


Abstract
Opioids have been shown to cause a potent inhibition of neurons in the locus ceruleus (LC) in vivo in brain slices and isolated neurons; however, the kinetics of opioid action have not been described. In this study, we used acutely isolated LC neurons to examine opioid and α2-adrenoceptor action on potassium and calcium currents. [Met]Enkephalin (ME), [d-Ser2,Leu5,Thr6]enkephalin, etorphine, and [d-Ala2,N-Me-Phe4,Gly-ol5]enkephalin increased potassium conductance, whereas morphine and naloxone were antagonists. The time constant of potassium channel activation was ∼0.7 sec and was the same for each agonist. The amplitude of the current and the time constant of decay were dependent on the agonist, suggesting that agonist efficacy and affinity, respectively, determined these parameters. The amplitude of potassium current induced by the α2-adrenoceptor agonist UK14304 was not significantly different from that induced by ME, but the time constant of current activation was half that of ME, and the decline was more rapid. When potassium conductances were blocked with the combination of internal cesium and external barium, opioid and α2 agonists had no effect at potentials more negative than −50 mV and decreased barium currents at potentials between −40 and +20 mV. Both morphine and clonidine caused a small inhibition of barium current. In dorsal root ganglion cells, morphine alone had small and inconsistent effects on the calcium current, but it always competitively antagonized the inhibition caused by [d-Ala2,N-Me-Phe4,Gly-ol5]enkephalin. The results in isolated LC neurons suggest 1) the amplitude and time course of the opioid-induced potassium current depend on agonist efficacy and affinity and 2) the coupling of both μ-opioid and α2-adrenoceptors to calcium channels seems to be more efficient than that to potassium channels.
Opioid inhibition of spontaneous activity in the LC was first observed using extracellular recordingin vivo (1). In brain slices, the inhibition by both μ-opioids and α2-adrenoceptor agonists results from an increase in the same inwardly rectifying potassium conductance (2-4). Experiments using cell-attached patch recording from acutely dissociated LC cells indicated that opioids increased potassium channel activity through a membrane-delimited pathway (5). Measurements of opioid receptor kinetics are limited in both cell-attached patch recording from isolated cells and whole-cell recordings in brain slices by the slow exchange of drugs, either at the tip of patch pipettes or by diffusion of drugs in brain slices.
Receptors, including the μ-opioid receptor and α2-adrenoceptors, that are linked with Gi/Go-subtype G proteins are known to activate inwardly rectifying potassium currents, decrease calcium currents, and inhibit adenylyl cyclase, all of which have been studied in acutely dissociated cells (6-9). Part of the opioid-sensitive current in LC cells in brain slices has also been suggested to result from either poor voltage control in the dendritic arbor (10) or a sodium-dependent conductance that is depressed by opioids (11). With acutely isolated neurons, the issue of voltage control is eliminated, and the control of the composition of both extracellular and intracellular solutions for the blockade of specific ion channels is improved compared with recordings in brain slices. Acutely isolated cells also offer the distinct advantage for the rapid application and washout of drugs (9). Although much is known about the pharmacology of the μ-opioid receptor and α2-adrenoceptor in the LC based on steady state analysis, nothing is known about kinetics of opioid actions. In this investigation, rapid applications of opioid receptor and α2-adrenoceptor agonists to acutely isolated LC neurons were made, and the effects of these agonists on potassium and calcium currents were measured. The results demonstrate the importance of agonist affinity and efficacy in sculpting the time course of opioid-mediated action.
Materials and Methods
The method used to isolate LC neurons was based on those described for acutely isolated hippocampal neurons (9). Cells were dissociated from slices taken from animals ranging in age from 5 to 14 days. The region of the LC was microdissected from slices using 18-gauge needles under a dissection microscope. Pieces containing the LC were treated with papain (20 units/ml) at 37° for 2 min, placed in a solution containing trypsin inhibitor and bovine serum albumin for 1 min, washed twice in the dissociation solution, and triturated, and LC cells were plated on uncoated plastic petri dishes. The dissociation solution contained 82 mm Na2SO4, 30 mm K2SO4, 10 mm HEPES, 5 mm MgCl2, and 10 mm glucose, pH adjusted to 7.4 and bubbled with 100% O2. The enzyme solution was made up from the dissociation solution with papain (20 units/ml) and l-cysteine (0.33 mg/ml). Inhibitor solution was also made from the dissociation solution with the addition of trypsin inhibitor and bovine serum albumen (1 mg/ml concentration of each).
Whole-cell recordings were made with a patch-clamp amplifier using appropriate series resistance compensation. Access resistance of ≤10 MΩ was considered acceptable and was monitored periodically throughout the experiment. Patch pipettes were filled with 115 mm KMeSO4, 20 mm KCl, 1.5 mm MgCl2, 5 mm EGTA, 2 mm ATP, 0.3 mm GTP, and 5 mm HEPES, pH 7.3. In some experiments, CsCl was substituted for KMeSO4 and KCl in the internal solution. The control external solution contained 146 mm NaCl, 5 mmKCl, 5 mm HEPES, 2 mm CaCl2, and 4 mm MgCl2, pH was adjusted to 7.4 with NaOH, and the osmolarity was adjusted to 320 mOsm using dextrose. Unless otherwise stated, agonist-induced currents (inward) were recorded in a high potassium solution containing 116 mmNaCl, 30 mm KCl, 5 mm HEPES, 2 mmCaCl2, and 4 mm MgCl2, pH adjusted to 7.4, and the osmolarity was adjusted to 320 mOsm using dextrose.
Drugs were applied by fast perfusion flow pipes. An array of four flow pipes (400-μm o.d.) fixed to a piezoelectric translator was used to change the solution perfusing the cell. Solution exchange using this method has a time constant of ∼50 msec measured using the inward current evoked by high potassium solution (Fig. 1A). Each of the flow pipes was connected to a reservoir containing extracellular solution with or without drug. Solutions are warmed in the flow pipes with a heat exchanger just before solutions enter the bath and run continuously through all the pipes during the experiment. Recordings were carried out at 32°. Results are presented as the mean ± standard error and statistical comparisons were made with paired t tests, with significance p < 0.05. Saturating concentrations of agonists (ME, DAMGO, DSLET) were determined by measuring the current induced by at least two concentrations (Table 1).

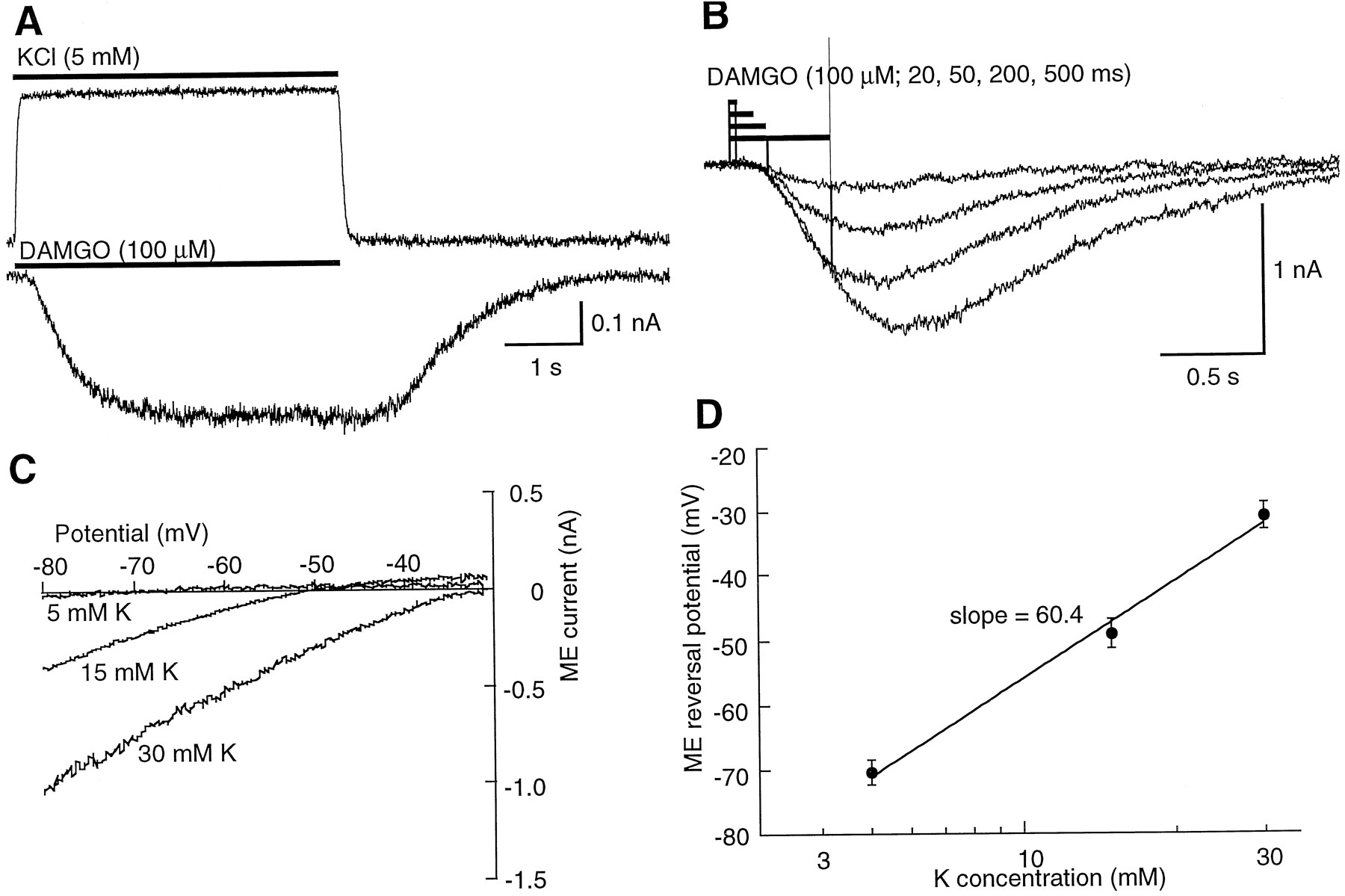
DAMGO activation of potassium conductance. A,Top trace, current induced by switching from a high potassium solution (30 mm) to one containing 5 mm potassium during the period indicated (bar). This current indicates the time course of solution exchange at the cell. Bottom trace, inward current that resulted from changing the superfusion solution from high potassium (30 mm) to high potassium with DAMGO (100 μm) during the period indicated (bar). B, Superimposed current traces of inward currents induced by DAMGO (100 μm) applied for increasing periods of time indicated (bars above the trace). C, Superimposed current-voltage plots of the ME current in solutions of different extracellular potassium. ME currents were obtained by subtraction of the control current from that obtained in the presence of ME (100 μm). Currents were recorded during application of a voltage ramp protocol from −80 to −30 mV (in 1 sec). D, Plot of the reversal potential of the ME-induced current as a function of the extracellular potassium concentration. The slope of this line approximates that predicted by the Nernst equation for a conductance selectively permeable to potassium.
Concentration dependence of opioid agonists
Results
Identification of LC cells.
The identification of dissociated LC neurons was based on their size and shape. Although several cell types could be distinguished in the preparation, the microdissection of the nucleus resulted in an enriched population of LC cells. These cells were large (30 × 50 μm) and multipolar with three to six major dendrites that branched into a few secondary processes. There usually were 5–15 neurons/plate that were suitable for recording. Normally, three to six plates were prepared from a single dissection.
Potassium conductance increase.
From a holding potential of −60 mV, inward potassium currents were evoked by changing the superfusion solution from control to one containing KCl (30 mm). The current induced by changing the extracellular potassium was used to determine the time course of solution exchange at the cell (time constant of exchange = 0.054 ± 0.007 sec, nine experiments; Fig. 1A). In Fig. 1A (top trace), the time course of solution exchange is shown. All agonist-induced currents were recorded at a membrane potential of −60 mV in the presence of 30 mm potassium. Fig. 1A (bottom trace) shows the inward current evoked when the solution was changed from the high potassium solution to a high potassium solution plus DAMGO (100 μm). After the application of DAMGO, there was a latency to the onset of inward current of 0.15 ± 0.02 sec (six experiments) and the current rose to a peak in 1 ± 0.16 sec (six experiments). Repeated short applications of DAMGO (1–5 sec) resulted in reproducible inward currents. Although there was little or no decline in current during a single application of DAMGO (15 sec), the current declined with repeated applications of DAMGO over a period of 5–15 min. The inward current caused by DAMGO was observed with applications that were as short as 20 msec, and the amplitude of the current was dependent on the duration of the application period (Fig.1B).
The reversal potential of the ME current was determined by using a voltage ramp protocol. The ME current was isolated by subtraction of the current obtained in control from that in the presence of ME. The ME current was obtained in three concentrations of potassium (Fig. 1, C and D), and reversal potential determined by this procedure was predicted by the Nernst equation for a potassium selective conductance. This result indicates that ME increases a potassium-selective conductance in isolated LC neurons.
Etorphine, ME, DAMGO, DSLET, and morphine.
The increase in potassium current induced by opioids was further investigated by determining the kinetics of activation and inactivation of DAMGO, ME, etorphine, DSLET, and morphine.
Agonists were applied at maximal (saturating) concentrations so that the kinetics of agonist binding would not limit the rate of onset and time to peak of the response. The concentration for each agonist was chosen based on steady state concentration response curves done in brain slices (4) and from preliminary experiments (Table 1). Despite the use of a saturating concentration of agonist, the amplitude of the potassium current was dependent on the period of application (Fig. 1B). The peak amplitude became larger with increasing duration of application. Currents could be detected with a 50-msec application, and a maximum steady state current was observed with application periods of 1–5 sec (DAMGO: 5 sec = −490 ± 155 pA, three experiments; 1 sec = −494 ± 156 pA, six experiments; 0.5 sec = −429 ± 149 pA, six experiments; 0.2 sec = −349 ± 115 pA, five experiments; 0.1 sec = −277 ± 98 pA, five experiments). Similar experiments with ME resulted in larger mean currents (1 sec = −817 ± 108 pA, five experiments; 0.5 sec = −723 ± 101 pA, five experiments; 0.2 sec = −610 ± 114 pA, five experiments; 0.1 sec = −539 ± 114 pA, five experiments; 0.05 sec = −375 ± 92 pA, five experiments).
In experiments in which ME and DAMGO were applied (1 sec) to the same cell, ME always caused an larger current than DAMGO (DAMGO = −357 ± 32, ME = −482 ± 49, seven experiments;p < 0.05 paired t test). A more complete analysis of the current induced by agonists was done using a 5-sec application of agonist so that the time course of activation (τ-on), the washout (τ-off), and the current amplitude could be measured in each cell (Table 2). To compare the effects of different agonists, ME was tested in each cell along with one of the following: DAMGO, DSLET, etorphine, or the α2-adrenoceptor agonist UK14304. The properties of the current induced by ME was used to make comparisons between each of the other agonists (Table 2). The activation of potassium current induced by each of the opioid agonists was the same (0.7 sec, Table 2). The washout, however, was different for each. The time course of washout was not influenced by rebinding of agonist to the receptor because the recovery on ME washout (1.26 ± 0.16 sec) was not different from that when the solution was stepped from ME to naloxone (1 μm, 1.12 ± 0.13 sec, five experiments; p > 0.05 paired t test).
Summary of kinetic results
The etorphine (1–10 μm) evoked current was very long lasting, requiring a period of ∼1–2 min to fully recover (Fig.2). The current induced by etorphine was smaller in amplitude from that induced by ME. This result suggests that the time course of this G protein-mediated response was dependent at least in part on the high affinity of etorphine for the receptor.

The time course of activation of potassium current is independent of the opioid agonist, whereas washout is agonist dependent. A, Currents induced by a 10-sec application of ME (100 μm) followed by a 10-sec application of etorphine (1 μm). B, Two superimposed current traces showing the activation of potassium current induced by DAMGO (100 μm) and etorphine (1 μm), each applied for 5 sec. The activation of current on agonist application is the same for each. The etorphine-induced current peaks and declines during the application period. All currents were measured at a holding potential of −60 mV in the presence of high potassium (30 mm).
DSLET binds with relatively low affinity to the μ-opioid receptor (K i = 5 μm) (19). The current induced by DSLET (100 μm) activated with the same time constant as that caused by ME, DAMGO, and etorphine (Table 2); however, the time constant of recovery after washout of DSLET was about half that of ME (Fig. 3A). This further suggests that the time course of opioid responses can be predicted from the affinity of agonist for receptor.
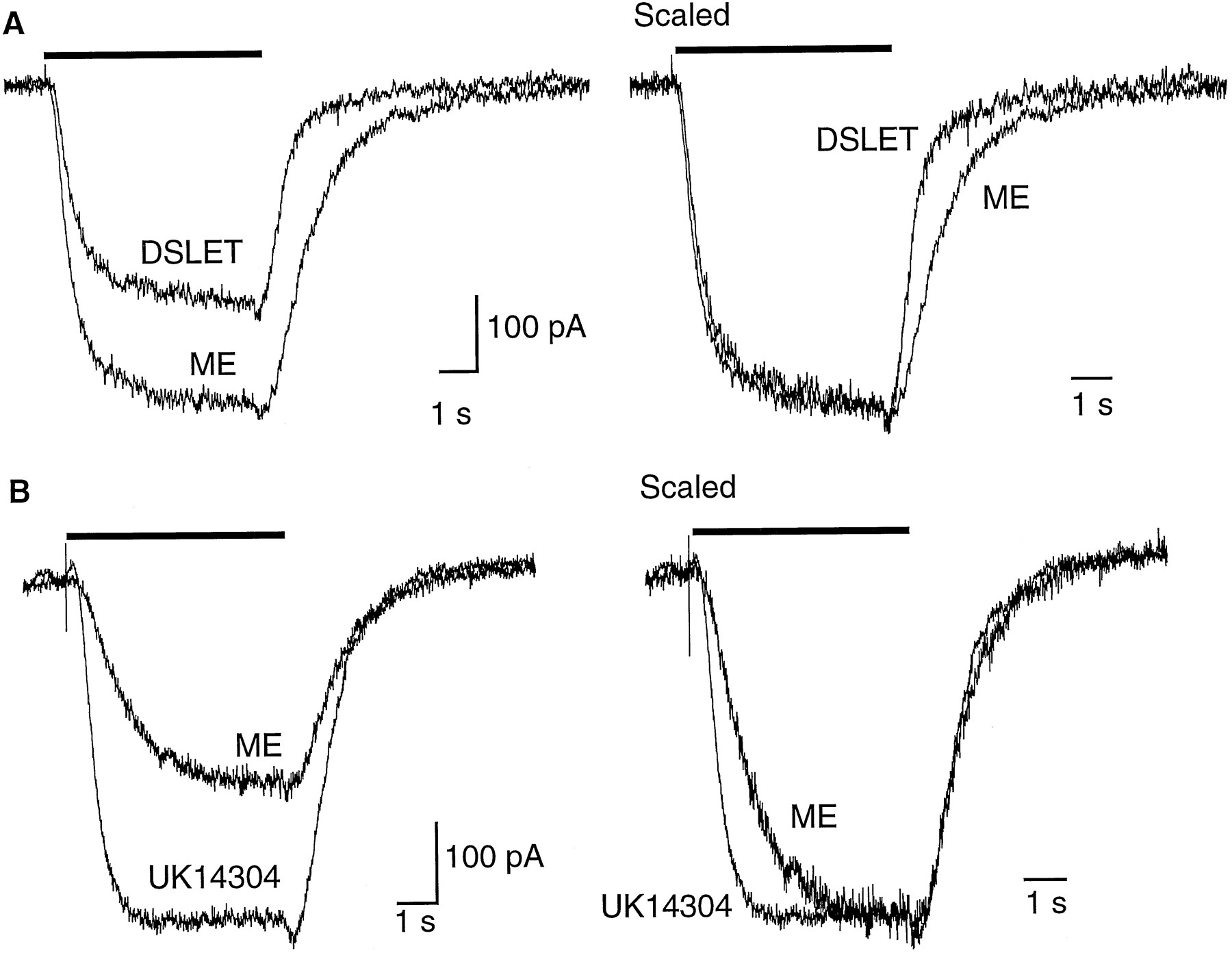
The time course of activation of potassium current is dependent on the receptor subtype. A, Left, superimposed current traces from the same cell illustrating different effects of DSLET and ME. Right, traces have been scaled to show that the rate of rise of current induced by both agonist is the same, whereas the decline of the DSLET current is faster than that of ME. B, Left, a similar experiment as shown in A from a different cell illustrating the currents induced by activation of opioid (ME, 100 μm) and α2-adrenoceptors (UK14304, 1 μm). Right, same traces scaled to illustrate the different time course of activation by UK14304 and ME.
The current induced by DAMGO was also smaller and washed out faster than that of ME (Table 2), suggesting that the affinity and efficacy of DAMGO were less than that of ME. This result was surprising based on experiments in brain slices, in which the EC50 value to DAMGO was ∼70 nm, and the washout was much slower than for ME even in the presence of peptidase inhibitors (20, 21).
Morphine (10–100 μm) never caused a potassium current (12 experiments; data not shown). It did, however, bind to the opioid receptor, as indicated by its ability to antagonize the effects of other agonists (see below).
α2-Adrenoceptors.
The α2-adrenoceptor agonist UK14304 evoked a potassium current that was similar in amplitude to that caused by ME. Unlike all of the opioid agonists tested, however, the activation of potassium current induced by UK14304 was significantly faster (Fig. 3B and Table2). This observation suggests a distinct difference between the coupling of μ-opioid receptors and α2-adrenoceptors to potassium conductance. This observation also supports the suggestion that the rate of current activation induced by opioid agonists was not dependent on agonist receptor binding kinetics. Clonidine has been recognized as a partial agonist at α2-adrenoceptors on LC cells in brain slices (12) and did not have any effect on potassium currents in acutely isolated cells (four experiments).
Calcium conductance decrease.
When potassium conductances were blocked with the combination internal cesium and the substitution of BaCl2 (4 mm) for CaCl2 (2.5 mm) in the extracellular solution, ME decreased the current evoked by stepping to 0 mV from −60 mV but did not change the current at −60 or −80 mV (Fig. 4A). Although the specific subtype of calcium conductance was not identified, the voltage dependence indicated that it was a high threshold conductance (Fig.4B). As has been observed elsewhere (13), the kinetics of current activation was slowed by opioids (Fig. 4A). In addition, the inhibition of the barium current by opioids was relieved by a prepulse to +60 mV (Fig. 4C), indicating that this action of opioids on LC neurons was similar to the voltage-dependent inhibition of calcium currents by G protein-linked receptors that has been observed in many different neurons (8).

Inhibition of calcium channels by ME. All experiments were carried out with cesium in the pipette and BaCl2 (4 mm) substituted for CaCl2in the extracellular solution. A, Top trace, voltage protocol used to measure current at three different potentials (−60, −80, and 0 mV). Bottom trace, three superimposed current traces illustrating the lack of effect of ME (100 μm) on the current measured at −60 and −80 mV and the inhibition of current measured at 0 mV. Below, time course of ME action and the blockade of the ME effect by naloxone (10 μm). B, Current-voltage plot of the current induced by depolarization from a holding potential of −60 mV. The threshold for the inward current was ∼−30 mV and peaked at ∼ 0 mV. ME (100 μm) decreased the inward current but did not change the shape of the current-voltage plot. C, Strong depolarization relieves the inhibition caused by ME. Top trace, protocol used. The current was measured at −60 mV, at 0 mV during the first step to 0 mV (0 pre), and then during the step to 0 from +60 mV (0 post). Bottom, time course of the inhibition caused by ME (100 μm) and the lack of any inhibition by ME after a strong depolarization.
The observation that morphine did not increase potassium conductance prompted an investigation on the action of morphine on calcium current. A protocol that allowed measurement of both potassium current and calcium currents at the same time was used. Inward potassium currents were measured at −60 and −80 mV in the presence of 30 mmKCl in the extracellular solution, and outward potassium currents were blocked with the use of intracellular solution containing CsCl and no potassium. Tetrodotoxin (1 μm) blocked sodium channels so that the step to 0 mV evoked only a calcium current (1.8 ± 0.26 nA, 10 experiments). The results illustrated in Fig. 5, A and B, show that ME caused an inward potassium current at −60 and −80 mV and decreased the calcium current measured at 0 mV. The time course of the activation of potassium current and the inhibition of calcium current were comparable; however, a detailed comparison of these kinetics was limited by the frequency of steps used to evoke calcium currents.

Naloxone and morphine block the increase in potassium conductance, but morphine only partially blocks the inhibition of calcium current. The voltage step protocol used in A and B is the same at that shown in Fig. 2A, in which the holding potential is −60 mV and is stepped to −80 mV and then to 0 mV. For these experiments, the intracellular solution contained CeCl to block outward potassium currents, and the extracellular solution contained 30 mm KCl and TTX (1 μm). The currents measured at −60 and −80 mV show the inward potassium current, and the current measured at 0 mV is a calcium current. ME increased the potassium current and decreased the calcium current. A, Naloxone (1 μm) blocked both the inward potassium current and the inhibition of the calcium current caused by ME (100 μm). B, Morphine (100 μm) blocked the inward potassium current but only part of the inhibition of the calcium current caused by ME (100 μm). C, Summary of results measuring barium currents (▪), calcium currents (░), and potassium currents (□). The inhibition of barium and calcium currents was plotted as the fractional inhibition of the control current [(Icontrol − Idrug)/Icontrol]. The potassium current data are presented as the fraction of the ME (100 μm) current induced in each cell. The results show that morphine (100 μm) decreased barium currents but had no effect on potassium conductance. Likewise, morphine (100 μm) partially blocked the inhibition of barium and calcium currents by ME (100 μm) but almost completely blocked the increase in potassium conductance. Naloxone (1 μm) blocked both the increase in potassium conductance and the inhibition of barium and calcium currents. UK14304 (UK, 10 μm) and clonidine (10 μm) both decreased barium currents. D, Summary of results obtained in experiments on dorsal root ganglion cells. Alone, DAMGO at both 1 and 10 μm caused a maximal inhibition. In the presence of morphine (1 μm), however, DAMGO (10 μm) caused a greater inhibition than DAMGO (1 μm). This indicates a shift in the concentration-response curve, which is consistent with competitive antagonism between morphine and DAMGO at the μ-opioid receptor.
Although naloxone (10 μm) completely blocked ME-induced effects on both potassium and calcium currents (Fig. 5A), morphine only partially antagonized the inhibition of calcium current (Fig. 5B). The washout of morphine was rapid compared with that of naloxone (Fig. 5, A and B). The time course of morphine washout was examined in a series of experiments in which the time course of the potassium current was examined when the superfusion solution was switched from one containing morphine (100 μm) alone directly into one containing ME (100 μm). The rise of the ME current had a time constant of 0.58 ± 0.11 sec (five experiments, data not shown) that was not different from the rise of current induced by the application of ME without prior treatment with morphine (τ = 0.56 ± 0.11 sec, five experiments). When the same protocol was used to investigate the inhibition caused by naloxone (1 μm), the rise time of the ME current was 0.5–1 min (11 experiments). Thus, the kinetics of antagonist action seem to be linked to the affinity for the receptor.
Results comparing the action of ME and morphine on calcium, barium, and potassium currents are summarized in Fig. 5C. Morphine had no effect on potassium current but decreased the barium current (by 8.8 ± 1.4%) and only partially antagonized the ME-induced inhibition of both calcium and barium currents. Naloxone (10 μm) completely blocked all actions of ME (100 μm).
Similar results with morphine were found for the inhibition of calcium currents in dorsal root ganglion cells. In dorsal root ganglion cells, morphine was a weak and inconsistent agonist. In a group of 10 cells, DAMGO (1 μm) caused an inhibition of calcium current in 6 cells (mean inhibition = 22 ± 5%), whereas morphine (50 μm) affected only 2 cells (6% and 3%). In cells that were affected by morphine, the EC50 value was ∼10 μm. Although morphine (1 μm) caused little or no inhibition of the calcium current itself, it reproducibly antagonized the inhibition caused by DAMGO (1 μm, Fig.5D). Antagonism seemed to be competitive because increasing the concentration of DAMGO 10-fold partially overcame the morphine block (Fig. 5D). Taken together, the results from both the LC and dorsal root ganglion indicate that morphine was a weak partial antagonist when measuring the opioid-induced inhibition of calcium conductance.
α2-Adrenoceptors.
The inhibition of barium current by α2 agonists was examined in another set of experiments (Fig. 5C). Clonidine caused a small inhibition of barium current measured at 0 mV (14 ± 1.1%, five experiments), whereas UK14304 caused a larger inhibition (28 ± 3%, seven experiments). Thus, as was found with the opioid receptor agonists, clonidine was a partial agonist at decreasing calcium currents but had no agonist activity at increasing potassium conductance. These results suggest that the coupling of both opioid receptor and α2-adrenoceptors to calcium channel inhibition may be more efficient than to activate potassium conductance.
Discussion
Potassium conductance.
The potassium conductance increase caused by G protein-linked receptors has been studied in several different preparations. In all studies, rapid application of agonist was followed by a latency of 50–150 msec before the onset of the potassium current, and the potassium current rose to a peak over a 0.5–2 sec period (9, 14-17). The latency and activation time course of potassium current evoked by opioid receptor and α2-adrenoceptor agonists in LC cells were similar to these other preparations. The time course of the current resulting from the activation of α2-adrenoceptors was, however, about twice as fast (τ = 300 msec) as that induced by opioid receptor agonists (τ = 700 msec). The difference in activation time seems to be receptor dependent rather than agonist dependent because the time course of activation was the same for all opioid agonists (Table 2). In isolated hippocampal cells, recorded at room temperature, the γ-aminobutyric acid B receptor-mediated potassium current activated with τ = 225 msec when either γ-aminobutyric acid or baclofen was applied in saturating concentrations (9). The latency and the rise of the current are an indication of the relative turnover rate of G proteins at receptor, diffusion of active G protein subunits, and channel activation. Without proposing physical segregation of specific receptors with potassium channels, the more rapid activation of current induced by α2-adrenoceptor activation suggests that G protein turnover at this receptor is greater than that at the μ-opioid receptor.
The decline of potassium current after washout of agonist could be dependent on second messenger processes or dissociation of bound ligand. If steps beyond receptor binding limit the rate of potassium channel closure, then recovery would be expected to be insensitive to agonist affinity. The results from this study suggest that agonist affinity determined the duration of response. The affinity of ligands for G protein-linked receptors is generally in the nanomolar range, which could account for the slow dissociation of agonist from the receptor and contribute to the decline of agonist response. Naloxone has an affinity for the μ-opioid receptor of ∼1 nm, as determined in both binding (18, 19) and functional (4) assays. The slow time course of naloxone washout observed in the current study is a further indication of the high affinity of this ligand. If the association rate for naloxone was assumed to be diffusion limited (107 m −1 sec−1), then the time course of recovery (60 sec) can be used to estimate the affinity (K d = k − 1/k + 1) to be ∼1–5 nm. The slow recovery after washout of etorphine also suggests a K dvalue in the low nanomolar range, whereas ME, DAMGO, DSLET, and morphine would be predicted to have affinities that are ≥100-fold lower. There is a reasonable correspondence of these values with steady state binding assays done under conditions in which low affinity states can be measured (in the presence of sodium and GTP or GTP analogs) (18,19). In membrane preparations of C6 cells expressing μ-receptors with guanosine-5′-O-(3-thio)triphosphate (18) and rabbit cerebellum with guanosine-5′-(β,γ-imido)triphosphate (19), theK i value for naloxone was 0.3 to 4 nm; for etorphine, 25 nm; for morphine, 132 nm to 1.4 μm; for DSLET, 5.4 μm; and for DAMGO, 279 nm to 1.6 μm. Thus, the kinetics of agonist effect observed in the current study reflect receptor affinity with limited interference caused by diffusion barriers, metabolism, or agonist rebinding that are present in brain slice experiments.
Subtle differences in the efficacy of agonists were identified based on the maximum current induced by opioid agonists. The peak potassium current amplitude was greatest for ME > DAMGO > etorphine = DSLET > morphine = naloxone. The observation that morphine was a pure antagonist suggests that the receptor reserve was dramatically diminished in isolated LC cells compared with brain slice preparations. This effect was not selective for opioid receptors because clonidine, a well known partial agonist at α2-adrenoceptors, also failed to activate a potassium current. Thus, the experimental conditions associated with recording from acutely dissociated cells favor the study of agonists with high efficacy. The removal of the dendritic arbor, enzymatic or mechanical disruption, or intracellular dialysis by the patch pipette could all play a role in the failure to observe the activation of potassium currents by morphine and clonidine.
The results suggest that ME has the highest efficacy, followed by DAMGO, etorphine, DSLET, and morphine. Experiments carried out in C6 cells expressing μ receptors indicated that the morphine-stimulated guanosine-5′-O-(3-thio)triphosphate binding was 0.83 times that of DAMGO (18), indicating a reduced efficacy of morphine over other agonists. A similar rank order of agonist efficacy is also known to occur at the δ-opioid receptor as reported in NG108–15 cells (22). In that study, most peptide agonists had a similar efficacy relative to etorphine (1), whereas morphine had a relative efficacy of 0.69. In each of these cell lines, high expression of opioid receptors seemed to render the effector limiting such that only agonists with very low efficacy, which do not saturate the effector, are distinguished. In brain slices, differences in agonist efficacy have been observed after only acute opioid desensitization (20) or chronic morphine treatment (21). Morphine normally activates a potassium current that is >90% of that induced by DAMGO, whereas after acute desensitization or chronic morphine treatment, the morphine current was reduced to 40–50% of maximum. Separation of agonists with high efficacy was not possible. The results in the current study using the activation of potassium conductance indicate that efficacies of ME, DAMGO, DSLET, and etorphine can be distinguished in dissociated cells.
Potassium and calcium conductances.
The time course of the opioid-induced inhibition of calcium current and the activation of potassium conductance in the present study were similar given that the measurement of calcium current inhibition was limited to a single point every 5 sec. A complete study of the time course of opioid-induced calcium channel inhibition was done using outside-out patches from primary afferent neurons (7). There was a latency of ≥150 msec between the application of DAMGO and the onset of calcium current inhibition, which rose to a maximum with a time constant of ∼1.3 sec (7). The one significant difference between the receptor coupling to potassium and calcium channels was the fact that morphine and clonidine were partial agonists at calcium channels and antagonists at potassium channels. This observation suggests that the coupling of receptors to calcium current inhibition was more efficient than the activation of potassium current. Both actions have been shown to result from membrane-delimited second messenger pathways (5, 7). The partial agonist activity of morphine on the inhibition of calcium currents in LC neurons was similar to that observed in dorsal root ganglion cells. The small and inconsistent action of morphine on calcium currents or action potentials in primary afferent neurons has been reported but largely ignored (23). This inconsistency has been suggested to result from nonselective actions or even a new opioid receptor, the ε-receptor in rat vas deferens; however, partial agonist properties of morphine may account for these observations (24, 25). The difference in regulation of potassium and calcium channels suggested by the partial agonist/antagonist action of morphine and clonidine may be exploited to further characterize the steps between G protein-linked receptor and these two effectors.
In summary, the results of the current study using acutely isolated LC cells indicate that opioids and α2-adrenoceptor agonists increase potassium and decrease calcium conductance. The kinetics of opioid activation of potassium channels was distinguishable (slower) from that induced by α2-adrenoceptors. Morphine was a partial agonist when measuring the inhibition of calcium conductance in both the LC and dorsal root ganglion cells and an antagonist when measuring the activation of potassium conductance. The rank order of agonist efficacy to cause potassium currents was ME > DAMGO > DSLET = etorphine > morphine. In addition, recovery after washout of ligand correlated with the affinity of agonists for the μ-opioid receptor.
Acknowledgments
We thank Vu Dang for preparation of the LC neurons and Drs. Bruce Bean, MacDonald Christie, and Neil Marrion for helpful discussions and comments on the manuscript.
Footnotes
- Received January 23, 1997.
- Accepted April 13, 1997.
-
Send reprint requests to: J. T. Williams, Vollum Institute, L474, Oregon Health Sciences University, 3181 S.W. Sam Jackson Park Road, Portland, OR 97201. E-mail:williamj{at}ohsu.edu
-
This work was supported by National Institutes of Health Grants DA01863 (J.T.W.) and DA07415 (E.W.M.).
Abbreviations
- LC
- locus ceruleus
- ME
- [Met]enkephalin
- DSLET
- [d-Ser2,Leu5,Thr6]enkephalin
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly-ol5]enkephalin
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}