


Abstract
The α4β2 nicotinic acetylcholine receptors (nAChRs), a major subtype in the brain, have been shown to be modulated by chronic treatment with nicotine. In this study, the regulation of recombinant human α4β2 nAChR subtype by (−)-nicotine and other cholinergic channel modulators was studied using human embryonic kidney 293 cells stably expressing this subunit combination. The treatment of transfected cells with (−)-nicotine and other activator ligands, including (−)-cytisine, 1,1-dimethyl-4-phenylpiperazinium, (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl)isoxazole, and (±)-epibatidine, resulted in concentration-dependent increases in the levels of α4β2 nAChRs. The increase in [3H]cytisine binding sites was initiated by low concentrations of (−)-nicotine (<100 nm); was maximal at 10 μm (15-fold), rapid (t 0.5 = 4.0 ± 0.5 hr), and totally reversible (t 0.5 = 11.7 ± 0.1 hr); and occurred with no change in ligand binding affinity. Antagonists, including dihydro-β-erythroidine, d-tubocurarine, and methyllycaconitine, also elicited significant increases in receptor levels. A good correlation was observed between theKi values for binding inhibition and the EC50 values for receptor up-regulation. Treatment of cells with mecamy-lamine, a noncompetitive antagonist, did not change receptor levels or alter (−)-nicotine-evoked up-regulation. (−)-Nicotine-evoked up-regulation was blocked by cycloheximide, suggesting a role for protein synthesis. Treatment of cells with (−)-nicotine or dihydro-β-erythroidine differentially modulated the efficacy of acetylcholine to activate cation efflux. Both 6-β-[β′(piperidino)propionyl]forskolin and phorbol-12-myristate-13-acetate increased [3H]cytisine binding sites and nAChR function and enhanced the effects of chronic (−)-nicotine treatment in a synergistic manner. These results collectively demonstrate that human α4β2 nAChRs can be differentially up-regulated by chronic treatment with nAChR ligands and activation of protein kinase A- and protein kinase C-dependent mechanisms.
Neuronal nAChRs, members of the excitatory ligand-gated cation channel family, are derived from ≥11 gene products termed α2–α9 and β2–β4 (1, 2). The hetero-oligomeric α4 and β2 combination represent one of the most abundant nAChRs in mammalian brain (3). This subtype embraces those neuronal nAChRs defined by high affinity binding of radiolabeled ligands such as (−)-nicotine, as suggested by the correspondence between agonist binding sites and brain regions expressing the α4 and β2 subunits (4, 5) and by studies in cell lines expressing these subunit combinations (6, 7). The elimination of high affinity [3H]nicotine binding in the mouse thalamus after deletion of the β2 gene further establishes the participation of this subunit in forming the high affinity agonist binding sites in vivo (8). These findings collectively suggest a fundamental role for the α4β2 nAChR subtype in mediating some of the neurochemical and behavioral effects of (−)-nicotine in the brain.
Drug- and disease-induced alterations in α4β2 nAChRs have been documented in various neuropathological conditions and implicated in the development of tolerance to some of the effects of (−)-nicotine (9). Altered function and/or number of nAChRs has been associated with Alzheimer’s disease, Parkinson’s disease, Tourette syndrome, and schizophrenia (10-12). For example, in patients with Alzheimer’s disease, [3H]nicotinic receptor density is markedly decreased in the brain, and chronic treatment with (−)-nicotine has been shown to improve cognitive impairment under such conditions. More recent studies have linked mutations in the human α4 subunit to pathology such as benign familial neonatal convulsions (13) and autosomal dominant nocturnal frontal lobe epilepsy (14). On the other hand, prolonged (−)-nicotine exposure is associated with an increase in the density of high affinity [3H]nicotine binding sites in certain brain regions (3, 15). This is generally associated with a paradoxical decline in receptor function, and these changes may parallel the development of tolerance to some of the behavioral and locomotor effects of nicotine as revealed by studies in rodents (16).
Despite such alterations in the expression and/or function in pathological and tolerant states, little information exists on the biochemical and pharmacological regulation of human α4β2 nAChRs. A detailed characterization of the regulation of human nAChR subtype is of importance because the use of cholinergic channel modulators in humans with degenerative diseases such as Alzheimer’s disease and Parkinson’s disease, attention processes, anxiety, and pain states could involve chronic treatment with these compounds (17). Novel cholinergic channel modulators that are in various stages of clinical development include analogs of (−)-nicotine such as ABT-418, ABT-089, SIB-1508Y, and RJR-1647 and those of anabaseine, including GTS-21. The availability of human α4β2 nAChRs stably expressed in a human cell line (7, 18) has facilitated the first detailed study of the regulation of this major subtype expressed in the human brain that is altered in disease states and by chronic treatment with cholinergic channel modulators.
Experimental Procedures
Materials.
Cell culture media, fetal bovine serum, geneticin (G418), and other antibiotics were purchased from Life Technologies (Grand Island, NY). Hygromycin was purchased from Boehringer-Mannheim Biochemicals (Indianapolis, IN). (−)-[3H]Cytisine (specific activity, 30.5 Ci/mmol) and 86Rb+(specific activity, 1.7 μCi/μg) were purchased from DuPont-New England Nuclear (Boston, MA). ACh chloride, (−)-nicotine hydrogen tartrate, (+)-nicotine di-p-toluoyltartrate,d-tubocurarine chloride, atropine methylnitrate, (−)-cytisine, DMPP, and staurosporine were obtained from Sigma Chemical (St. Louis, MO). Methyllycaconitine citrate, mecamylamine hydrochloride, DHβE, (±)-epibatidine dihydrochloride, IBMX, dibutyl cAMP, 6-β-[β′(piperidino)propionyl]forskolin HCl, PMA, and 4-α-PMA were purchased from Research Biochemicals (Natick, MA). ABT-418 [(S)-3-methyl-5-(1-methyl-2-pyrrolidinyl) isoxazole] and A-85380 [3-(2(S)-azetidinylmethoxy) pyridine] were synthesized at Abbott Laboratories (Abbott Park, IL).
Cell culture.
HEK 293 cells stably expressing the human α4β2 nAChRs were maintained in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, 250 μg/ml geneticin, and 100 μg/ml hygromycin in a humidified atmosphere (5% CO2/95% air) at 37° as previously described (K177 cell line; Ref. 7). Briefly, this cell line was obtained after cotransfection of HEK 293 cells with the human α4 and β2 nAChR subunits, both of which were subcloned in the expression vector pRcCMV, followed by appropriate antibiotic selection and propagation. During initial evaluation, this cell line was found to express high levels of specific [3H]cytisine binding and hence chosen for further studies, including [3H]cytisine binding, mRNA analysis, cation efflux, and patch-clamp. Previous studies have demonstrated stable expression of the α4β2 nAChR subtype in this cell line with appropriate pharmacological and biophysical properties (7, 18).
Measurement of α4β2 nAChR expression.
[3H]Cytisine binding was used to measure α4β2 nAChR expression. Cells were plated onto six-well culture dishes or 75 cm2 flasks and incubated with various test compounds for the duration indicated. At the end of the treatment period, cells were rinsed twice with ice-cold assay buffer (composed of 50 mm Tris·HCl, 120 mm NaCl, 5 mm KCl, 1 mm MgCl2, and 2.5 mm CaCl2, pH 7.4 at 4°), scraped, and homogenized using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY) for 10 sec. The homogenate was centrifuged at 45,000 × g for 20 min at 4°, and the pellet washed two times by repeated centrifugation and kept frozen at −80°. Radioligand binding was carried out as previously described (7) in a total volume of 0.5 ml using a single concentration of [3H]cytisine (6 nm) and 15–25 μg of protein/tube. Nonspecific binding was defined by the addition of 10 μm unlabeled (−)-nicotine to a duplicate set of tubes. In some studies, the cell homogenate itself was used in binding assays. Bound radioactivity was quantified by liquid scintillation spectroscopy at an efficiency of 45% (LS 5000 TD; Beckman Instruments, Somerset, NJ).
Measurement of α4β2 nAChR activity.
An isotopic86Rb+ efflux assay was used to assess the functional activity of α4β2 nAChRs. Assays were carried out using cells grown attached to 24-well culture dishes (Nunc, Naperville, IL) in serum-free medium at 21° as previously described (7, 19). Briefly, cells were plated in poly-l-lysine-coated 24-well culture dishes at a density of 2.5 × 105 cells/well. When cells were 60–80% confluent, the culture media was replaced with fresh media containing the test compounds and incubated in a cell incubator at 37°. Ligand-containing medium was removed at the end of 20 hr and replaced with 86Rb+ (2 μCi/well)-containing medium supplemented with ligands at the appropriate concentrations to initiate the86Rb+ loading process for an additional 4 hr. After chronic treatments, washout of the ligand was performed as previously described (20) with some modifications to minimize variations as a result of culture conditions,86Rb+ loading, incubation temperature, and so on that may influence the magnitude of the efflux response. Briefly, at the end of the 24-hr treatment period, ligand-containing medium was removed by gentle aspiration, and cells were subjected to two cycles of rinses with 250 μl of DMEM at 37°. This wash procedure was repeated two more times at 15-min intervals. After the final cell rinse,86Rb+ efflux was assessed by incubating the cells with 250 μl of DMEM containing ACh for 5 min. This protocol was chosen because initial time course studies showed no appreciable recovery from down-regulation during this washout period. Atropine (1.5 μm) was included in the assay medium to eliminate any confounding responses evoked by activation of endogenous muscarinic receptors. After exposure to the agonist, radioactivity in the assay medium was detected by γ-counting (Gamma 5500; Beckman Instruments, Fullerton, CA).
The nAChR ligands used included (−)-nicotine, (+)-nicotine, DMPP, (−)-cytisine, (±)-epibatidine, ABT-418, A-85380, DHβE, mecamylamine, d-tubocurarine, and methyllycaconitine. In radioligand binding studies, the concentration dependence of effects were examined, although in some cases, the maximum concentrations of the compounds used were limited by their solubility in culture medium or by adverse effects on cell survival. Fresh media-containing ligands were replaced every 48 hr wherever appropriate. In some studies, cells were also treated with forskolin, dibutyl cAMP, IBMX, PMA, and staurosporine. Compounds were dissolved in water or 100% dimethylsulfoxide, and equal volumes of solvents were added to control cultures as appropriate.
Data and statistical analysis.
Significant differences between groups of means were assessed by the unpaired Student’st test using Instat (GraphPAD Software, San Diego, CA). A value of p < 0.05 was considered significant. The concentration dependence of up-regulation of [3H]cytisine binding and for ACh-stimulated86Rb+ efflux and the time course data were analyzed by nonlinear least-squares regression analysis using Inplot (GraphPAD). In cases in which maximal responses were not attainable due to limits of solubility or adverse effects on cell viability, the estimated EC50 values were calculated by nonlinear least-squares regression analysis using variable maximal response. The correlation coefficient values were determined by linear regression analysis. Values are expressed as mean ± standard error unless otherwise indicated.
Results
Characterization of (−)-nicotine-evoked up-regulation of human α4β2 nAChRs.
Treatment of HEK 293 cells stably expressing the human α4β2 nAChRs with (−)-nicotine resulted in an increase in the levels [3H]cytisine binding. Fig.1A shows the time-dependency of the (−)-nicotine-evoked increase in [3H]cytisine binding levels. Cells were incubated with 1 μm(−)-nicotine for 2–168 hr, and [3H]cytisine binding levels were assessed at the time intervals indicated. A significant increase in [3H]cytisine binding was observed as early as 2 hr after treatment of cells with 1 μm (−)-nicotine (control, 1410 ± 293 fmol/mg; 2 hr (−)-nicotine, 3636 ± 155 fmol/mg). Nonlinear regression analysis of the data revealed that the time course kinetics did not differ significantly when fitted for one or two sites (p < 0.0.5), indicating that the response to (−)-nicotine could be a monophasic process. Thet 0.5 value of (−)-nicotine-evoked up-regulation was determined to be 4.0 ± 0.5 hr (four experiments). The maximal increase in binding levels was sustained in the continuous presence of (−)-nicotine for ≥168 hr (Fig. 1A). The reversibility of this effect was examined by treating the cells with 1 μm nicotine for 168 hr, followed by removal of the ligand by washout at different time intervals. As shown in Fig. 1B, the recovery from (−)-nicotine-evoked up-regulation was a relatively slower process, with a t 0.5 value of 11.7 ± 0.1 hr (four experiments), and was complete with a return to pretreatment values by 48 hr after removal of (−)-nicotine from the media.
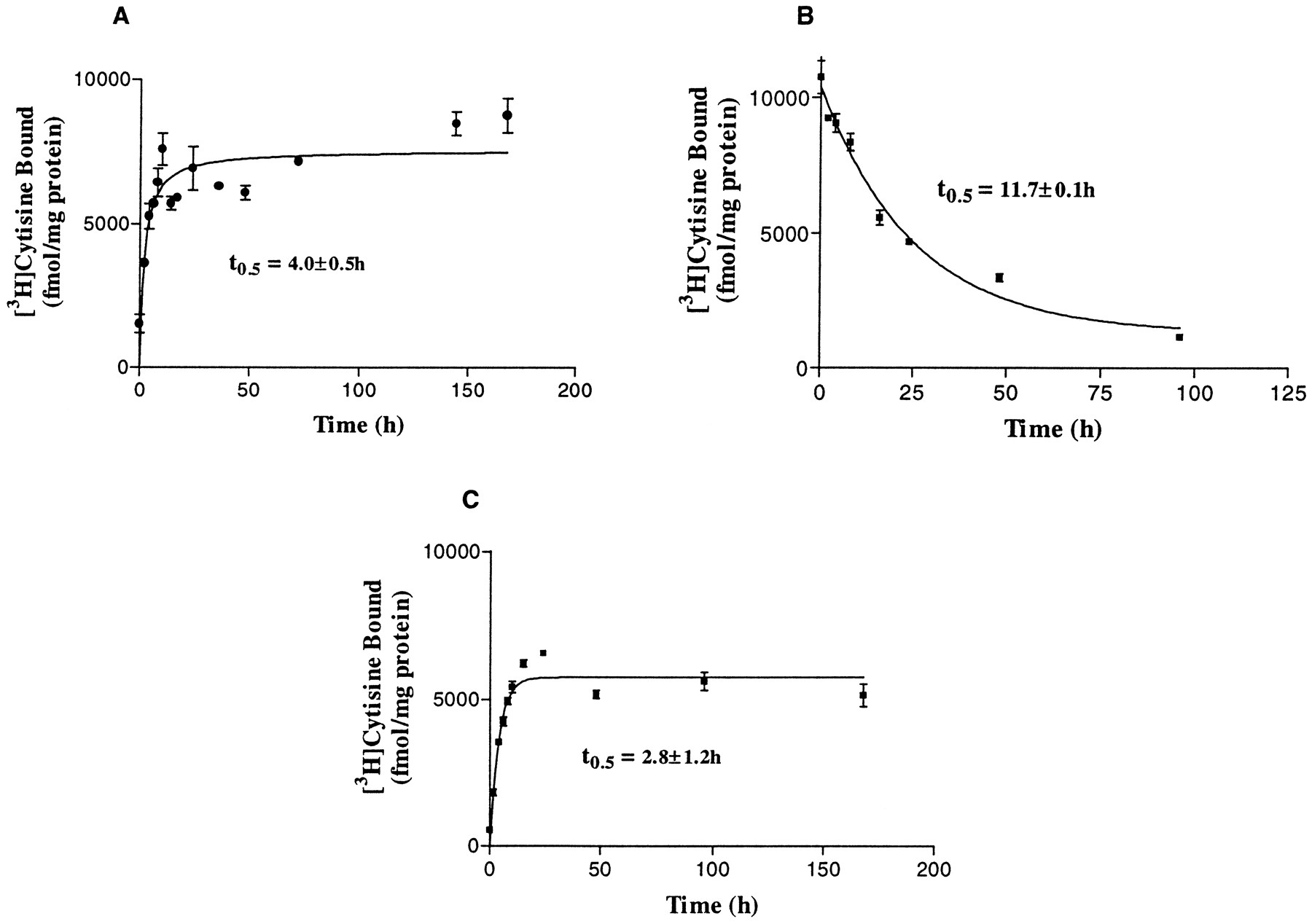
Time course of human α4β2 nAChR regulation. A, On-rate of up-regulation by (−)-nicotine. HEK 293 cells stably expressing human α4β2 nAChRs were treated with 1 μm (−)-nicotine for 2–168 hr. B, Time course of recovery from (−)-nicotine-evoked nAChR up-regulation. Cells were treated for 168 hr with 1 μm (−)-nicotine, after which nicotine was removed for the time intervals indicated. C, Time course of up-regulation of nAChRs by methyllycaconitine. Cells were treated with 100 μm methyllycaconitine for the time periods indicated. Binding assays were carried out with 6 nm[3H]cytisine using cell membranes prepared from each sample as described in Experimental Procedures. Error bars, standard error of three or four experiments.
The effects of (−)-nicotine were concentration dependent. As shown in Fig. 2A, a significant increase in [3H]cytisine binding levels was observed at concentrations as low as 100 nm, and maximal 15-fold increase from control values was observed at 10 μm(−)-nicotine. The EC50 value for (−)-nicotine-evoked up-regulation was 0.49 ± 0.1 μm (four experiments). Saturation studies revealed that this increase in binding sites was due to an increase in theB max value of [3H]cytisine bound with no significant effect on the KD value of the radioligand (1 μm (−)-nicotine:B max = 7660 ± 1196 fmol/mg;KD = 0.60 ± 0.15 nm; control, B max = 1480 ± 191 fmol/mg; KD = 0.21 ± 0.08 nm; three experiments). Treatment of cells with the (+)-enantiomer of nicotine also elicited significant increases in [3H]cytisine binding (EC50 = 4.6 ± 1.1 μm; five experiments), although the maximal increase attained was only 5-fold over that of untreated cells (Fig.2A).

Concentration-dependence of up-regulation of human α4β2 nAChRs in HEK 293 cells by (A) activator [(−)-nicotine, (+)-nicotine, (±)-epibatidine, DMPP, (−)-cytisine] or (B) antagonist (DHβE, d-tubocurarine). Cells were treated with varying concentrations of ligands as indicated for 168 hr [3H]Cytisine (6 nm) binding to membranes were performed as described in Experimental Procedures. Values are mean ± standard error of three experiments, each assayed in quadruplicate. The estimated EC50 values are summarized in Table 1. C, Correlation between binding affinities (Ki values) and up-regulation potencies (EC50 values) of cholinergic channel ligands in cells stably transfected with human α4β2 nAChRs. TheKi values are from Ref. 7, except for A-83580, which is from Ref. 21. Solid line, linear regression through the data points. Dashed line, 1:1 correlation.
Regulation of human α4β2 nAChRs by other activator ligands.
To examine whether the increase in receptor levels was unique to nicotine, [3H]cytisine binding levels were examined after treatment with other activator ligands; treatment with (−)-cytisine, DMPP, (±)-epibatidine, ABT-418, and A-85380 for 168 hr resulted in concentration-dependent increases in [3H]cytisine binding levels (Fig. 2A). The estimated EC50 values for up-regulation for these ligands are summarized in Table 1 with binding affinities at this nAChR subtype. (+)-Epibatidine and A-85380, two potent ligands (Ki = 40–50 pm) that are known to interact with the human α4β2 nAChRs (21), increased [3H]cytisine binding levels with EC50 values of 10.2 ± 0.6 nm (three experiments) and 8.2 ± 0.1 nm (three experiments), respectively. The maximal increase in receptor levels observed with (+)-epibatidine was significantly lower than those elicited by (−)-nicotine. Although both (−)-cytisine and DMPP are partial agonists at this subtype (7), these compounds increased [3H]cytisine binding to levels equal to those maximally elicited by (−)-nicotine. The relative potencies for the various activators for up-regulation of the human α4β2 subtype are (±)-epibatidine ∼ A-85380 > (−)-nicotine > (+)-nicotine > (−)-cytisine > ABT-418 ∼ DMPP.
Regulation of human α4β2 nAChRs in HEK 293 cells by cholinergic channel ligands
Effects of antagonists on human α4β2 nAChRs.
In a previous study, Peng et al. (23) showed that (−)-nicotine-evoked up-regulation of the avian α4β2 nAChRs could be attenuated by nAChR antagonists such as d-tubocurarine. In the current study, when cells were treated with d-tubocurarine (100 μm) in presence of 1 μm (−)-nicotine, no blockade of (−)-nicotine-evoked up-regulation was observed. Interestingly, treatment of cells with d-tubocurarine alone resulted in significant up-regulation of α4β2 nAChRs (Fig. 2B). Treatment of cells for 168 hr with 100 and 300 μm d-tubocurarine increased [3H]cytisine binding by 72 ± 9% (three experiments) and 185 ± 76% (three experiments), respectively, over control values. To confirm that these effects were not unique to d-tubocurarine, we examined the effect of DHβE, a competitive antagonist at this subtype (6). As shown in Fig.2B, a concentration-dependent increase in [3H]cytisine binding levels were observed after treatment of cells with DHβE. Furthermore, treatment of cells with methyllycaconitine, a potent α7 nAChR antagonist (22) that inhibits the α4β2 subtype at 1–2 μm concentrations (18), also significantly increased [3H]cytisine binding levels (100 μm methyllycaconitine: 6826 ± 67 fmol/mg; control, 940 ± 19 fmol/mg; three experiments).
To examine whether the patterns of up-regulation of antagonists differed from those observed with (−)-nicotine, we studied the time course of up-regulation by methyllycaconitine. Cells were treated with 100 μm methyllycaconitine for 2–168 hr, and [3H]cytisine binding was assessed. Thet 0.5 value of methyllycaconitine-evoked up-regulation, 4.4 ± 0.5 hr (three experiments), was not significantly different from that observed with (−)-nicotine (Fig.1C). In addition, the time course of up-regulation did not differ significantly when fitted to a one- or two-site model, indicating that the response to chronic antagonist treatment could be a monophasic process similar to that observed with (−)-nicotine.
A comparison of the logarithms of the EC50 values for evoking up-regulation of α4β2 nAChRs and the corresponding [3H]cytisine binding site affinities for the series of activator and antagonist nAChR ligands is depicted in Fig.2C. Analysis of the data reveal a linear correlation with a coefficient value (r) of 0.91.
No significant change in [3H]cytisine binding levels were observed after treatment of cells with the open channel blocker mecamylamine (100 μm; Table2). Mecamylamine treatment also did not alter the up-regulation elicited by 1 μm (−)-nicotine, which is in contrast to the effects reported for the avian homologs (23). Furthermore, no significant change in [3H]cytisine binding was observed when mecamylamine was used over a wider concentration range (i.e., 0.01–100 μm).
Effect of mecamylamine and d-tubocurarine on (−)-nicotine-evoked up-regulation of α4β2 nAChRs in HEK 293 cells
Effect of nAChR modulators on human α4β2 nAChRs.
Considerable evidence points to the existence of binding sites distinct from those defined classically by ACh on α4β2 nAChRs that can be activated by ligands such as physostigmine (24). Accordingly, it was of interest to examine whether physostigmine or other cholinesterase agents, such as tacrine, elicit effects similar to those of (−)-nicotine. The treatment of cells with physostigmine (0.01–100 μm) or tacrine (0.01–1 μm) did not result in up-regulation of nAChRs. Furthermore, methoxyverapamil (D600) and nimodipine, both of which are antagonists of the L-type voltage-sensitive calcium channels but have been shown to interact with certain neuronal nAChRs (31), did not significantly alter [3H]cytisine binding after chronic treatment of the cells. The muscarinic cholinergic antagonist atropine failed to elicit changes in receptor levels, indicating that up-regulation of α4β2 nAChRs is not a consequence of heterologous influences.
Role of protein synthesis.
To investigate whether (−)-nicotine-evoked up-regulation of the human α4β2 nAChRs involves de novo protein synthesis, we examined the effect of protein synthesis inhibition on [3H]cytisine binding. In the presence of 7 μm cycloheximide, a concentration previously shown to completely inhibit [3H]leucine incorporation into HEK 293 cells (25), the treatment of cells with (−)-nicotine (1 μm) for 24 hr failed to produce an increase in [3H]cytisine binding (control, 1436 ± 51 fmol/mg; (−)-nicotine, 6094 ± 89 fmol/mg; cycloheximide and nicotine, 1703 ± 53 fmol/mg; Fig.3). It was observed that treatment of cells with cycloheximide alone led to a small, but significant, decline of 39% (875 ± 38 fmol/mg) in the basal level of [3H]cytisine binding, which was absent when (−)-nicotine was present.
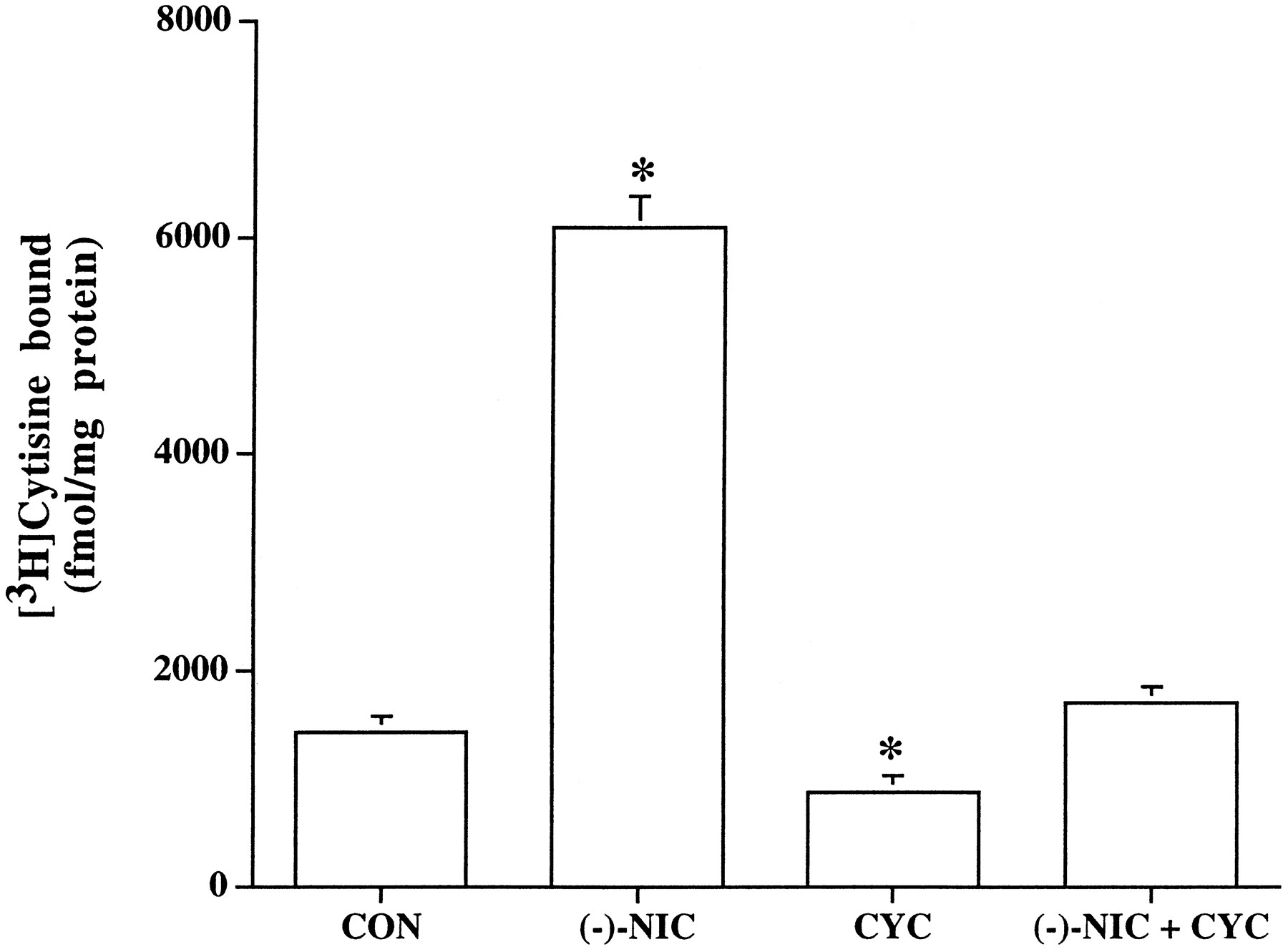
Influence of protein synthesis inhibition on (−)-nicotine-evoked nAChR up-regulation. α4β2 nAChR-expressing HEK 293 cells were treated for 24 hr without (CON) or with (NIC) 1 μm (−)-nicotine and with 7 μm cycloheximide alone (CYC) or in the presence of nicotine 1 μm (NIC + CYC). ∗, Significantly different from untreated cells. Values are mean ± standard error from triplicate culture dishes, each assayed in quadruplicate.
Membrane localization of nAChRs up-regulated by (−)-nicotine.
After exposure of cells to 1 μm nicotine, [3H]cytisine binding was measured in both the heavy membrane fraction (40,000 × g) and the total cell homogenate. The number of binding sites measured in the membrane preparation was increased 5.3-fold; the total number of binding sites measured in the homogenate increased similarly (5.8-fold). The increase in binding sites measured by [3H]cytisine could therefore be largely attributed to an increase in the presence of receptors in the membranes.
Role of PKA and PKC pathways.
Compounds interacting with protein kinase pathways were evaluated for their effects on the up-regulation of α4β2 nAChRs. It was found that treatment of cells with either forskolin (10 μm) or nicotine (1 μm) for 24 hr elicited increases in [3H]cytisine binding sites of ∼2- and ∼4-fold, respectively, compared with untreated cells (control, 1065 fmol/mg, forskolin, 1870 fmol/mg; nicotine, 4254 fmol/mg; Fig.4). When cells were coincubated with both (−)-nicotine (1 μm) and forskolin (10 μm) for 24 hr, it was found that the resulting increase in [3H]cytisine binding was enhanced ∼8-fold compared with control values. This indicates that the observed effects are synergistic and not just additive (Fig. 4). Other agents that activate PKA, including the membrane-permeant cAMP analog dibutyl cAMP and the phosphodiesterase inhibitor IBMX, elicited similar effects, indicating that this phenomenon was not unique to forskolin. Thus, treatment of cells with dibutyl cAMP (100 μm) alone for 24 hr led to a significant increase in [3H]cytisine binding of 38% (1574 ± 145 fmol/mg; control, 1139 ± 112 fmol/mg; three experiments), respectively, whereas treatment with both dibutyl cAMP and (−)-nicotine significantly enhanced the response of (−)-nicotine by ∼87% (7898 ± 172 fmol/mg; nicotine, 4212 ± 284 fmol/mg; three experiments) respectively. Similar results were obtained when studies were carried out for 168 hr (data not shown).
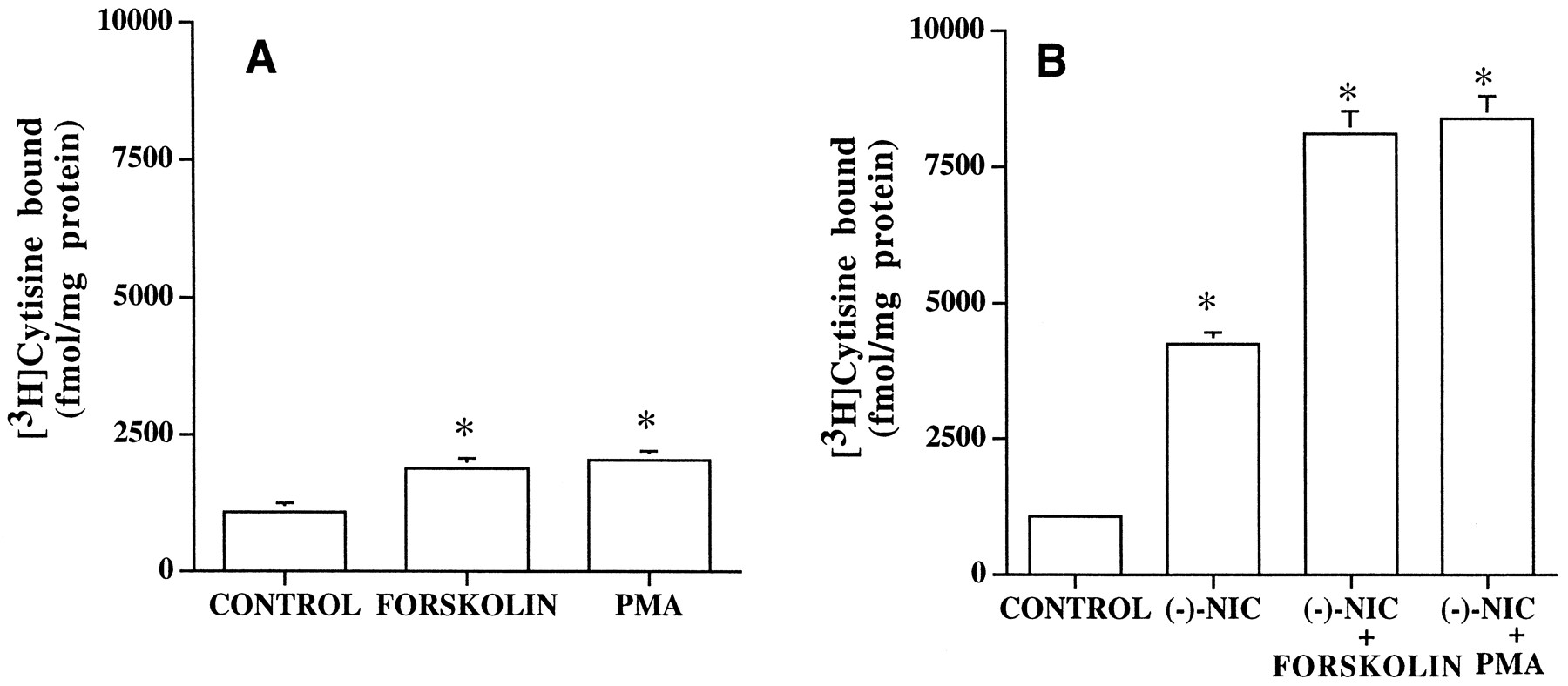
Influence of PKA and PKC activation on α4β2 nAChR levels stably expressed in HEK 293 cells. A, Effects on [3H]cytisine binding after treatment of cells for 24 hr with piperidino-propionyl forskolin (10 μm) or PMA (100 nm). [3H]Cytisine binding was assessed at a radioligand concentration of 6 nm. B, Effect of PKA and PKC activation on the up-regulation evoked by (−)-nicotine (NIC). Cells were treated with 1 μm(−)-nicotine alone or in the presence of 10 μmpiperidino-propionyl forskolin or 100 nm PMA for 24 hr, after which [3H]cytisine binding was assessed as described. Values are mean ± standard error of three or four determinations, each performed in duplicate. ∗, Significant (p < 0.05) differences with respect to untreated cells.
Because it was found that (−)-nicotine and PKA activation were synergistic in causing substantial increases in the density of α4β2 nAChRs, antagonist-induced increase was examined in presence of forskolin to determine whether the pattern differed from those observed with (−)-nicotine. The treatment of cells with both methyllycaconitine and forskolin for 24 hr increased [3H]cytisine binding levels in a synergistic manner, by ∼8-fold compared with those observed with untreated cells, although the use of each compound alone evoked only a 4- and 2-fold increase in nAChR levels, respectively (untreated, 1015 ± 46 fmol/mg; 100 μmmethyllycaconitine, 4935 ± 249 fmol/mg; methyllycaconitine and forskolin, 7982 ± 472 fmol/mg; three experiments). These results are quite similar to the effects observed with (−)-nicotine.
We next examined the effects of PMA on nAChR up-regulation. The treatment of transfected cells with PMA (100 nm) for 24 hr elicited a 2-fold increase in [3H]cytisine binding (Fig. 4A). However, when cells were treated with both PMA and (−)-nicotine, nicotine-evoked up-regulation of α4β2 nAChRs was found to be enhanced by ∼8-fold with respect to control (Fig. 4B). This synergistic effect is similar to that observed with forskolin. The treatment of cells with the inactive phorbol ester α-PMA (100 nm) did not elicit up-regulation alone (control, 1231 ± 36 fmol/mg; α-PMA, 1380 ± 22 fmol/mg; three experiments) or modify the effects of (−)-nicotine, 1 μm (nicotine, 4682 ± 156 fmol/kg; α-PMA, 4948 ± 200 fmol/mg; three experiments). This lack of effect of 4-α-PMA, the stereoisomer of PMA that neither binds nor activates PKC, on α4β2 nAChR levels indicates specificity of the observed effects. Because the possibility exists that exposure to PMA could desensitize/down-regulate PKC levels and thus inhibit PKC, the effects of the PKC inhibitor staurosporine were examined. Staurosporine at 20 nm was previously shown to be effective in inhibiting PKC-mediated changes in nicotinic receptor levels in neuroblastoma cells (26). The treatment of cells with 50 nm staurosporine for 24 hr did not significantly alter (−)-nicotine-evoked up-regulation (nicotine, 7337 ± 450 fmol/mg; nicotine and staurosporine, 7589 ± 203 fmol/mg; three experiments). Similarly, (−)-nicotine-evoked increase in binding levels were not affected by two other PKC inhibitors: sphingosine (100 nm sphingosine and nicotine, 7565 ± 254 fmol/mg) and chelerythrine HCl (10 μm chelerythrine and nicotine 7512 ± 199 fmol/mg).
Functional consequences of human α4β2 nAChR regulation.
We assessed α4β2 nAChR function after chronic treatments by ACh-evoked cation efflux. In initial experiments, it was established that ligand washout was effective and the observed effect could not be attributed to residual ligand. Previous studies by Lukas (20) have shown that momentary exposure of cells to nicotine has no effect on functional inactivation provided removal of the ligand-containing medium was followed by two cycles of superficial rinses of the culture dish. In the current study, the washout protocol was more rigorous, with cells subjected to two cycles of rinses with 250 μl of fresh ligand-free medium-lacking serum followed by two similar rinses at 15-min intervals before ACh-evoked cation efflux was initiated. We used a radioreceptor assay to measure residual levels of (−)-nicotine present, if any, after washout. The evidence that (−)-nicotine was effectively removed is derived from these experiments, in which the supernatant collected after centrifugation of the cell homogenate after the final rinse was tested for its effect on dose inhibition by nicotine of [3H]cytisine binding. No shift was observed in the dose- response curve (data not shown). Had residual nicotine been present in the supernatant (at a concentration >1 nm), the dose response should have shifted to the left, which did not occur. In addition, had residual (−)-nicotine been present in the assay, a change in the EC50 value of ACh should also have been observed, which was not the case (vide infra).
ACh activated cation efflux from transfected cells with an EC50 value of 14.3 ± 1.4 μm(five experiments) and a maximum efficacy at 1 mm, which is consistent with previous observations (7). When cation flux was assessed after the treatment of cells with 0.1 and 1 μm (−)-nicotine for 24 hr and washout, a significant increase in the maximal efficacy of ACh (1 mm) to activate86Rb+ efflux was observed compared with untreated cells. Under these conditions, the EC50 values for ACh, however, did not significantly differ after treatment with (−)-nicotine (1 μm (−)-nicotine, EC50 = 16 ± 1.9 μm; six experiments) compared with control (EC50 = 14.3 ± 1.4 μm; five experiments). When cells were treated with higher concentrations of (−)-nicotine (i.e., 10, 100, and 1000 μm), the maximal ACh-evoked efflux showed a concentration-dependent decrease compared with untreated cells, although [3H]cytisine binding levels were elevated (Fig. 5A). Interestingly, a significant increase in the efficacy of ACh (1 mm) to activate cation efflux was observed when cells were chronically treated with the competitive antagonist DHβE. Importantly, this increase was observed at all concentrations of DHβE examined (1–1000 μm). As shown in Fig. 5B, significant increases in efflux were observed after treatment with 1 μm DHβE, and a maximal enhancement in efficacy of 90 ± 5% (four experiments) was observed at a concentration of 100 μm DHβE.

Influence of (−)-nicotine (A) and DHβE (B) on the functional activity and [3H]cytisine binding levels in HEK 293 cells stably expressing α4β2 nAChRs. Cells were treated with varying concentrations of (−)-nicotine (A) or DHβE (B), as indicated. Solid lines, functional activity of α4β2 nAChRs is expressed as percent cation efflux relative to that evoked by 1 mm ACh from untreated cells (taken as 100%).Vertical bars, [3H]cytisine binding values expressed in fmol/mg of protein.
The significant increase in receptor density observed after treatment with PMA and forskolin was accompanied by an enhancement in the effect of ACh to evoke cation efflux. As depicted in Fig.6, cells treated with PMA (100 nm) or forskolin (10 μm) elicited a significant increase in the maximal ACh-evoked ion efflux response by 49 ± 7.6% (five experiments) and 78 ± 19% (n = 6), respectively, compared with untreated cells without a significant change in the EC50 value of ACh (Table 3). The treatment of cells with (−)-nicotine (1 μm) and PMA (100 nm) or forskolin (10 μm) resulted in an enhancement in the maximal ACh-evoked efflux by 100 ± 3% and 45 ± 9%, respectively. When cells were treated with DHβE (300 μm) and PMA or forskolin, the efficacy of ACh to evoke cation efflux was significantly increased by 189 ± 7% and 112 ± 22%, respectively. The results from86Rb+ efflux studies are summarized in Table 3.

Influence of nicotinic cholinergic ligands and protein kinase activators on the functional activity of α4β2 nAChR levels in HEK 293 cells stably expressing α4β2 nAChRs. Cells were treated with 100 nm PMA or 10 μm forskolin (A), 1 μm (−)-nicotine or 300 μm DHβE (B), and combinations of forskolin (C) or PMA (D) with nicotine or DHβE for 24 hr, after which functional activity was assessed by86Rb+ efflux as described in Experimental Procedures The responses evoked under various conditions were expressed as the function of efflux evoked by a maximally effective concentration of ACh (1 mm) in untreated cells.
Effect of chronic ligand treatment on cation efflux in HEK 293 cells stably expressing human α4β2 nAChRs
Discussion
Results of the current study confirm, as previously reported (23,27, 28), that the elements necessary for nicotine-evoked up-regulation of α4β2 nAChRs are constitutively contained in cells of non-neuronal origin; the transfected cells that we used were derived from the human kidney cell line HEK 293. Expression studies in mammalian cell lines have previously shown that the recombinant α4 and β2 subunits coassemble to form functional ion channels exhibiting [3H]agonist ligand binding, pharmacological and biophysical properties that are consistent with those of native brain nAChRs (6, 7, 18).
Previous studies have shown that chronic (−)-nicotine treatment elicits increased levels of high affinity nicotinic receptors in the mammalian brain as measured by radioligands or antibodies (3, 5, 29). The current study extends these findings to human recombinant α4β2 nAChRs and provides clear evidence that this nAChR subtype stably expressed in a human cell line is up-regulated by treatment with both activator and competitive antagonist ligands. In this study, the α4β2 nAChRs were up-regulated maximally ∼15-fold by (−)-nicotine in a rapid (within ∼10–12 hr) and totally recoverable manner. Moreover, the threshold level for significant up-regulation (100 nm) is consistent with circulating levels of (−)-nicotine present in smokers (150 nm; Ref. 30). The EC50 value of (−)-nicotine (0.49 μm) is in excellent agreement with those reported previously for up-regulation of the chick homologs stably expressed in mouse fibroblasts (M10 cells, 0.2 μm; Ref.23). (−)-Nicotine-evoked up-regulation is homologous because parallel treatment with muscarinic or voltage-sensitive calcium channel ligands failed to alter [3H]cytisine binding sites. It is also interesting to note that (−)-nicotine-evoked up-regulation in transfected HEK 293 cells occurs in the absence of any transcriptional factors or receptor gene promoter elements that are likely associated with the expression of these subunits in intact neurons. Therefore, the minimal structural elements involved in the up-regulation process resides within the α4β2 nAChR subunits expressed in these cells. The maximal up-regulation by (−)-nicotine in transfected HEK 293 cells of ∼15-fold above control levels is higher than those reported with native tissues or endogenously in cell and ≥10-fold higher than those reported with the M10 cells (27). Because we have not examined nAChR up-regulation in other isolated α4β2 clones, the possibility remains that the magnitude of up-regulation observed here is somehow a consequence of the particular clone that was isolated. In another cell line stably expressing the α7 nAChR subtype (22), a 6-fold increase in [125I]α-bungarotoxin binding levels was observed after treatment with nicotine (1000 μm).1
Examination of the pharmacology of up-regulation revealed that other activator ligands, including DMPP, (−)-cytisine, ABT-418, A-85380, and (±)-epibatidine, elicited up-regulation in a concentration-dependent manner. Competitive antagonists at the α4β2 nAChR, such as DHβE and d-tubocurarine, were also capable of receptor up-regulation, although with a lower magnitude than (−)-nicotine within the concentration range examined. The EC50 values for up-regulation by both activators and antagonists showed a good correlation with their binding affinities (Fig. 2C), although the EC50 values were generally ∼100-fold higher than their corresponding binding affinities at the α4β2 nAChRs (7). Such close correlation between the EC50 values and the binding affinities suggests that receptor up-regulation may be related to the interaction of these ligands with the high affinity desensitized state of the nAChRs. However, in a comparison of the efficacies of up-regulation, (+)-nicotine and (+)-epibatidine showed lower maximal effects relative to the other activator ligands evaluated, indicating that the degree of up-regulation may not be related to the binding affinities of these ligands.
Up-regulation elicited by (−)-nicotine and other activator ligands could not be prevented by treatment with competitive antagonists such as d-tubocurarine, which is in contrast to the previous observations of Peng et al. (23), who reported attenuation of nicotine-evoked up-regulation of avian α4β2 nAChRs byd-tubocurarine. Although concentrations ofd-tubocurarine and nicotine used in our study are similar to that used by Peng et al., no attenuation of the effect of nicotine was observed in our study. The inability of this compound to attenuate the effects of (−)-nicotine (Table 2) may be related to the differences in α4β2 nAChR binding affinities (∼1000-fold) of these two ligands (6, 7). Interestingly, in contrast to previous observations (23), treatment of cells with d-tubocurarine alone elicited a modest increase in binding levels. Similarly, increases in [3H]cytisine binding sites observed after chronic treatment with DHβE or methyllycaconitine indicate that antagonist-evoked up-regulation is not unique tod-tubocurarine. Both DHβE and methyllycaconitine have been shown to inhibit ACh-evoked currents or cation efflux in this cell line, with IC50 values in the low micromolar range (7, 18). The up-regulation evoked by antagonists was smaller than that of (−)-nicotine at the concentrations tested. The fact that up-regulation is observed regardless of whether agonists or antagonists were used for chronic treatment indicates that receptor occupancy, by either activator or competitive antagonist, may be sufficient to trigger an increase in receptor levels. The observation thatd-tubocurarine failed to attenuate the effect of (−)-nicotine is also consistent with this hypothesis. The membrane-impermeable quaternary amines DMPP andd-tubocurarine also evoked up-regulation of nAChRs, supporting an extracellular site of action, which is consistent with previous studies (23). In the current study, it was also found that treatment of cells with the open channel blocker mecamylamine did not elicit changes in the level of nAChRs or modify (−)-nicotine-evoked up-regulation. This observation is in agreement with in vivostudies using chlorisondamine, another noncompetitive antagonist, which did not elicit up-regulation by itself and did not prevent (−)-nicotine-evoked up-regulation of [3H]nicotine binding sites in rat brain (29). Our results differ from the observations of Peng et al.(23), who reported a 2-fold increase in [3H]nicotine binding by 50 μmmecamylamine in M10 cells stably expressing the avian subunits. In addition to mecamylamine, noncompetitive activator ligands such as physostigmine and tacrine, whose binding sites are distinct from those for ACh or ACh-competitive ligands (allosteric modulators; Ref. 24), also failed to modify nAChR levels in our study.
The similar increase in the levels of [3H]cytisine binding sites observed in both homogenate and membrane preparations after chronic (−)-nicotine treatment indicates there is no contribution from cytosolic components to the up-regulation process. However, [3H]cytisine binding does not distinguish between nAChRs present on cell surface or in cellular membrane components because the intracellular receptors present in the membranes of endoplasmic reticulum or of vesicular bodies would also sediment with the plasma membrane when homogenized. Other approaches to direct quantification of receptors on cell surface that use labeled ligands that do not cross the cell membrane or antibody probes (23) will be necessary to address the issue of intracellular versus cell surface localization of up-regulated nAChRs.
To examine the mechanisms by which nicotine induces a rapid increase in receptor number, studies were performed using cycloheximide to prevent protein synthesis while measuring the existing receptor levels with and without exposure to (−)-nicotine. The observation that (−)-nicotine treatment prevented the steady state decline in receptor levels observed in cells treated with cycloheximide alone (Fig. 3) is consistent with previous studies (23) in which nicotine treatment of dexamethasone-induced M10 cells decreased the rate of degradation of α4β2 nAChRs in the presence of protein synthesis inhibition. However, the (−)-nicotine-induced increase in [3H]cytisine binding was also inhibited by cycloheximide, suggesting that the up-regulation process may in part require de novo α4β2 nAChR synthesis (27). It is unlikely that (−)-nicotine regulates nAChR expression by altering the transcriptional activities because both the α4 and β2 subunit genes lack transcriptional regulatory elements and are under the control of the cytomegalovirus promoter, which is constitutively active. This, together with the previously documented lack of effects on steady state mRNA levels after chronic nicotine treatment both in vivo(5) and in vitro (23, 27, 28), indicates that post-transcriptional mechanisms are involved in nAChR up-regulation. These processes may involve, for example, altered translation rates, increased efficiency of receptor assembly from its constituent subunits as reported for the muscle nAChRs (32), and/or altered rates of receptor degradation (23; current study).
We then assessed the roles of cAMP and PKC, two signaling pathways that have been known to regulate neuronal nAChR function (33). The increase in [3H]cytisine binding sites seen after chronic treatment with cholinergic channel ligands was mimicked by forskolin (also dibutyl cAMP and phosphodiesterase inhibitor IBMX) and PMA. The lack of effect of 4-α-PMA, a stereoisomer of PMA that neither binds nor activates PKC, on [3H]cytisine binding levels indicates specificity of the observed effects. It was also found that cotreatment of cells with (−)-nicotine (or DHβE) plus forskolin or PMA led to a synergistic enhancement in the up-regulation compared with those elicited by nAChR ligands alone. These synergistic (or multiplicative) effects on α4β2 nAChR levels indicate that distinct pathways may exist for protein kinases and nicotinic ligands. This is further supported by the observation that staurosporine, sphingosine, or chelerythrine, all inhibitors of PKC, failed to alter nicotine-evoked up-regulation. Our results suggest that there are at least two mechanisms by which α4β2 nAChR levels can be regulated: one is mediated by ligand interaction at the nAChR, and the other occurs in response to activation of PKA and PKC. It is possible that these two pathways may be linked as, for example, the phosphorylation of the nAChR by PKA and PKC. Previous studies of the peripheral-type nAChRs from muscle and electric organ have provided evidence that alterations in cAMP levels could lead to receptor phosphorylation, a process involved in desensitization (33). cAMP, through activation of PKA, has been shown to up-regulate muscle-type nAChRs by increasing the efficiency of receptor assembly from its constituent subunits through phosphorylation of the γ subunit (32) and through prevention of receptor degradation (32, 34). Agents that activate the phosphoinositide pathway, such as substance P, have been shown to alter nAChR desensitization in both chick ciliary and sympathetic ganglion neurons (35). It is noteworthy that the cytoplasmic loop connecting transmembrane segments III and IV in the human α4 subunit possesses consensus sequence for phosphorylation by both PKA (Ser362) and PKC (Ser334, Ser421, Thr532, Thr545, Ser550, and Ser586). The existence of these multiple consensus phosphorylation sites raises the issues of whether phosphorylation of these sites by kinases, including PKA and PKC, could be important to nAChR turnover and localization (33) and whether phosphorylation of these sites is required for receptor up-regulation. Previous studies in chick ciliary ganglion neurons have shown that cAMP analogs regulate the levels of functional nAChRs and increase the phosphorylation of the α3 subunit (35, 36). More recently, phosphorylation of rat brain α4 subunit by PKA in vitro has been directly demonstrated (37). Future site-directed mutagenesis experiments will be needed to investigate the role of these residues in the regulation of α4β2 nAChR expression and function.
Up-regulation of nAChRs has been shown to be accompanied by decreased nicotinic functional response in rodents receiving repeated, long term treatment with nicotine. In the current study, despite the substantial increases in α4β2 nAChR levels, a consistent decline in maximal ACh-evoked ion flux was observed when cells were treated with concentrations of (−)-nicotine of >1 μm. This is consistent with previous studies showing inactivation of nicotinic functional responses after pretreatment with nicotine measured biochemically as attenuation of hormone/neurotransmitter release or behaviorally as tolerance to some of the effects of (−)-nicotine, including behavioral effects, decreases in locomotor activity, and decreases in body temperature (9, 16). Although nAChR levels remain elevated ∼10–15-fold by (−)-nicotine treatment, the observed decline in functional response, perhaps indicative of the failure to recover from a desensitized state, may underlie the development of tolerance seen in vivo after long term (−)-nicotine treatment. However, when cells were treated with low concentrations of (−)-nicotine (100 nm and 1 μm), although unitary flux per nAChR was diminished, the overall net cation flux evoked by ACh showed a significant increase with respect to untreated cells, with a maximal increase observed with 1 μm(−)-nicotine. It is interesting to note that the range of concentrations of (−)-nicotine that elicited enhancement in nAChR function is comparable with the levels present in the serum of smokers (150 nm; Ref. 30). It is tempting to speculate that such enhancement in nAChR function after treatment with low concentrations of (−)-nicotine mediates some of the beneficial neurochemical and behavioral effects of (−)-nicotine, whereas the down-regulation of function observed after treatment with high concentrations of (−)-nicotine could underlie the development of tolerance (16).
In contrast to (−)-nicotine, treatment with DHβE showed a somewhat differential profile in modulating the functional activity of ACh. The maximal efficacy of ACh to activate cation efflux showed significant increases compared with untreated cells at all concentrations of DHβE tested (Fig. 5B). Although DHβE, unlike (−)-nicotine, does not produce inactivation, the net maximal increase in ion flux was only ∼90% (i.e., <2-fold) compared with the nearly 7-fold maximal increase in binding levels. This lack of correlation between the receptor density and function may be attributed to the fact that much of the up-regulated [3H]cytisine binding sites may represent nAChRs located in intracellular compartments and, accordingly, unavailable to contribute to ion flux measurements. Chronic DHβE treatment in vivo has been shown to increase the number of [3H]cytisine binding sites without concomitant development of behavioral tolerance as opposed to nicotine, and it has been suggested that receptor up-regulation by chronic agonist, but not antagonist, administration is associated with behavioral desensitization or tolerance (38). Such a differential modulation of α4β2 nAChR function by chronic drug treatment, as observed in the current study, may underlie some of the beneficial effects of cholinergic channel ligands in neurodegenerative diseases such as Alzheimer’s disease, in which one of the most consistent neurochemical abnormality is a decrease in cholinergic transmission arising from the degeneration of the basal forebrain cholinergic system (39).
The availability of functional human α4β2 nAChRs stably expressed in a mammalian cell line has facilitated the study of the regulation of this major neuronal nAChR in the human brain. Results of the current study demonstrate that treatment with cholinergic channel ligands can rapidly up-regulate human α4β2 nAChRs and differentially modulate their functional activity by processes that do not require receptor activation or ion flow through the channel. Clearly, differences are apparent between human and avian homologs, especially in the regulation by antagonist ligands. The magnitude and kinetics of up-regulation with the human α4β2 subtype also seem to be different from those observed in dexamethasone-induced M10 cells expressing the avian α4β2 nAChRs, in which a maximal 2–2.5-fold increase over 4 days of (−)-nicotine treatment has been reported (23, 27). Whether these variations arises from the choice of different expression systems (e.g., constitutive versus inducible) or cell line types or are due to differences in the amino acid sequences of the avian and human subunits remains to be investigated. For example, the M10 cells require induction with the glucocorticoid dexamethasone for α4β2 nAChR expression (6), and it is possible that dexamethasone alone modifies receptor expression, as reported for the muscle-type nAChRs expressed in myotubes (40). In addition, differences are noted at the amino acid level between the chick and the human α4 sequences adjacent to the highly conserved disulfide loop (Cys161–Cys175) and in the long cytoplasmic domain of the α4 subunit, which contains multiple phosphorylation sites and shows only ∼55% identity between human and avian homologs. For example, compared with the human sequence, the chick α4 subunit possesses additional potential PKC phosphorylation sites.
There is emerging evidence that activation of nAChRs mediates a diversity of responses, including neurotransmitter release, neurogenic control of cerebral blood flow, cognitive enhancement, neuroprotection, and analgesia (1, 17). Although it is not clear which subunit combinations form nAChRs in situ to mediate these diverse effects (for a discussion, see Ref. 2), the identification of compounds that selectively modulate heterologously expressed nAChR subtypes has been a major focus of research for the past decade. Such efforts have led to the development of novel cholinergic channel ligands, including ABT-418, ABT-089, RJR-1647, SIB-1508Y, and GTS-21, with beneficial effects on cognition, attention processes, anxiety, and neurodegenerative diseases (17). It is possible that chronic treatment with subtype-selective ligands may differentially regulate various nAChR combinations. Further understanding of the regulation of nAChRs by the various novel cholinergic channel modulators could considerably strengthen the basis for the development of novel cholinergic therapeutics lacking tolerance liabilities and with potential for long term use in ameliorating some of the deficiencies associated with degenerative and other diseases.
Acknowledgments
We thank Drs. Stephen P. Arneric, Diana Donnelly-Roberts, and Theresa Kuntzweiler for helpful discussions and comments on the manuscript.
Footnotes
- Received October 22, 1996.
- Accepted May 20, 1997.
-
Send reprint requests to: Murali Gopalakrishnan, Ph.D., Neuroscience Research (D-47C; Bldg. AP10LL), Pharmaceutical Products Division, Abbott Laboratories, 100 Abbott Park Road, Abbott Park, IL 60064-3500. E-mail:murali.gopalakrishnan{at}abbott.com
-
↵1 M. Gopalakrishnan, E. J. Molinari and J. P. Sullivan, unpublished observations.
Abbreviations
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- DHβE
- dihydro-β-erythroidine
- DMPP
- 1,1-dimethyl-4-phenylpiperazinium iodide
- DMEM
- Dulbecco’s modified Eagle’s medium
- HEK
- human embryonic kidney
- PKC
- protein kinase C
- PKA
- cAMP-dependent protein kinase
- PMA
- phorbol-12-myristate-13-acetate
- IBMX
- 3-isobutyl-l-methylxanthine
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}