


Abstract
All ionotropic glutamate receptor (iGluR) subunits analyzed so far are heavily N-glycosylated at multiple sites on their amino-terminal extracellular domains. Although the exact functional significance of this glycosylation remains to be determined, it has been suggested that N-glycosylation may be a precondition for the formation of functional ion channels. In particular, it has been argued that N-glycosylation is required for the formation of functional ligand binding sites. We analyzed heterologously expressed recombinant glutamate receptors (GluRs) of all three pharmacological subclasses of glutamate receptors,N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, and kainate receptors. By expressing the GluR subunits in tunicamycin-treated, nonglycosylating Xenopus laevis oocytes, we determined that in neither case is N-glycosylation required for ion channel function, although for NMDA receptors, functional expression in the absence of N-glycosylation is very low. Furthermore, we analyzed and compared the interaction of the desensitization-inhibiting lectin concanavalin A (ConA) with all functional GluR subunits. We show that although ConA has its most pronounced effects on kainate receptors, it potentiates currents at most other receptor subtypes as well, including certain NMDA receptor subunits, although to a much lesser extent. One notable exception is the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor GluR2, which is not affected by ConA. Furthermore, we show that ConA acts directly via binding to the carbohydrate side chains of the receptor protein.
iGluRs are the prevalent excitatory neurotransmitter receptors in the central nervous system of vertebrates (1). They can be classified into three major subfamilies on the basis of pharmacological and electrophysiological profiles: NMDA, AMPA, and KA receptors. Being the main mediators of cell-to-cell signaling, GluRs are regulated and functionally modulated by a multitude of post-transcriptional and post-translational mechanisms such as alternative splicing, RNA editing, protein phosphorylation, palmitoylation, andN-glycosylation (for a review, see Ref. 2). iGluRs are thought to consist of five subunits and may form heteromeric receptors containing different subunits, in many cases resulting in a receptor complex with different properties than their constituent subunits. For some receptor subunits, function can be demonstrated only on coexpression with another subunit of the same subfamily (for a review, see Ref. 2).
The amino acid sequences of iGluR subunits contain 4–12 potential extracellular sites for N-glycosylation, which conform to the universal consensus sequence N-X-S/T, with X ≠ P. These sites are marked in Fig. 1 and occur in the two large domains of iGluRs that according to the recently proposed three-transmembrane domain model, are located in the two putatively extracellular domains, the amino terminus and the loop between transmembrane domains B and C (3, 4). Many but not all of the sites are conserved within or even across subfamilies; however, no site is conserved across all iGluRs. The use of at least some of these consensus sites has been confirmed (5). For GluR1 alone, a systematic investigation of all existing consensus sites demonstrated that each of the six sites is glycosylated, at least in oocytes (3); however, the functional role of the carbohydrate moieties of iGluRs is not clear. Carbohydrate side chains have been implicated in many diverse functions, including protection from proteases, correct assembly of the subunits, formation of ligand binding sites, receptor targeting to the cell surface, and recognition by extracellular modulators (for a review, see Ref. 6). Importantly, the role ofN-glycosylation in receptor function seems to vary widely among different receptors.

Structure of all iGluR subunits investigated in this study. Arrowheads, position of all extracellularN-glycosylation consensus sites (N-X-S/T, X ≠ P).Right of bars, total number of sites. Stippled vertical lines, connect conserved consensus sites at homologous positions in different receptors. Black squares, Position of transmembrane domains A, B, and C. Hairpin loop between domains A and B, position of the ion channel-forming domain. Hatched rectangles, alternatively spliced domains (only indicated for splice variants that were investigated).
There have been contradictory reports regarding whetherN-glycosylation is essential for GluR function. Several groups have claimed that glycosylation is required for channel function (7, 8) or ligand binding (9-11), whereas other studies found no such requirements (3, 12). We therefore reinvestigated this question to determine whether N-glycosylation is necessary for channel function.
Carbohydrate side chains attached to N-glycosylation consensus sites of iGluRs most likely are the sites of interaction with lectins such as ConA. ConA has been found to potentiate current responses of native (13-17) and recombinant (18-21) iGluRs, with a strong selectivity for KA receptors over AMPA receptors. It is assumed that ConA acts by inhibiting receptor desensitization (14, 15). Several studies have reported the potentiation of selected iGluR subunits by ConA (18-20, 22), but not all of the functional subunits have been examined, and no study has compared the effects of ConA at all members of the iGluR family. Specifically, NMDA receptors have not been investigated in great detail, and the two existing reports on ConA modulation of these receptors are contradictory (18, 19). NMDA receptor splice variants have not been examined at all. Because the potentiating effect of ConA is increasingly used as a pharmacological tool to identify subsets of iGluRs in tissue slices or cell culture, it is important to know which effects ConA exerts on each of the known functional iGluR subunits or combinations; therefore, we systematically investigated the modulatory properties of ConA for all cloned subtypes of iGluRs.
This study shows that (a) N-glycosylation is not strictly essential for functional expression of any iGluR subunit combination but may affect current amplitudes and (b) receptor modulation by ConA is a direct effect of the lectin on the receptor protein and is subunit and splice variant dependent.
Materials and Methods
ConA (grade VI) was obtained from Sigma Chemie (Munich, Germany). Tunicamycin was purchased from Boehringer-Mannheim Biochemica (Mannheim, Germany). Other drugs were purchased from Sigma unless noted otherwise. Adult female frogs (Xenopus laevis) were obtained from Nasco (Fort Atkinson, WI).
cRNA synthesis.
Template was prepared from circular plasmid cDNA by linearization of each clone with a suitable restriction enzyme. cRNA was prepared from 1 μg of linearized template using an in vitro transcription kit (Stratagene, Heidelberg, Germany) with a modified standard protocol that uses each of the nucleotides at 800 μm (except GTP, for which 200 μm was used), 800 μm m7GpppG (Pharmacia, Vienna, Austria) for capping, and an extended reaction time of 3 hr with T3 or T7 RNA polymerase. All cRNAs were trace-labeled with [32P]UTP (Amersham, Braunschweig, Germany) to allow quality checks by gel electrophoresis and calculation of the yield.
Electrophysiological recordings from X. laevisoocytes.
Frog oocytes of stages V or VI were obtained by surgical removal of parts of the ovaries of X. laevis anesthetized with 3-aminobenzoic acid ethyl ester (2 g/liter). The removed ovaries were chopped and incubated with 815 units/ml (2.8 mg/ml) collagenase type I (Worthington Biochemicals, Freehold, NJ) and 2200 units/ml (0.15 mg/ml) trypsin at 20° for 2 hr in calcium-free Barth’s solution (see below) with slow agitation to remove the follicular cell layer and then washed extensively with Barth’s solution [88 mm NaCl, 1.1 mm KCl, 2.4 mm NaHCO3, 0.3 mm Ca(NO)3, 0.3 mmCaCl2, 0.8 mmMgCl2, 15 mm HEPES, pH 7.6 with NaOH]. Oocytes were maintained in Barth’s solution supplemented with 100 μg/ml gentamycin, 40 μg/ml streptomycin, and 63 μg/ml penicillin. Oocytes were injected with 10 ng of cRNA for homomeric receptors and 5 ng of cRNA for each subunit of heteromeric receptors 24 hr after collagenase treatment using a 10-μl Drummond (Broomall, PA) microdispenser. At 5–6 days after RNA injection, oocytes were recorded in amphibian Ringer’s solution (115 mm NaCl, 2.5 mm KCl, 1.8 mm CaCl2, 10 mm HEPES, pH 7.2 with NaOH) under voltage clamp at a holding potential of −70 mV, with a Turbo Tec-10CD amplifier (NPI, Tamm, Germany). Voltage electrodes had resistances of 1–3 MΩ and were filled with 3 m KCl; current electrodes had resistances of ∼1 MΩ and were filled with 3m CsCl. Agonist was applied by superfusion at a flow rate of 6 ml/min in a 50-μl recording chamber. For each oocyte, steady state currents were recorded before and after ConA treatment. Lectin treatment was carried out by pipetting 100 μl of 10 μmConA in amphibian Ringer’s solution (calculated for the tetramer of 102 kDa) directly into the recording chamber while perfusion was stopped. Oocytes were incubated for 8 min in the lectin solution; then, perfusion was restarted for 1 min before the next agonist application. To estimate EC50 values, eight different agonist concentrations (A) were applied, and steady state values of the evoked currents (I) were measured and fitted with the SigmaPlot program (Jandel Scientific, San Rafael, CA) to the equation I = Imax/[1 + (EC50/A)nH], where Imax is the maximal current, EC50 is the agonist concentration giving half-maximal currents, and nH is the Hill coefficient.
Inhibition of N-glycosylation by tunicamycin.
To express nonglycosylated receptors in oocytes, we used tunicamycin, a potent inhibitor of N-glycosylation (23). At 1 day before injection with cRNA, oocytes were preinjected with 50 nl of 400 μg/ml tunicamycin (20 ng/oocyte, or ∼22 μg/ml for an oocyte of 1.2-mm diameter). Immediately before injection, a stock solution of 10 mg/ml tunicamycin in DMSO was diluted to 4% DMSO with amphibian Ringer’s solution; 4% DMSO did not adversely affect the oocytes. Bath treatment of oocytes with tunicamycin turned out to be largely ineffective in inhibiting N-glycosylation (data not shown).
Site-directed mutagenesis.
Single nucleotide exchanges were introduced by polymerase chain reaction-mediated site-directed mutagenesis using mutagenetic primers as described previously (3). We mutated wild-type GluR2 at the Q/R editing site to GluR2(R586Q) [hereafter referred to as GluR2(Q)] for both the flip and flop splice variants. To create N-glycosylation site mutants of GluR1, we deleted the first N-glycosylation consensus site in the GluR1 sequence to obtain GluR1(N45S), which we called GluR1-ΔNG1. We also created the double-mutant GluR1-ΔNG1/3 by also deleting the third N-glycosylation consensus site [mutation GluR1(N239S)], and we engineered the mutant GluR1-ΔNG3 by introducing solely the mutation N239S. GluR1-ΔNG2/4/5/6 was engineered by deleting N-glycosylation sites 2 and 4–6 by introducing the mutations N231S, N345S, N383S, and N388S, respectively. GluR2(Q) with two ectopic N-glycosylation sites (GluR2-EG1/2) was engineered by introducing the mutations E45N and A47T (to create EG1) and D243N and D245T (to create EG2). All mutations were verified by chain-termination method sequencing using the Sequenase kit from United States Biochemical Corp. (Cleveland, OH).
Labeling of cell surface glycoproteins with biotinylated ConA.
To identify only the fraction of receptor protein inserted in the plasma membrane, surface proteins were tagged with biotin and isolated by streptavidin/Sepharose-mediated precipitation of the labeled protein. Briefly, intact oocytes were incubated in 1 mg/ml NHS-SS-Biotin (Pierce, Indianapolis, IN) solution for 2 hr at 4°. After five 10-min washes in frog Ringer’s solution, 10 oocytes were homogenized with a Teflon pestle in 200 μl of buffer H [100 mm NaCl, 20 mm Tris·HCl, pH 7.4, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, and a cocktail of additional proteinase inhibitors (2.5 μg/ml leupeptin, 20 μg/ml aprotinin, 2.5 μg/ml pepstatin, and 20 μg/ml benzamidine hydrochloride)]. The homogenate was kept on ice for 60 min. After centrifugation for 60 sec at 16,000 × g to remove yolk platelets and the melanin pigment granula, the supernatants were supplemented with 20 μl of streptavidin/Sepharose beads (Sigma) and incubated for 3 hr at 4° on a rotator. The streptavidin/Sepharose beads were pelleted by a 60-sec spin and washed three times with buffer H, and the washed pellets were boiled in 40 μl of SDS-polyacrylamide gel loading buffer (0.8 m β-mercaptoethanol, 6% SDS, 20% glycerol, 25 mm Tris·HCl, pH 6.8, 0.1% bromphenol blue).
Crude membrane preparation, gel electrophoresis, and Western blotting.
At 3–6 days after RNA injection, a crude membrane fraction was prepared from oocytes. Briefly, 20 oocytes were homogenized with a Teflon pestle in 400 μl of buffer H as described above. The homogenate was kept in a shaker at 4° for 15 min and then spun down for 1 min at 16,000 × g. The supernatants containing the cytosolic proteins as well as the solubilized membrane proteins were separated at 4° on discontinuous SDS-polyacrylamide gels (3) with a 5% stacking gel and a 7.5% separating gel. The gel was blotted (3) onto Hybond ECL nylon membranes (Amersham). Membranes were blocked with 1X Roti-block (Roth) and probed (4° overnight) with the primary anti-GluR antibody diluted in antibody incubation buffer (0.1X Roti-block, 0.1% Triton X-100, 20 mm Tris·HCl, pH 7.6, 140 mm NaCl). Immunoreactive bands were detected by peroxidase-labeled donkey anti-rabbit IgG antibodies (Jackson Laboratories, Bar Harbor, ME) in the case of polyclonal primary antibodies or by peroxidase-labeled donkey anti-mouse IgG antibodies (Jackson Laboratories, Bar Harbor, ME) in the case of monoclonal primary antibodies. All antibodies were diluted in antibody incubation buffer and visualized with the enhanced chemiluminescence method (ECL Kit, Amersham).
Deglycosylation.
Crude membranes were enzymatically deglycosylated with N-Glycosidase F (Boehringer-Mannheim) at 37° for 4 hr in the presence of protease inhibitors (see above) after denaturation for 10 min at 100° in 1% SDS, following the manufacturer’s protocol.
Results
N-Glycosylation is not required for receptor function.
To investigate the requirement forN-glycosylation of GluRs, we expressed recombinant rat GluR subunits in X. laevis oocytes as homomeric proteins as well as in various heteromeric combinations. Oocytes were either not pretreated at all or preinjected with the antibiotic tunicamycin. Tunicamycin, a hydrophobic analog of UDP-N-acetylglucosamine, specifically inhibitsN-glycosylation by blocking the addition ofN-acetylglucosamine to the sugar carrier dolicholphosphate, which is the first step in the formation of the core oligosaccharide inN-linked glycosylation events. We then compared the steady state current amplitudes of receptor subunits expressed in untreated, glycosylating oocytes with those in tunicamycin-preinjected, nonglycosylating oocytes.
For AMPA receptors, function is preserved in the nonglycosylated state for all homomeric and heteromeric subunit combinations tested (Table1). Interestingly, the flop and flip versions of each of the four AMPA receptor subunits behave quite differently: although lack of N-glycosylation increases Glu-evoked currents at all flip versions except for GluR3flip, amplitudes at flop versions are generally reduced, except in the case of GluR4flop (Fig. 2, A and B, and Table 1). The behavior of heteromeric AMPA receptor combinations cannot be easily predicted on the basis of that of the constituting subunits, except for a tendency to follow the behavior of the flop subunit in the combination rather than the flip subunit.
Modulation of steady state current responses of iGluRs by tunicamycin or ConA pretreatment

Tunicamycin pretreatment of oocytes differentially affects currents of expressed iGluRs. Representative current traces obtained with either KA or Glu as agonist are shown for untreated (left) and tunicamycin-pretreated (right) oocytes from the same frog. GluR1flop (A), GluR1flip (B), GluR6(Q)flop (C), GluR6(Q)/KA1 (D), NR1–4b (E), and NR1–1b/2B (F) are shown as examples of subunits with reduced (A and F), increased (B and D), unchanged (C), and abolished (E) responses after tunicamycin treatment. Note different scale bars for KA- and Glu-evoked currents at AMPA receptors.
When currents observed in tunicamycin-treated oocytes were normalized to currents in untreated control oocytes, a comparison of Glu- and KA-evoked normalized currents at 16 AMPA receptor subunits or combinations revealed unexpected differences between the two agonists. KA-evoked responses in tunicamycin-treated oocytes were either smaller (11 of 16 subunit combinations) or about the same size (five of 16) as those in untreated oocytes. In contrast, with Glu as the agonist, seven of the 16 receptors examined displayed significantly larger currents compared with glycosylating controls (Table 1). However, regardless of the glycosylation state of the receptor, absolute current amplitudes are always considerably larger for KA-evoked responses than for Glu-evoked responses, with the sole exception of homomeric GluR2flips.
At KA receptors, N-glycosylation is not required for KA- or Glu-evoked steady state currents at any of the subunit combinations that give measurable currents in oocytes (Fig. 2C and Table 1). In fact, the heteromeric subunit combinations GluR6(Q)/KA1 and GluR6(Q)/KA2 give even larger steady state currents in the absence ofN-glycosylation (Fig. 2D and Table 1). In contrast to steady state currents, however, the rapidly desensitizing initial peak currents are significantly decreased or even lacking at nonglycosylated receptors (Fig. 2C).
At NMDA receptors, lack of N-glycosylation causes a large reduction (≥99%) in steady state currents in heteromeric subunit combinations containing NR2A or NR2B as the structural subunit but does not seem to abolish channel function entirely (Fig. 2F and Table 1). For all homomeric subunits tested and for heteromeric combinations containing either NR2C or NR2D, we failed to detect any currents in nonglycosylating oocytes (Fig. 2E and Table 1). However, because these particular subunits and subunit combinations comprise the NMDA receptors with the smallest currents, it is possible that channel function was not entirely abolished but merely dropped below our detection level. If we assume that for the subunits with low levels of expression, lack of N-glycosylation reduces currents to ≤1% of control values measured in glycosylating oocytes, as it is for the better-expressing NMDA receptor subunits, then maximal amplitudes would have dropped to ≤∼1 nA and thus could not have been detected in our expression system.
To rule out that the small NMDA receptor currents observed in tunicamycin-treated oocytes are due to incomplete inhibition ofN-glycosylation, the absence of carbohydrate side chains must be confirmed. Two lines of evidence argue against residualN-glycosylation. First, we found a total lack of ConA-mediated current potentiation on tunicamycin treatment for all subunits tested, including NR1–4a/NR2B, which normally will be potentiated when glycosylated (Table 1). A second line of evidence was provided by Western blots of Triton X-100-solubilized crude oocyte membranes separated on polyacrylamide gels. Because all receptor subunits have at least four consensus sites forN-glycosylation (Fig. 1) and all or most of these sites are likely to be used (3), a substantial difference in molecular weight can be expected between N-glycosylated and non-N-glycosylated proteins, which is easily resolved on a polyacrylamide gel. Western blots of such gels revealed a downward shift in the molecular weight of receptor proteins expressed in tunicamycin-treated oocytes. No bands equivalent to theN-glycosylated protein could be detected inN-glycosylation-incompetent oocytes (Fig.3). This confirms our previous findings for GluR1 that up to 12 days after cRNA injection into tunicamycin-treated oocytes, no N-glycosylated receptor protein is being synthesized (3). Furthermore, N-glycosidase F treatment of membrane preparations demonstrated that the molecular weight of nonglycosylated receptor protein obtained from tunicamycin-pretreated oocytes was identical to that obtained by enzymatic deglycosylation of receptor protein from control oocytes (Fig. 3B). Moreover, N-glycosidase F treatment of receptor protein from tunicamycin-pretreated oocytes did not show any further downward shift in the molecular weight (Fig. 3B).

Western blots of total membrane preparations (A–F, H, and I) or surface receptors (G; see text for details) from untreated (−) or tunicamycin-treated (+) oocytes expressing various iGluR subunits as noted. Lanes marked E, samples enzymatically deglycosylated with N-glycosidase F. Blots were probed with polyclonal, affinity-purified carboxyl-terminal antibodies against GluR1 (A and B) or GluR6 (C and D) (provided by Bob Wenthold), a monoclonal anti-NR1 antibody (E–H) (provided by Nils Brose), and a polyclonal carboxyl-terminal anti-NR2B antibody (I) (Chemicon International, Temecula, CA). cRNA (5 ng/oocyte) had been injected, except for blot E (10 ng), and the protein equivalent to two oocytes was loaded per lane, except for blots F (1 oocyte) and G (10 oocytes).Filled arrows, glycosylated receptor proteins.Open arrows, smaller unglycosylated variants. ∗, Band not related to the heterologously expressed protein; it is recognized by the antibody even in uninjected oocytes. This band varies considerably even between oocytes from the same frog. 83, 119, And 213 kDa, positions of prestained protein markers.
The amount of protein produced in glycosylating versus nonglycosylating oocytes depended on the receptor subfamily. AMPA receptors seem to express equally well, regardless of N-glycosylation (Fig.3A), whereas KA receptors such as GluR6(Q) (Fig. 3, B and C) and GluR6(R) (data not shown) may show some reduction in protein expression. The editing variants of GluR6 express equally well. In contrast, the NR1 subunit of the NMDA receptor is expressed only very weakly in glycosylation-deficient oocytes compared with normal, glycosylating oocytes (Fig. 3, D–G). This observation most likely explains the absence of currents at homomeric NR1 receptors and the low levels of currents observed for heteromeric NR1/NR2B receptors. Reduced NR1 subunit expression in nonglycosylating oocytes is observed for both major NR1 splice variants, NR1–1a (Fig. 3D) and NR1–1b (Fig. 3, E and G), and is found to a similar extent in total expressed protein (Fig.3E) and surface protein (Fig. 3F). Interestingly, the reduction in NR1 protein expression in nonglycosylating oocytes is not accompanied by a reduction in the expression level of the NR2B subunit coexpressed in the same oocyte (compare Fig. 3, G and H). Similarly, coexpression of NR1–1a and GluR6 does not lead to a reduction in GluR6 expression (compare Fig. 3, B and C). Thus, the reduced expression level of NR1 in tunicamycin-treated oocytes does not reflect a generalized down-regulation of protein synthesis.
The lack of N-glycosylation did not dramatically alter the EC50 values for the agonists KA or Glu. EC50 values were either unchanged [GluR1flop (Fig.4A) and GluR6(Q) (Fig. 4C)], slightly decreased [NR1–1b/NR2B (Fig. 4D)], or slightly increased [GluR1flip (Fig. 4B)]. The very modest changes in EC50 values cannot account for the observed changes in current amplitude. In fact, for NR1–1b/NR2B, the current is decreased, whereas the affinity for Glu is increased. No changes innH values were seen for any of the subunits tested, indicating that the number of ligand binding sites per receptor complex remained unaltered (Fig. 4).
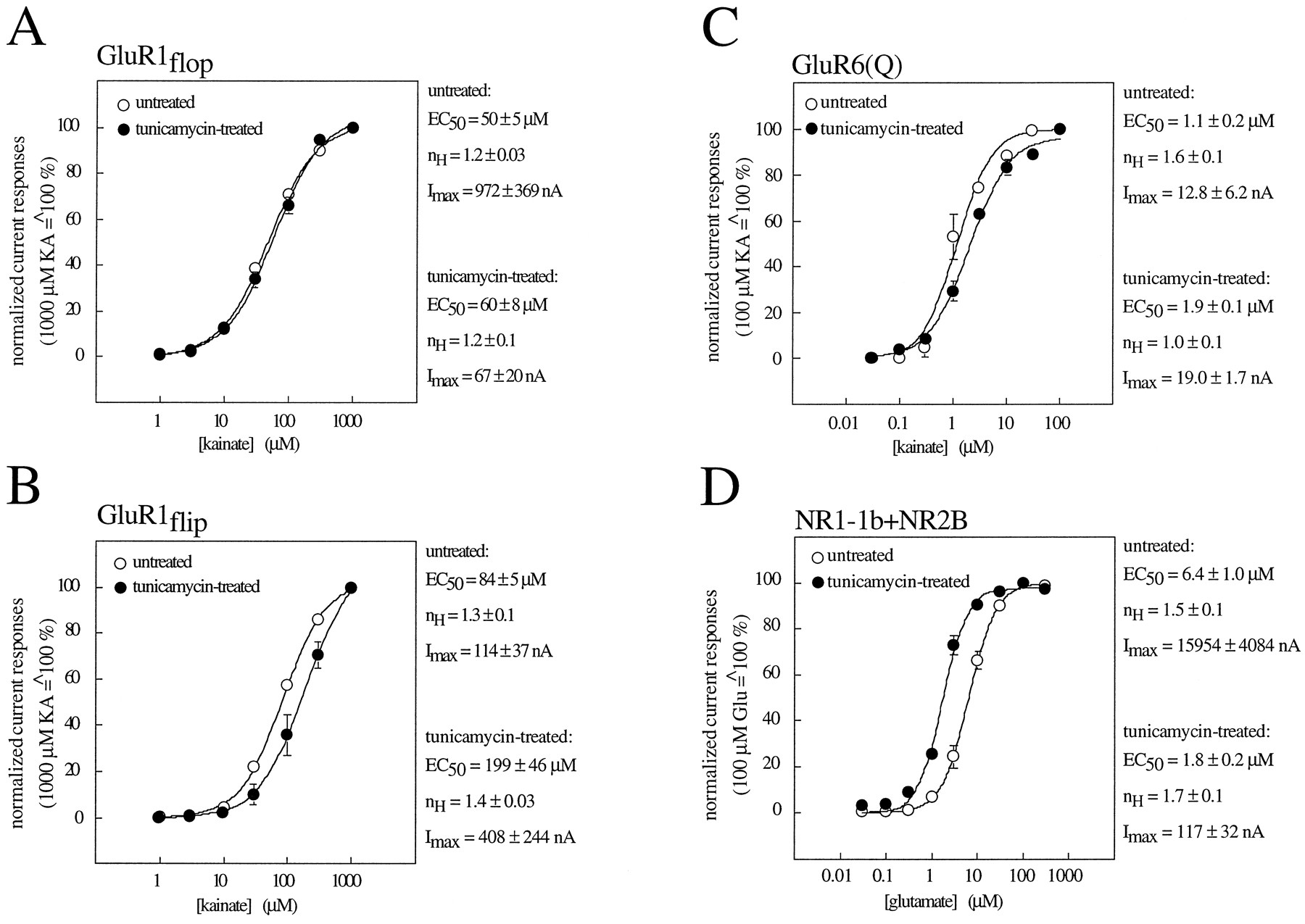
Dose-response curves for GluR1flop (A), GluR1flip (B), GluR6(Q) (C), and NR1–1b/NR2B (D) expressed in untreated (○) and tunicamycin-treated (•) oocytes. EC50 values, n Hvalues, and maximal currents (Imax) were estimated with KA as the agonist for GluR1 and GluR6 and with Glu in the presence of 10 μm glycine as the agonist for NR1–1b/NR2B. Values represent the mean ± standard error of three individually measured dose-response curves.
In summary, our data suggest that N-glycosylation is not a prerequisite for the expression of functional ion channels in any of the three major subfamilies of ionotropic GluRs. Although not essential, N-glycosylation is, however, necessary for the efficient expression of NMDA receptor channels. In this case,N-glycosylation seems to be specifically required to ensure high-level expression of the NR1 subunit. In contrast, no such requirement was found for AMPA or KA receptor subunits.
Lectins potentiate current amplitudes at GluRs in a highly subunit-specific manner.
To analyze lectin-mediated modulation of AMPA receptors, we examined all eight homomeric flop/flip splice variants (flop and flip variants each of GluR1 to GluR4) as well as six heteromeric combinations of these subunits. With 300 μmGlu as the agonist, all receptors except homomeric GluR2 subunits showed small but significant potentiations of steady state currents after an 8-min pretreatment with 10 μm ConA. Potentiations of Glu-evoked currents ranged from 3.6 ± 1.2 for GluR4flop/GluR2flop to 13 ± 4.0 for GluR4flip and were always larger than potentiations of KA-evoked currents (Table1). Notably, we found no significant differences in potentiation between flop and flip splice variants, regardless of whether homomeric or heteromeric receptors were analyzed (Fig.5A). Heteromeric combinations of GluR1, GluR3, or GluR4 with GluR2 (flip or flop) generally displayed ConA-mediated current potentiations similar to those of the respective homomeric receptors (Table 1), indicating that the nonpotentiated GluR2 subunit does not dominate the ConA effect the way it dominates the rectification properties and the calcium permeability when expressed in heteromeric assemblies (2).

ConA pretreatment differentially affects currents of iGluRs in an N-glycosylation-dependent manner. Representative current traces recorded with Glu or KA are shown for untreated oocytes as well as oocytes from the same batch incubated in 10 μm ConA for 8 min. GluR1flop (A), GluR2(Q)flop (B), and GluR6(Q) (C) are shown as examples of subunits with slightly increased, unchanged, and hugely potentiated responses, respectively, after ConA treatment. NR1–4a (D) and NR1–4b (E) are shown as examples for splice variant-specific potentiation: NR1–4a is potentiated but NR1–4b is unaffected by ConA. Note that after tunicamycin pretreatment (right), no more ConA potentiation is seen, regardless of the subunit analyzed.
Because homomeric GluR2 subunits seemed to be the only AMPA receptors not affected by ConA (Fig. 5B), we investigated these receptors more closely. Homomeric GluR2 subunits yield only minute currents, which makes it very difficult to assess reliably the effect of ConA. Therefore, we used site-directed mutagenesis on both GluR2flop and GluR2flip to construct mutants containing a glutamine residue (Q) instead of an arginine residue (R) at the Q/R editing site in the ion channel (position 586). This mutation is known to increase the maximal amplitudes of GluR2 channels considerably, and it also changes the calcium permeability and rectification properties (24, 25). The large currents of the mutant GluR2(Q) channels allowed easy quantification of the ConA effect, and we confirmed that GluR2 indeed is not potentiated by ConA, regardless of the editing status at the Q/R site (Fig. 5B and Table 1).
GluR2 has the lowest number of N-glycosylation sites of all iGluRs, with only four consensus sites. It differs from other AMPA receptors in lacking two of the six sites present in GluR1 and one of the five sites found in both GluR3 and GluR4. We set out to test whether those lacking N-glycosylation sites could potentially account for the absence of ConA-mediated current potentiation at GluR2 receptors. We used site-directed mutagenesis to alter the consensus site asparagine residues at the first and thirdN-glycosylation sites of GluR1 to serine residues. We created mutants lacking only the first site (GluR1-ΔNG1) or the third site (GluR1-ΔNG3) and a double-mutant lacking both the first and third sites (GluR1-ΔNG1/3). When these mutants were tested for ConA-mediated current potentiation, GluR1-ΔNG1 and GluR1-ΔNG3 still showed potentiation, although Glu-evoked currents were significantly less increased than in wild-type GluR1 (Fig.6). However, the double-mutant GluR1-ΔNG1/3 could not be lectin-potentiated any more, regardless of the agonist used for channel activation (Fig. 6). The complementary quadruple mutant GluR1-ΔNG2/4/5/6, which lacksN-glycosylation sites 2 and 4–6, on the other hand, was still potentiated (Fig. 6), demonstrating that sites 1 and 3 are sufficient to allow potentiation. This mutant was difficult to analyze, however, because the currents were quite small; in some cases, currents could be seen only after ConA treatment. These data suggest that the GluR2-like N-glycosylation pattern of GluR1-ΔNG1/3 is linked to a GluR2-like phenotype in terms of lectin interaction and the two sites are both necessary and sufficient. We conclude that the lack of ConA-mediated current potentiation of GluR2 is a consequence of its particular configuration of N-glycosylation sites rather than differences in the amino acid sequence between GluR2 and the other AMPA receptors elsewhere in the protein. To further support this conclusion, we engineered a mutant GluR2(Q), which has two additional, ectopic N-glycosylation sites at positions equivalent to sites 1 and 3 of GluR1 (see Fig. 1). This “GluR1-like” mutant of GluR2(Q) indeed regained some ConA-mediated current potentiation, although it did not approach wild-type GluR1 levels (Fig. 6).
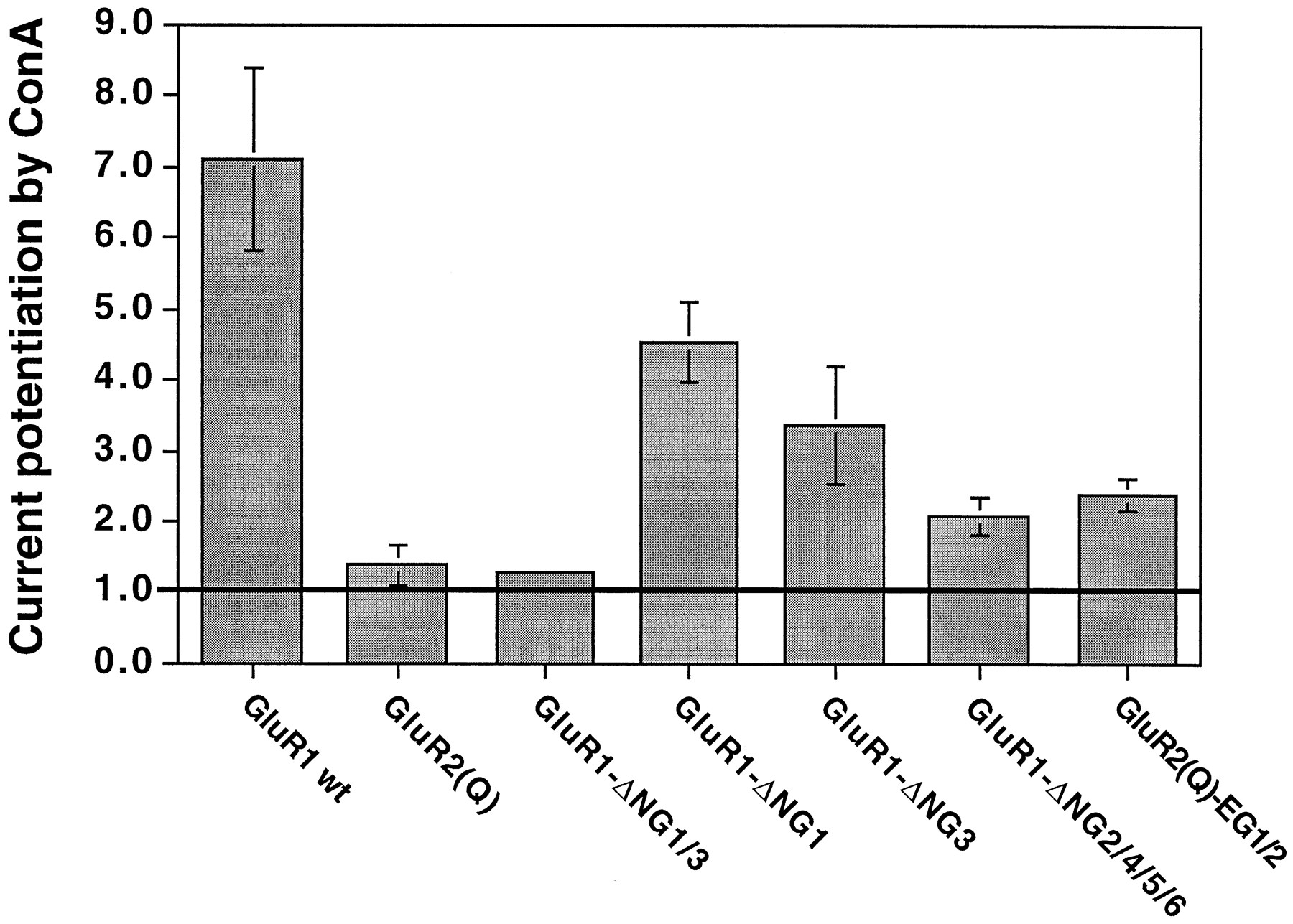
Simultaneous removal ofN-glycosylation sites 1 and 3 from GluR1, which leaves a GluR2-like N-glycosylation pattern (Fig. 1), abolishes ConA-mediated potentiation of Glu-evoked currents. GluR1 wt, GluR1 wild-type (n = 19); GluR2(Q) shown for comparison (n = 16);GluR1-ΔNG1/3, GluR1 lackingN-glycosylation sites 1 and 3 (n = 7); GluR1-ΔNG1, GluR1 lackingN-glycosylation site 1 (n = 9);GluR1-ΔNG3, GluR1 lackingN-glycosylation site 3 (n = 6);GluR1-ΔNG2/4/5/6, GluR1 lackingN-glycosylation sites 2 and 4–6 (n= 7); GluR2(Q)-EG1/2, GluR2(Q) carrying two additional ectopic N-glycosylation sites at positions equivalent to sites 1 and 3 (see Fig. 1) in GluR1 (n = 14).Error bars, ± standard error; those of GluR1-ΔNG1/3 are too small to show. Horizontal line, control current level (no ConA potentiation).
KA receptors displayed the largest ConA effects, which could easily reach values of several thousand-fold, for GluR5 as well as for GluR6 and for heteromeric assemblies of these subunits with KA1 and KA2 (Fig.5C and Table 1). To achieve fully developed potentiation, incubation with the lectin had to be performed for ≥8 min at 10 μmConA (5 min gave half-maximal potentiation; data not shown). Unexpectedly, we found a marked difference in ConA potentiation of GluR6 receptors of different Q/R-site editing status. Although the unedited Q form gave huge potentiations (>1000-fold, Fig. 5C), the effect was much more modest with the edited R form (∼100-fold). This was counterintuitive because the extracellular ligand binding sites as well as the N-glycosylation sites presumably targeted by ConA should be identical in both these editing variants (see Discussion).
An additional difference between Q and R editing variants was revealed by the agonist-dependency of the degree of ConA potentiation: Glu-evoked currents at Q variants are potentiated to a larger degree than KA-evoked currents, whereas at R variants, there is no obvious difference. The agonist-dependent differences in ConA potentiation of KA receptor variants extended to other agonists as well, such as domoate and methylglutamate. Interestingly, there is an inverse relation between ConA-mediated current potentiation and the amplitude of agonist-evoked basal currents. Although agonist-evoked steady state currents at GluR6(Q) follow the sequence domoate ≫ KA > methylglutamate > Glu, ConA-mediated current potentiation follows the reverse sequence Glu > methylglutamate > KA ≫ domoate (data not shown).
The GluR7 subunit of the KA receptor subfamily could not be analyzed because it did not give any measurable currents, whether expressed homomerically nor in heteromeric combinations with KA1, KA2, or both (Table 1). This confirms previous observations by other researchers (26, 27) .
NMDA receptors revealed an interesting pattern of small (≤2-fold) ConA-mediated current potentiations of certain subunits (Fig. 5, D and E, and Table 1). We compared all eight splice variants of the homomeric NR1 subunit, comprising four progressively shorter carboxyl-terminally spliced forms (NR1–1-4) that each can occur without (“a”) or with (“b”) an amino-terminal insertion of 21 amino acids (28, 29). All splice variants lacking the amino-terminal insertion (NR1–1a, NR1–2a, NR1–3a, and NR1–4a) showed potentiation, whereas the corresponding “b” splice variants (NR1–1b, NR1–2b, NR1–3b, and NR1–4b) did not (Fig. 5, D and E, Table 1). Instead, there was a small apparent reduction in maximal amplitudes in “b” splice variants. This reduction, however, was due to a slow run-down of NMDA receptor currents seen in most oocytes over time. This run-down occurred during the 8-min ConA incubation period and did not depend on the presence of ConA (data not shown).
On examination of heteromeric combinations of NR1–1a or NR1–1b with each of the four structural NMDA receptor subunits, NR2A, NR2B, NR2C, and NR2D (30), no potentiation was seen in any combination containing a “b” splice variant of NR1 (Table 1). However, when heteromeric combinations included an “a” splice variant of NR1, a small ConA-mediated potentiation was observed in cases in which the structural subunit was either NR2B or NR2D but not NR2A or NR2C. Thus, ConA-mediated current potentiation at NMDA receptors is subunit specific as well as splice variant dependent.
ConA acts via direct binding to the carbohydrate side chains of GluRs but does not affect the ligand binding site.
Current potentiation seen after ConA treatment is not due to an increase in the affinity for the agonists KA and Glu; EC50 values were virtually unchanged by ConA treatment. On treatment with lectin, they were entirely unaffected (NR1–1b/NR2B; Fig.7C), marginally decreased [GluR6(Q); Fig. 7B], or even slightly increased (GluR1flop; Fig. 7A). This finding was quite unexpected because binding of a large molecule such as ConA to the extracellular domain of a receptor protein might be expected to severely affect the ligand binding site; after all, ConA is a tetramer with a molecular weight of ∼102 kDa, which is in the same range as that of one glycosylated GluR subunit (∼105 kDa for GluR1, ∼118 kDa for GluR6, and ∼117 kDa for NR1).

Dose-response curves for GluR1flop (A), GluR6(Q) (B), and NR1–1b/NR2B (C) expressed in untreated (○) and ConA-treated (•) oocytes. EC50 values,n H values, and maximal currents (Imax) were estimated with KA as the agonist for GluR1 and GluR6 and Glu in the presence of 10 μm glycine as the agonist for NR1–1b/NR2B. Values represent the mean ± standard error of three individually measured dose-response curves.
It is well known that benzothiadiazines such as cyclothiazide act as potent and highly selective inhibitors of desensitization of AMPA receptors, and it has been suggested that they act by stabilizing a nondesensitized agonist-bound closed state of the receptor complex (31). To test whether ConA might act via the same mechanism as cyclothiazide, we compared KA- and Glu-evoked currents at GluR1flop-expressing oocytes that had been sequentially treated with cyclothiazide alone, ConA alone, and ConA plus cyclothiazide. As can be seen in Fig.8A, the effects of ConA and cyclothiazide are additive and not mutually exclusive regardless of which agonist is used. In another presentation of the same data (Fig. 8B), ConA-mediated potentiation factors are identical in the absence and presence of cyclothiazide. Thus, ConA and cyclothiazide clearly act via different molecular mechanisms.

ConA and cyclothiazide act via different mechanisms. A, ConA-mediated potentiation of AMPA receptors is independent of cyclothiazide-induced current potentiation. Modulatory effects of 10 μm ConA (2, open bars), 100 μm cyclothiazide (3, hatched bars), and 10 μm ConA plus 100 μmcyclothiazide (4, filled bars) on KA- and Glu-evoked currents of GluR1flop-expressing oocytes [1 (controls), stippled bars]. B, ConA-mediated current potentiation has the same size in control and cyclothiazide-treated oocytes. Potentiation of KA- and Glu-evoked currents at GluR1flop in the absence (contr., open bars) and presence (cyclo, filled bars) of 100 μm cyclothiazide. Note that for both agonists, ConA potentiation factors are identical regardless of the presence of cyclothiazide. Values are mean ± standard error for four oocytes.
The potentiating effect of ConA could be caused by direct interaction of the lectin and the receptor protein or by some indirect mechanism in which the lectin binds to some protein that in turn modulates the receptor. To distinguish between these two possibilities, we tested the ConA effect on each GluR subunit in tunicamycin-pretreated oocytes that will produce only nonglycosylated receptor protein (see Fig. 3). Under these conditions, ConA potentiation was completely abolished for all receptors, even in the case of GluR6, for which a current increase of several thousand-fold is seen without the tunicamycin pretreatment (Fig. 5C and Table 1). Thus, receptor N-glycosylation is an absolute requirement for ConA potentiation. These data demonstrate that for all subunits examined, the lectin must bind directly to the receptor via its carbohydrate side chains to elicit current potentiation.
Discussion
The functional importance of N-glycosylation.
Our data show that N-glycosylation in general is not required for GluR function. The analysis did not includeO-glycosylation, which is known to occur in GluRs (5) and persists in tunicamycin-treated oocytes. For AMPA and KA receptors, the lack of N-glycosylation does not impair the formation of fully functional ion channels. Certain subunits, such as the flip splice variants of the AMPA receptor or heteromeric KA receptors, pass even larger currents in the absence of N-glycosylation. The lack of N-glycosylation does not significantly alter the EC50 values for agonists at AMPA or KA receptors, indicating that carbohydrate side chains are not part of the ligand binding pocket and do not play a significant role in establishing the protein conformation required for the formation of that pocket. We observed that at many AMPA and KA receptor subunits, inhibition ofN-glycosylation causes an increase in Glu-evoked relative to KA-evoked steady state currents regardless of how the absence ofN-glycosylation affects the absolute amplitudes of the currents. Because the smaller steady state currents seen with Glu are usually attributed to Glu being a more strongly desensitizing agonist than KA (22), it seems that Glu is less desensitizing at nonglycosylated receptors. This suggests thatN-glycosylation has a role in setting the maximal desensitization levels of AMPA and KA receptors.
The lack of N-glycosylation does not prevent AMPA or KA receptor protein synthesis, transport, subunit assembly, or insertion into the plasma membrane, as demonstrated by the perfectly functional ion channels generated in nonglycosylating oocytes. Our data on recombinant receptors are in agreement with the report by Sumikawaet al. (12), who found that KA currents expressed from total brain RNA were not abolished by tunicamycin.
Our data partially contradict those of Kawamoto et al. (9,10), who claimed that N-glycosylation is essential for ligand binding of GluR1 and GluR2. The fact that we observed functional receptors that could be activated by various glutamatergic agonists (Glu and KA; Fig. 2, A and B) in tunicamycin-treated oocytes that showed no biochemical (Fig. 3) or pharmacological (Table 1) evidence of residual N-glycosylated receptors demonstrates conclusively that ligands must have bound to the nonglycosylated receptor. This is especially obvious for the flip splice variants of GluR1 and GluR2, which are even potentiated by lack of N-glycosylation. Muβhoff et al. (7) used tunicamycin-treated oocytes to investigate the effect of inhibition of N-glycosylation on AMPA, KA, and quisqualate responses expressed from total rat brain RNA and reported a total lack of responses in the absence ofN-glycosylation. This finding contradicts our data as well as those of Sumikawa et al. (12). The reason for this discrepancy remains unclear; however, it might be speculated that the RNA preparation that Muβhoff et al. used selectively expressed the more N-glycosylation-sensitive GluR subunits (Table 1) or an N-glycosylation-dependent subunit that has not yet been cloned.
Non-N-glycosylated NMDA receptors, unlike AMPA and KA receptors, undergo large reductions in their current amplitudes but no change in the EC50 values for the agonist, demonstrating that ligand binding site formation is not dependent on the direct or indirect participation of N-linked carbohydrate side chains. Western blots reveal a significant reduction in NR1 (but, interestingly, not NR2) protein expression (Fig. 3, D–H). This reduction is not an artifact caused by our use of a monoclonal anti-NR1 antibody that might not bind to the nonglycosylated form of the receptor because we obtained similar results with a polyclonal anti-NR1 antiserum (data not shown). The reduction in NR1 expression seems to be the likely cause of the marked reduction (>99%) in maximal current amplitudes. The small fraction of residual channel activity is not due to an incomplete block of glycosylation, as indicated by two independent lines of evidence. First, on Western blots of total membranes from tunicamycin-treated oocytes, we do not detect the N-glycosylated receptor, whereas we see a distinct band at a lower molecular weight, which is consistent with the nonglycosylated receptor protein (Fig. 3). Second, the small residual currents detected for the subunit combinations NR1–1a/NR2B and NR1–4a/NR2B in tunicamycin-treated oocytes were not potentiated by ConA, which they should have been if they had somehow escaped tunicamycin inhibition.
The observed general decrease in NMDA receptor function on block ofN-glycosylation is consistent with the reported lack of binding of the glycine site antagonist 5,7-dichlorokynurenate to NR1 subunits expressed in tunicamycin-treated Sf9 insect cells (11). However, Kawamoto et al. (11) observed no concomitant decrease in protein levels. Our Western blot data are also at odds with reports that the number of human embryonic kidney 293 cells expressing nonglycosylated NR1/NR2A receptors on their surface is similar to the number expressing glycosylated receptors (8). However, the immunoblot data shown by these authors indicate a large decrease in the amount of NR1 protein (8). Furthermore, although the same number of cells may be labeled, the expression level in each cell might be reduced. The drastic decrease in functional receptors as measured by a decrease in NMDA receptor-mediated excitotoxic cell death by these authors matches our finding that current amplitudes are reduced to 0.3% of glycosylated controls (Table 1). Based on our observation that EC50 values do not increase in nonglycosylated NMDA receptors but that NR1 expression levels drop precipitously, we conclude that although the channel structure remains intact, the decrease in receptor function is due to the lack of sufficient NR1 subunits.
We do not know whether the observed large reduction in protein expression of NR1 subunits is due to a down-regulation of protein synthesis or increased degradation of nonglycosylated NR1 subunits, but we suspect the latter to be the case because protein synthesis itself clearly is not compromised by tunicamycin treatment, as demonstrated by our data for KA or AMPA receptor expression as well as the fact that NR2 or GluR6 subunits, when coexpressed with NR1 in the same oocyte, are not reduced (Fig. 3, H and C). Such an increase in degradation might be caused by failure of nonglycosylated subunits to assume their correct folding, assemble correctly with other subunits, or be transported correctly to the Golgi apparatus and/or the plasma membrane. Our data do not allow us to distinguish among these possibilities. However, the similarity between the observed overall decrease in NR1 receptor protein in total cell homogenates (Fig. 3E) and the reduction seen in NR1 surface protein (Fig. 3F) rules out that nonglycosylated receptor subunits are merely prevented from reaching the plasma membrane and instead accumulate intracellularly without degradation.
Previous studies on the functional importance ofN-glycosylation for various receptor proteins using tunicamycin as a tool have uncovered a wide variety of effects. In the case of acetylcholine receptors, N-glycosylation was found to be required for correct subunit assembly (32) and for protection from intracellular degradation (33, 34). Similarly, subunit assembly and membrane insertion of Na+ channels were shown to be dependent on proper N-glycosylation (35). On the other hand, expressions of the two-subunit protein Na+/K+-ATPase (36), human erythrocyte anion transporter (37), m2 muscarinic acetylcholine receptor (38), and voltage-gated potassium channel RCK1 (7, 12) were found to be independent of N-glycosylation.
This multitude of seemingly contradictory observations indicates thatN-glycosylation has distinct effects on specific receptor systems and that no general predictions can be made for the functional consequences (if any) of a lack of N-glycosylation. As our data show, this multifaceted significance of N-glycosylation is true not only for membrane proteins in general but also within the closely related family of iGluRs, in which theN-glycosylation state can have profoundly different functional consequences, depending on the subunit composition. This mechanism adds one more tool to the modulatory arsenal of neuronal cells.
ConA-mediated current potentiation.
We cannot confirm the conclusion by Partin et al. (20, 22) that ConA potentiates AMPA receptors only weakly (<2-fold). Their conclusion was based solely on the examination of KA-evoked currents. With KA as the agonist, we also failed to see significant ConA-mediated potentiation, except for GluR1flop. When Glu is used as the agonist, a significant potentiation is revealed, ranging from 5.7- to 13-fold (Table 1). ConA did not differentially potentiate either one of the AMPA receptor splice variants, regardless whether Glu or KA was used as the agonist. Interestingly, GluR2 could not be potentiated, a feature we showed to be due to its particular arrangement ofN-glycosylation sites (Fig. 6). However, this property is restricted to homomeric GluR2 receptors, and it is thus questionable whether it could have any physiological significance.
KA receptors in our study showed huge ConA-mediated potentiation (ltequ]6000-fold). The considerably smaller potentiation factors reported by Partin et al. (20) and Yue et al.(19) most likely stem from shorter incubation periods (2–3 min) and the lower concentrations of ConA used in those studies. Unlike Partinet al. (20), we found that ConA-mediated potentiation of homomeric GluR6 as well as heteromeric combinations of GluR6 with KA1 or KA2 depends on the editing status of the receptor. The highly potentiated Q variant and the modestly potentiated R variant differ merely by one amino acid located in the middle of the ion channel. Because ConA acts by inhibiting desensitization, this finding implies that desensitization may not simply be determined by a certain agonist-evoked conformational change of the extracellular ligand binding site but rather may reflect a complex interaction of that site with the ion pore itself.
Before our study, NMDA receptor potentiation by ConA had been examined for only one splice variant of NR1 (NR1–1a) and for one heteromeric subunit combination (NR1–1a/NR2A), and no potentiation had been observed in either case (19). However, ConA treatment in the study by Yue et al. (1995) was carried out for only 2 min, which might have prevented full potentiation to be achieved. In the same study, succinyl-ConA was shown to cause a 2-fold potentiation of NR1–1a, presumably because the smaller, dimeric succinyl-ConA has a faster time course of action than the tetrameric ConA. In keeping with this interpretation, NMDA receptor currents expressed from total rat brain RNA have been shown to be potentiated by a very long (30 min) treatment with ConA (18). The data presented in our study demonstrate that all NMDA receptors made up of homomeric NR1 subunits of the “a” splice type can be modestly potentiated by ConA, and the same is true for certain heteromeric receptors containing NR1 “a” splice variants. Thus, the amino-terminal exon of 21 amino acids that is located between potential N-glycosylation sites 1 and 2 (see Fig. 1) and is absent from “a” splice variants but present in “b” splice variants seems to control the ConA effect.
Plant lectins such as ConA do not occur in the mammalian central nervous system. Given the modulatory power of heterologous lectins, especially when acting on KA receptors, it is tempting to speculate that endogenous mammalian homologs of ConA might exist that could serve as highly potent and subunit- and even splice variant-specific modulators of GluR function under physiological conditions.
Acknowledgments
We would like to thank Dr. Robert Wenthold (Laboratory of Neurochemisrty, National Institute on Deafness and Other Communication Disorders, Bethesda, MD) for his kind gift of affinity-purified anti-GluR1 and anti-GluR6 antibodies, Dr. Nils Brose (Dept. of Molecular Neurobiology, Max-Planck-Institute) for the monoclonal anti-NR1 antibody, and Dr. Peter Seeburg (Dept. of Molecular Neuroendocrinology, ZMBH, University of Heidelberg, Germany) for the GluR5(Q) cDNA clone.
Footnotes
- Received April 7, 1997.
- Accepted July 16, 1997.
-
Send reprint requests to: Dr. Michael Hollmann, Glutamate Receptor Laboratory, Max-Planck-Institute for Experimental Medicine, Hermann-Rein-Str. 3, D-37075 Göttingen, Germany. E-mail:hollman{at}mail.mpiem.gwdg.de
-
This work was supported by Grant SFB 406 and a Heisenberg Fellowship of the Deutsche Forschungsgemeinschaft (M.H.).
Abbreviations
- iGluR
- inotropic glutamate receptor
- GluR
- glutamate receptor
- DMSO
- dimethylsulfoxide
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- Glu
- glutamate
- SDS
- sodium dodecyl sulfate
- KA
- kainate, NMDA
- N-methyl-d-aspartate
- ConA, concanavalin A
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}