnotes
-
This work was supported by the Biotechnology and Biological Sciences Research Council UK New Investigator Grant BB/C507237/1 (to J.N.J.) and a Novartis-funded PhD studentship (to D.G.). We thank Dr. Tom McAvoy, Laura Maruca, and Anya Sihra-Jovanovic for technical assistance, and Dr. Talvinder Sihra for critical reading of this manuscript.
- Correspondence should be addressed to Dr. Jasmina N. Jovanovic, Department of Pharmacology, The School of Pharmacy, University of London, Brunswick Square, London WC1N 1AX, UK. jasmina.jovanovic{at}pharmacy.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
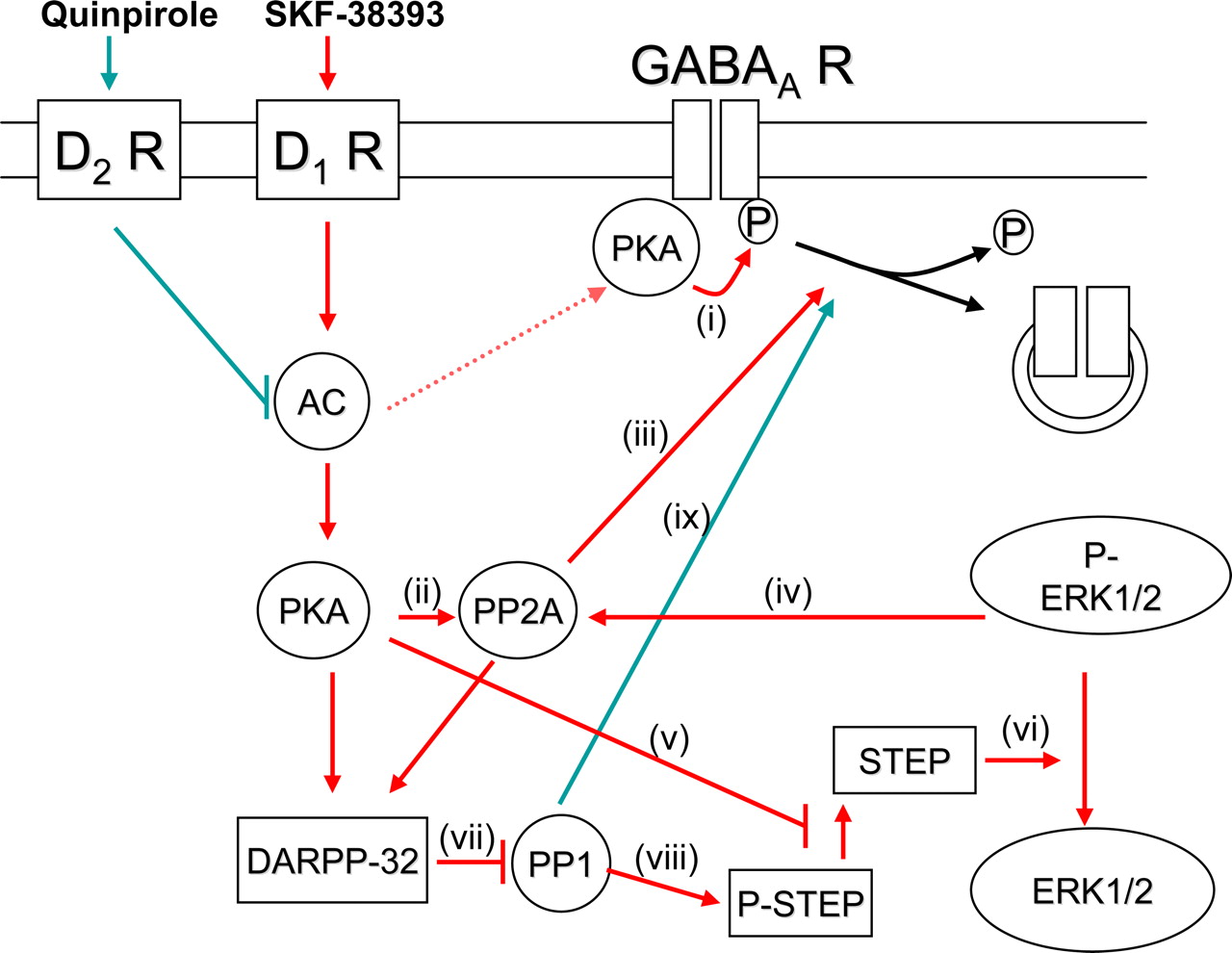{kind=link}