


Abstract
A role for phosphoinositides in the endocytosis of muscarinic cholinergic receptors (mAChRs) has been investigated via inhibition of the activity of phosphatidylinositol-4-kinase (PI4K). Pretreatment of SH-SY5Y neuroblastoma cells with micromolar concentrations of wortmannin (WT), LY-294002, or phenylarsine oxide (PAO), three chemically distinct agents known to inhibit PI4K, resulted in both an inhibition of agonist-induced endocytosis of mAChRs and a selective reduction in the 32P-labeling of phosphatidylinositol-4-phosphate. PAO-mediated inhibition of both receptor endocytosis and phosphoinositide synthesis could be fully reversed by inclusion of the bifunctional thiol 2,3-dimercaptopropanol. The requirement for phosphoinositide synthesis in mAChR endocytosis was independent of a role for these lipids in the maintenance of the cytoskeleton because disruption of the latter with cytochalasin D, ML-7, or colchicine failed to inhibit receptor internalization. Determination of PI4K activity in subcellular fractions of SH-SY5Y cells indicated that enzyme activity in fractions enriched in endocytic vesicles and cytosol was preferentially inhibited by WT, LY-294002, and PAO, a profile consistent with the subcellular distribution of the 110-kDa β isoform of PI4K, as determined by Western blot analysis. Activity of PI4Kβ present in immunoprecipitated cell lysates was inhibited >75% by inclusion of each of the three inhibitors. These results indicate that ongoing synthesis of phosphoinositides is necessary for mAChR endocytosis and that the activity of a WT-sensitive form of PI4K, such as PI4Kβ, is required.
A common adaptive response to the continuous occupancy of many GPCRs is the endocytosis of receptors present at the cell surface into an endosomal compartment that is inaccessible to hydrophilic ligands. The endocytosis of GPCRs may serve to either limit the duration of receptor activation (Sorensen et al., 1997) or provide a mechanism by which the receptor can be resensitized (Pippig et al., 1995). Although the endocytosis of GPCRs has frequently been documented, only recently have details of the underlying mechanism become available. In this context, evidence has been obtained to suggest that some GPCRs undergo endocytosis via a clathrin-coated pit mechanism (Chuang et al., 1986; Von Zastrow and Kobilka, 1992; Slowiejko et al., 1996; Tolbert and Lameh, 1996), in a process that may involve arrestins (Goodman et al., 1997) and the GTPase dynamin (Zhang et al., 1996). However, the molecular events that trigger receptor endocytosis remain to be defined. Although the possibility has been considered that second messenger production constitutes the initiating event, this seems unlikely for the following reasons: first, mutations of β2-adrenergic, mAChR and angiotensin receptors have permitted a dissociation of receptor-effector coupling events and endocytosis (Cheung et al., 1990; Campbell et al., 1991; Hunyady et al., 1994; Moro et al., 1994); second, the endocytosis of mAChRs can be monitored in permeabilized SH-SY5Y neuroblastoma cells under conditions in which second messenger production is prevented (Slowiejko et al., 1994); third, an antibody-bound nonsignaling form of the thyrotropin-releasing hormone receptor undergoes endocytosis at the same rate as the agonist-bound Gq-coupled receptor (Petrou et al., 1993); and fourth, the endocytosis of cholecystokinin and 5-hydroxytryptamine2Areceptors in response to antagonist addition has been reported (Berryet al., 1996; Roettger et al., 1997).
Factors necessary for the maintenance of receptor endocytosis also remain to be established. In this context, the polyphosphoinositides have recently been demonstrated to play an essential role in membrane trafficking events that is distinct from their classic role as precursor molecules for the generation of second messengers. For example, the involvement of 3-phosphoinositides in membrane trafficking from Golgi to lysosomes has been proposed (for reviews, see De Camilliet al., 1996; Martin, 1997). A direct role for the quantitatively major polyphosphoinositides PIP and PIP2 in membrane trafficking was first proposed by Eberhard et al. (1990), who observed that the ATP requirement for Ca2+-regulated exocytosis in adrenal chromaffin cells could be attributed to the need to generate PIP2. Subsequently, three cytosolic proteins (PEPs) were shown to be required for exocytosis. PEP1 and PEP3 have been identified as PIP 5-kinase and the PI-transfer protein, respectively (Martin, 1997). Recently, Wiedemann et al. (1996) demonstrated that inhibition of PIP synthesis by PAO prevents catecholamine release from chromaffin cells, a result which suggests PI4K may represent PEP2. Thus, for catecholamine exocytosis to occur, a series of reactions culminating in the synthesis of PIP and PIP2 is required.
In the current study, we investigated, by pharmacological and biochemical means, the possibility that inositol lipids may also play a key role in the endocytosis of GPCRs. The cell system chosen to address this question was the SH-SY5Y human neuroblastoma; these cells possess a high density of mAChRs (of the m3 subtype) that couple to phosphoinositide-specific PLC and are internalized via a clathrin-coated pit mechanism (Slowiejko et al., 1996). Modulation of phosphoinositide synthesis in these cells has been accomplished via the inhibition of PI4K activity. The rationale behind this approach was 2-fold. First, we have previously demonstrated that PI4K activity is enriched in endosomal fractions that contain endocytosed mAChRs (Sorensen et al., 1997). Second, recent studies have identified two isoforms of PI4K [110 kDa (β) and 230 kDa] that are readily susceptible to inhibition by either WT or LY-294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one] at concentrations in excess of those required for inhibition of PI3K (Downing et al., 1996; Nakagawa et al., 1996a,1996b; Balla et al., 1997; Meyers and Cantley, 1997). The data indicate that when PIP synthesis in SH-SY5Y cells is prevented by inclusion of WT, LY-294002 or PAO, the agonist-induced endocytosis of mAChRs is similarly inhibited. All three chemically distinct inhibitors of PI4K activity preferentially inhibit a form of the enzyme that is present in subcellular fractions enriched in endocytic vesicles and cytosol, a profile of inhibition that parallels the distribution of an immunologically distinct 110-kDa isoform of PI4K, PI4Kβ. The results demonstrate that phosphoinositide synthesis is a prerequisite for the endocytosis of mAChRs and point to the involvement of a WT-sensitive isoform of PI4K, such as PI4Kβ, in receptor internalization.
Experimental Procedures
Materials.
[3H]NMS (87 Ci/mmol), [γ-32P]ATP (6,000 Ci/mmol), [32P]orthophosphoric acid (10 mCi/ml), and detection reagents for enhanced chemiluminescence (ECL kit) were from Amersham (Arlington Heights, IL). [3H]QNB (45.4 Ci/mmol) was obtained from New England Nuclear Research Products (Boston, MA). PI, ATP, atropine, WT, PAO, BAL, and colchicine were from Sigma Chemical (St. Louis, MO). Oxo-M was purchased from Research Biochemicals (Natick, MA). Tissue culture supplies were purchased from Corning Glass Works (Corning, NY) and Sarstedt (Newton, NC). Powdered Dulbecco’s modified Eagle’s medium and fetal calf serum were obtained from GIBCO (Grand Island, NY). Protein A/G-agarose, peroxidase-conjugated anti-rabbit IgG and anti-goat IgG, and polyclonal anti-PI4Kα were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies to PI4Kβ, the p85 subunit of PI3K and monoclonal antibody 4C5G were from Upstate Biotechnology (Lake Placid, NY). LY-294002, cytochalasin D, and ML-7 were obtained from Calbiochem (La Jolla, CA). Human SH-SY5Y neuroblastoma cells were obtained from Dr. June Biedler (Sloan-Kettering Institute, New York, NY). L929-m3 cells (fibroblasts transfected with the cDNA for the m3 mAChR) were obtained from Dr. M. Uhler (University of Michigan, Ann Arbor, MI).
Cell culture conditions.
SH-SY5Y cells (passage 68–78) were grown in tissue culture flasks (75 cm2/250 ml) in 20 ml of Dulbecco’s modified Eagle’s medium supplemented with 10% (v) fetal calf serum. Cells were grown for 7–14 days at 37° in a humidified atmosphere containing 10% CO2. Cells were isolated after aspiration of the medium and incubation with a modified Pucks D1 solution. Cells were then resuspended in buffer A (142 mm NaCl, 5.6 mmKCl, 2.2 mm CaCl2, 3.6 mmNaHCO3, 1 mmMgCl2, 5.6 mmd-glucose, and 30 mm HEPES, pH 7.4). In experiments using attached cells, the cells were subcultured into 35-mm, six-well culture plates (Becton Dickinson Labware, Lincoln Park, NJ) for 2–4 days before treatment. All experiments were performed on cells that had reached confluency.
Subcellular fractionation.
Crude plasma membrane (P1), a membrane fraction enriched in endocytosed mAChRs (V1) and cytosol (S2), was obtained essentially as described previously (Slowiejko et al., 1996). The one exception was that the cells were hypotonically lysed in 5 ml of TE buffer (10 mm Tris·HCl, pH 7.4, 2 mm EDTA) containing 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm Pefabloc, to obtain a more concentrated high speed supernatant fraction (S2: 200,000 × g/90 min). V1 and P1 fractions were resuspended in TE buffer or KGEH (139 mm potassium glutamate, 4 mm MgCl2, 10 mm EGTA, 30 mm HEPES, pH 7.4) at a final protein concentration of 1–2 mg/ml.
Radioligand binding.
Agonist-induced endocytosis of mAChRs was routinely monitored as the appearance of receptors in the V1, as determined by an increase in [3H]QNB binding (Slowiejko et al., 1996; Sorensen et al., 1997). Cells were pretreated with PI4K inhibitors, or vehicle alone, before incubation with 1 mm Oxo-M or buffer alone. After subcellular fractionation, V1 fractions were incubated in TE buffer with 1 nm [3H]QNB at 37° for 90 min. Nonspecific binding was determined as that unaffected by inclusion of 25 μm atropine. Reactions were rapidly terminated by filtration through Whatman GF/B glass-fiber filters, and radioactivity was determined after the addition of 5 ml of Universol scintillation fluid. In some experiments, the agonist-induced sequestration of mAChRs was monitored in intact cells by means of a loss of [3H]NMS binding sites, as described previously (Slowiejko et al., 1994). Oxo-M/[3H]NMS competition studies were performed using the P1 membrane fraction. Membranes were incubated (in buffer A) with [3H]NMS with or without Oxo-M for 90 min at 37°. Reactions were terminated as described above.
32P-Phospholipid labeling.
Attached SH-SY5Y cells were labeled with 5–20 μCi of [32P]orthophosphoric acid in 1.0 ml of buffer A for 2 hr. Cells were washed once with 1.0 ml buffer A and then incubated with PI4K inhibitors or vehicle (with or without agonist) as indicated. Reactions were stopped by aspiration of the treatment solution followed by the addition of 0.5 ml of ice-cold 5% TCA. Cells were scraped from the wells and transferred to glass tubes. The wells were washed with an additional 1 ml of 5% TCA, and the washes were combined. The cells were then left on ice for 1 hr to allow tissue precipitation. TCA pellets were obtained by low speed centrifugation at 4° and washed once with 2 ml of ice-cold distilled water, and the final pellet resuspended in 0.5 ml of water. Lipids were extracted, separated, and quantified as described previously (Sorensen et al., 1997).
SDS-polyacrylamide gel electrophoresis.
Aliquots (25 μg) of whole-cell lysates or subcellular fractions of SH-SY5Y cells were boiled in SDS-polyacrylamide gel electrophoresis sample buffer for 5 min and electrophoresed through 7.5% or 10% SDS-polyacrylamide gels. Proteins were transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA) and processed for immunoblot analysis.
Immunoblot analysis.
Nonspecific binding sites were blocked in phosphate-buffered saline, pH 7.4, containing 0.1% Tween 20 (PBS-T) and 1% bovine serum albumin for 1 hr at room temperature. Primary antibodies were diluted in blocking solution (final concentration, 0.5–1.0 μg/ml) and incubated with the membranes for 1 hr. Excess primary antibody was removed by washing the membranes three times in PBS-T. The blots were then incubated in the appropriate peroxidase-conjugated secondary antibody diluted in PBS-T (1:10,000–15,000) for 1 hr and subsequently washed three additional times in PBS-T. Immunoreactive proteins were detected by ECL. Quantitative analysis of autoluminograms was performed by computer-assisted imaging densitometry (MCID; Imaging Research, St. Catherine’s, Ontario, Canada).
Immunoprecipitation.
After removal of the plating medium, confluent 35-mm cultures of SH-SY5Y cells were washed once with 2 ml of ice-cold PBS. Cells were then incubated on ice and scraped into lysis buffer (200 μl) containing 20 mm HEPES, pH 7.4, 1% Triton X-100, 50 mm NaCl, 1 mm EGTA, 5 mm β-glycerophosphate, 30 mm sodium pyrophosphate, 100 μm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. Cell debris was removed by centrifugation at 12,000 × g for 5 min at 4°, and the protein concentration of the supernatant was determined using a commercially available protein assay kit (Pierce Chemical, Rockford, IL). Aliquots (∼500 μg) of detergent-soluble cellular protein were transferred to tubes containing 2 μg of PI4Kβ antibody (200 μl final volume), and incubated at 4° for 16–20 hr with continuous mixing. Protein A/G-agarose (20 μl) was added for an additional 4 hr with mixing. Immune complexes were pelleted by centrifugation and washed three times with ice-cold MOPS buffer (50 mm MOPS, pH 7.25, 18 mm MgCl2, 1 mmdithiothreitol) and resuspended in the same buffer for assay of PI4K activity.
PI4K activity.
The activity of PI4K was determined by measuring the incorporation of 32P from [32P]ATP into PI. Immunoprecipitates of PI4Kβ, hypotonic cell lysate, P1, V1, or S2 fractions (∼10 μg of protein) were incubated in 50 mm MOPS buffer (pH 7.25) containing 4 mm PI and 0.17% Triton X-100 in the absence or presence of PI4K inhibitors for 1 min before the addition of 10 μCi of 32P-ATP (450 μm final concentration). In some experiments, subcellular fractions (200 μg of protein) were incubated overnight at 4° with 50 μg/ml of the monoclonal antibody 4C5G and PI4K activity monitored in a 25-μg aliquot. Reactions were allowed to proceed in the presence of 0.2% Triton X-100 and 18 mm MgCl2 for 5–10 min at 34° in a final volume of 100 μl, according to the method of MacDonald et al. (1987). Reactions were terminated by the addition of 0.75 ml of chloroform/methanol (1:2), and lipids were extracted in acidified chloroform/methanol, separated by thin layer chromatography, and quantified as described previously (Sorensenet al., 1997).
PI3K activity.
PI3K activity was monitored in p85 antibody immunoprecipitates of SH-SY5Y cells grown in 75-cm2 flasks according to the method of Lavie and Agranoff (1996). Immunoprecipitates were incubated with lipid substrate for 15 min before the addition of either vehicle alone or WT for 1 min. Reactions were then initiated by the addition of [32P]ATP.
Other measurements.
Cells were permeabilized by the addition of 20 μm digitonin, as described previously (Slowiejkoet al., 1994). Protein content was measured with a Pierce BCA protein assay reagent (Rockford, IL).
Data analysis.
Data are expressed as mean ± standard error (or range for two experiments) for the number of separate experiments performed. Student’s two-tailed t tests were used to evaluate the statistical differences between the mean values of paired or unpaired sets of data.
Results
WT or LY-294002 inhibits agonist-induced mAChR endocytosis and phosphoinositide synthesis.
Throughout this study, receptor endocytosis is reported as the agonist-induced appearance of mAChRs, as monitored by an increase in [3H]QNB binding, into a “light” vesicular membrane fraction (V1) obtained by the differential centrifugation of hypotonic lysates of SH-SY5Y cells (Slowiejko et al., 1996). When this paradigm is utilized, V1fractions obtained from quiescent cells contain ∼2–4% of the complement of mAChRs and a similar number of [3H]ouabain binding sites, a plasma membrane marker for Na+/K+-ATPase. Addition of Oxo-M, a muscarinic agonist, results in a 4–5-fold enrichment of mAChRs in the V1 but no increase in [3H]ouabain binding (Sorensen et al., 1997). Preincubation of SH-SY5Y cells with 10 μm WT, a concentration reported to inhibit PI4K (seeDowning et al., 1996; Balla et al., 1997; Meyers and Cantley, 1997), resulted in a >90% inhibition of the agonist-induced translocation of mAChRs into the V1 fraction. However, WT had no effect on mAChR densities in the V1 fractions obtained from control cells incubated in the absence of agonist (see Fig.1A). Preincubation of the cells with 100 μm LY-294002, a structurally unrelated inhibitor of PI4K (Downing et al., 1996), also markedly inhibited agonist-induced mAChR endocytosis but had little or no effect on mAChR densities in the V1 fractions obtained from control cells (Fig. 1A). The IC50 values for WT- and LY-294002-mediated inhibition of mAChR endocytosis were determined to be 300 nm and 30 μm, respectively (Fig.1B). The agonist-induced internalization of mAChRs involves two kinetically distinct phases. The first is a loss of mAChRs from the cell surface (sequestration), which is then followed by a redistribution of these receptors to intracellular sites (endocytosis). In two separate experiments, the ability of WT to inhibit mAChR sequestration (i.e., the agonist-induced loss of cell surface mAChRs, as monitored by a reduction in [3H]NMS binding) in SH-SY5Y cells and L929-m3 fibroblasts was monitored. The addition of Oxo-M resulted in a 39–54% reduction in the number of cell surface mAChRs in both cell lines, losses that were significantly attenuated by pretreatment with WT (Table1).
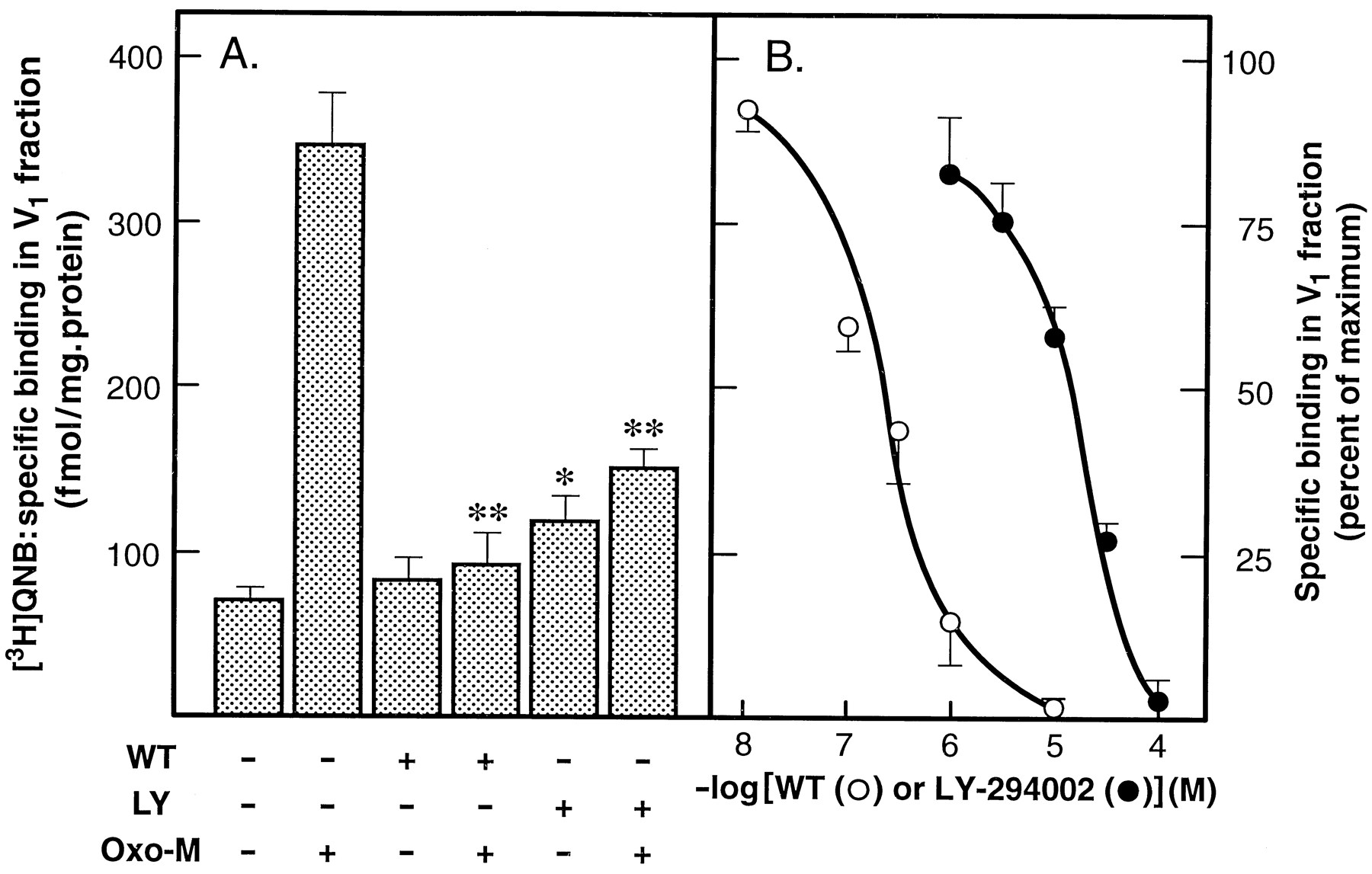
WT or LY-294002 inhibits agonist-induced mAChR endocytosis. A, SH-SY5Y cells were pretreated with vehicle alone, 10 μm WT, or 100 μm LY-294002 for 15 min before the addition of 1 mm Oxo-M or buffer alone for 30 min as indicated. Reactions were terminated by the addition of ice-cold buffer A, and cells were hypotonically lysed before subcellular fractionation as described in the text. mAChRs present in the V1 fractions were then monitored by means of [3H]QNB binding. Values are expressed as the specific binding of [3H]QNB. Results shown are mean ± standard error for five to eight separate experiments. ∗, Different from vehicle alone, p <0.05. ∗∗, Different from Oxo-M alone, p < 0.001. B, Cells were pretreated with WT (○) or LY-294002 (•) at the concentrations indicated for 15 min before the addition of Oxo-M. mAChRs present in the V1were monitored as described in A. Values are expressed as the specific binding of [3H]QNB (percent of maximum; mean ± standard error, three experiments).
WT inhibits the agonist-induced loss of cell surface mAChRs in SH-SY5Y neuroblastoma and L929-m3 fibroblast cells
Exposure of intact SH-SY5Y cells to either WT (10 μm) or LY-294002 (100 μm) resulted in a selective reduction in the 32P-labeling of PIP (80 ± 2% and 60 ± 4%, respectively, three or four experiments), whereas the labeling of the other lipids, such as PA and PIP2, was either unaltered or slightly increased (Fig. 2A). The addition of Oxo-M alone to SH-SY5Y cells resulted in a 50–60% reduction in both PIP and PIP2 labeling due to PLC activation. In the presence of either WT or LY-294002, the32P-labeling of these lipids was further reduced (Fig. 2B). The concentration of WT required for 50% inhibition of [32P]PIP labeling under basal conditions was ∼1 μm (Fig. 3), a value similar to that obtained for inhibition of receptor endocytosis (300 nm; Fig. 1B). In contrast, a much lower concentration of WT (10 nm) was required for the complete inhibition of PI3K activity (Fig. 3). The latter concentration of WT had no effect on either mAChR endocytosis or [32P]PIP labeling.

WT or LY-294002 inhibits the synthesis of phosphoinositides. Cells were prelabeled with32Pi for 2 hr and then incubated for 10–15 min with vehicle (0.2% DMSO), 10 μm WT, or 100 μm LY-294002. A, Reactions were terminated by the addition of TCA. B, Cells were incubated with either vehicle or 1 mm Oxo-M for an additional 10 min before the addition of TCA. Lipids were extracted, separated, and quantified as described in the text. Values are expressed as lipid labeling relative to control cells (vehicle alone). The results shown are the mean ± standard error for three or four separate experiments. ∗, Different from control (−Oxo-M), p <0.05. ∗∗, Different from control (−Oxo-M), p <0.05; ∗∗∗, different from Oxo-M alone, p < 0.05.

WT-mediated inhibition of 32P-PIP labeling can be dissociated from inhibition of PI3K activity. Cells were prelabeled with 32Pi for 2 hr before the addition of either vehicle alone or WT (•) for 15 min. Reactions were terminated by the addition of TCA, and [32P]PIP was isolated and quantified as outlined in the text. For determination of PI3K activity, p85 immunoprecipitates of cell lysates were treated with vehicle alone or WT at the concentrations indicated (○) before determination of enzyme activity as described in the text. Values are expressed as percent of maximum inhibition (∼80% for PIP labeling and 99% for PI3K). The results shown are mean ± standard error for three experiments (or range for two experiments).
As a consequence of their ability to inhibit the synthesis of PIP and PIP2, both WT and LY-294002 also prevent the sustained production of phosphoinositide-derived second messengers (Nakanishi et al., 1995; Linseman et al., 1998). To address the possibility that the inhibitory effects of WT on mAChR internalization were secondary to its inhibition of second messenger production, mAChR endocytosis was monitored in a digitonin-permeabilized cell preparation under conditions in which PLC activity is essentially abolished ([Ca2+]<10 nm, see Fisher et al., 1989). mAChR endocytosis was readily monitored in permeabilized SH-SY5Y cells, and inclusion of 10 μm WT inhibited receptor internalization by >75% (Fig. 4). These results are consistent with our previous observation that second messenger production is not a prerequisite for mAChR sequestration in these cells (Slowiejko et al., 1994). Furthermore, the data indicate that the effects of WT on mAChR trafficking can be dissociated from its ability to inhibit sustained PLC activity.

Inhibition of agonist-induced mAChR endocytosis by WT persists in permeabilized SH-SY5Y cells. Cells were permeabilized by the addition of digitonin and preincubated in KGEH buffer ([Ca2+] <10 nm) for 15 min in the absence or presence of 10 μm WT before the addition of 1 mm Oxo-M or KGEH buffer for an additional 30 min. Cells were then washed with KGEH, hypotonically lysed, and fractionated as described in the text. Values are expressed as specific binding of [3H]QNB (mean ± standard error, three experiments). ∗, Different from vehicle (−Oxo-M), p < 0.01, ∗∗, Different from Oxo-M (−WT), p < 0.02. In parallel experiments, mAChR-stimulated phosphoinositide hydrolysis in permeabilized cells, as monitored by the release of [3H]inositol phosphates (Slowiejko et al., 1994), was <6% of that observed for intact cells.
Receptor endocytosis occurs independently of the cytoskeleton.
Phosphoinositides have been shown to be required for maintenance of the actin cytoskeleton (Chong et al., 1994; Hartwig et al., 1995). Therefore, it is conceivable that the depletion of PIP, and subsequently PIP2, could indirectly attenuate receptor endocytosis via a disruption of the cytoskeleton. However, preincubation of SH-SY5Y cells with either 1 μmcytochalasin D (a treatment that disrupts actin microfilaments and, as a consequence, the activation of focal adhesion kinase in these cells; see Linseman et al., 1998), 10 μm ML-7 (an inhibitor of myosin light chain kinase, a key enzyme involved in the regulation of actin/myosin interactions and cell contractility), or 10 μm colchicine (which disrupts microtubules) had no significant effect on mAChR translocation (Fig.5). These results suggest that an intact cytoskeleton is not required for mAChR endocytosis.

Disruption of the actin cytoskeleton does not impair receptor endocytosis. Cells were pretreated with either buffer A, 1 μm cytochalasin D (Cyt D), 10 μm ML-7, or 10 μm colchicine (Colch) for 30 min at 37° before the addition of 1 mm Oxo-M for an additional 30 min. Reactions were terminated, and receptor endocytosis was monitored as the appearance of mAChRs in the V1 fraction as described in the legend to Fig. 1. Results are expressed as the specific binding of [3H]QNB in the V1 fractions (mean ± standard error, three experiments).
PAO-mediated inhibition of mAChR endocytosis and phosphoinositide synthesis is reversed by BAL.
To further probe the relationship between inhibition of phosphoinositide synthesis and attenuation of mAChR endocytosis, cells were preincubated with PAO, an inhibitor of PIP synthesis (Wiedemann et al., 1996). When control SH-SY5Y cells were pretreated with 20 μm PAO for 15 min, an increased accumulation of mAChRs in the V1fraction was observed. However, pretreatment of cells with PAO also resulted in a marked attenuation of the Oxo-M-mediated increase in mAChR density in the V1 fraction. Thus, in control cells treated with Oxo-M, a net increase in mAChR density of 300 fmol/mg of protein was observed, whereas in PAO-pretreated cells, this value declined to 100 fmol/mg of protein (Fig.6, A and B). Inclusion of 2.5 mm BAL, a bifunctional thiol, also resulted in a small increase in mAChR density in the V1 under basal conditions but had no effect on the agonist-induced translocation of mAChRs (330 fmol/mg of protein). When SH-SY5Y cells that had been pretreated with PAO were washed free of the arsenical and incubated with 2.5 mm BAL (which forms a stable ring structure with the arsenical moiety of PAO), the inhibitory effect of PAO was fully reversed (Fig. 6B). Similar results were obtained with a lower concentration of BAL (250 μm). In contrast, inclusion of 250 μm β-mercaptoethanol, a monofunctional thiol, did not reverse the effects of PAO and the addition of a higher concentration of β-mercaptoethanol (2.5 mm) resulted in only a partial (∼50%) reversal of inhibition. Two additional series of experiments were conducted to test the specificity of action of PAO. First, although PAO can act as a protein tyrosine phosphatase inhibitor (Singh and Aggarwal, 1995), involvement of this class of enzyme in receptor internalization can be discounted because the addition of 100 μm sodium orthovanadate, an agent that also inhibits the phosphatase, had no effect on mAChR endocytosis. Second, because the addition of PAO (and BAL) increased mAChR densities in V1 fractions obtained from quiescent cells, we tested the possibility that these agents were able to induce receptor internalization. However, when receptor internalization was monitored as a loss of [3H]NMS binding sites from intact cells, no reduction in the number of cell surface mAChRs was observed in the presence of either PAO or BAL. The addition of PAO blocked the agonist-induced loss of mAChRs by >80%, an effect that was again readily reversible by BAL. It thus seems possible that altered fractionation properties of SH-SY5Y cells pretreated with PAO or BAL account for the increased recovery of mAChRs in V1 fractions.

PAO inhibits agonist-induced mAChR endocytosis and is readily reversed by BAL. Cells were first incubated with either vehicle alone (0.1% DMSO) or 20 μm PAO for 15 min and washed once with buffer A. Vehicle- and PAO-pretreated cells were then incubated with vehicle alone (1.0% DMSO) or 2.5 mm BAL for 15 min before the addition of vehicle or Oxo-M. Incubations were allowed to proceed for an additional 30 min, followed by quantification of mAChR endocytosis as described in the legend to Fig. 1. A, Specific binding of [3H]QNB in V1 fractions obtained under each condition. B, Receptor endocytosis is expressed as the net agonist-induced increase in the specific binding of [3H]QNB in the V1 fractions (specific binding of [3H]QNB in agonist-treated cells minus that obtained in absence of agonist). Results shown are the mean ± standard error for three separate experiments. ∗, Different from vehicle (−Oxo-M), p < 0.05; ∗∗, different from vehicle (+Oxo-M), p < 0.05; ∗∗∗, different from vehicle alone, p < 0.05.
The addition of PAO also resulted in a marked reduction in the32P-labeling of PIP in intact cells, whereas little or no effect on the 32P-labeling of either PIP2 or PA was observed. Although BAL, when added alone, had no effect on PIP labeling, its inclusion fully reversed the inhibitory effect of PAO on [32P]PIP labeling (Fig. 7).

The inhibition of phosphoinositide synthesis by PAO can be fully reversed by BAL. 32Pi-prelabeled cells were treated with 20 μm PAO and/or 2.5 mm BAL as described in the legend to Fig. 6. Reactions were stopped by the addition of TCA, and lipids were extracted and quantified as described in the text. Values are expressed as (percentage) lipid labeling relative to control cells (vehicle alone). The results shown are the mean ± standard error for three separate experiments. ∗, Different from control,p < 0.05.
WT, LY-294002 or PAO do not significantly modulate agonist binding to mAChRs.
To address the possibility that the inhibitory effects of WT, LY-294002, or PAO on mAChR endocytosis were secondary to a disruption of agonist binding to the mAChR, the ability of these agents to interfere with Oxo-M binding to a membrane preparation of SH-SY5Y cells was evaluated. In competition studies, Oxo-M was able to fully displace [3H]NMS bound to the mAChR in the absence or presence of inhibitors. Although the inclusion of 10 μm WT had little or no effect on the [3H]NMS/Oxo-M competition curve, the presence of 100 μm LY-294002 resulted in a small rightward shift (IC50 values = 125 and 240 μmfor control and LY-294002, respectively). Conversely, inclusion of 20 μm PAO resulted in a leftward shift in the agonist competition curve (IC50 value = 37 μm), indicating that an increase in agonist affinity occurs in the presence of the arsenical (Fig.8). Because the concentration of Oxo-M used to induce endocytosis (1 mm) is 20–50-fold higher than the EC50 value (Sorensen et al., 1997), the impact of a 2–3-fold increase/decrease in agonist affinity on receptor occupancy is expected to be minimal. Thus, the inhibitory effects of WT, LY-294002 and PAO on mAChR endocytosis occur at a step that is distal to the initial ligand/receptor interaction.
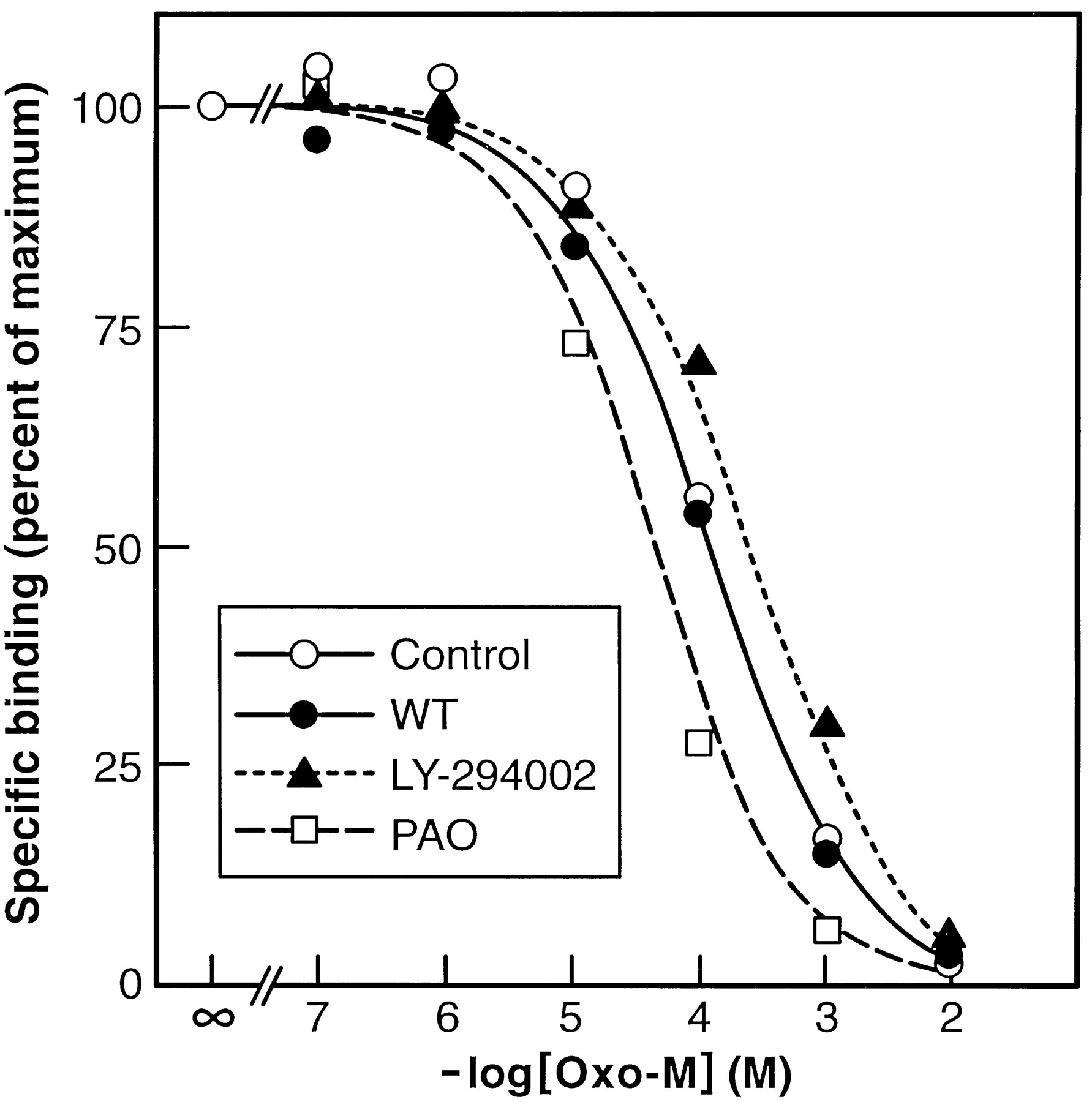
Competition for specific [3H]NMS binding sites by Oxo-M in the absence or presence of WT, LY-294002, or PAO. SH-SY5Y cells were hypotonically lysed, and a P1membrane fraction was isolated. Membranes were then resuspended in buffer A and incubated for 90 min at 37° with 1 nm[3H]NMS (Kd = 0.6 nm) in the presence of increasing concentrations of Oxo-M. In some experiments, WT (10 μm), LY-294002 (100 μm), or PAO (20 μm) was present during the incubations. For the sake of clarity, a single line is drawn for [3H]NMS displacement in control and WT-containing incubations. Results shown are mean of six (control) or three (WT/LY-294002/PAO) replicates from one experiment. In a second experiment performed under the same conditions, the IC50values for control, WT, LY-294002, and PAO incubations were 99, 132, 275, and 29 μm, respectively. The binding of [3H]NMS to the membrane preparations in the presence of WT, LY-294002 or PAO alone was 113 ± 7%, 73 ± 4%, and 108 ± 0% of control, respectively (mean ± range, two experiments).
WT-, LY-294002-, and PAO-sensitive PI4K activity is localized predominantly to vesicular and cytosolic fractions.
We previously demonstrated that a major fraction of PI4K activity present in SH-SY5Y cells is recovered from the crude plasma membrane (P1) fraction, whereas on the basis of specific activity, the enzyme is most enriched in the V1fraction (Sorensen et al., 1997). To determine whether WT, LY-294002, or PAO preferentially inhibited PI4K activity in a specific subcellular fraction, two series of experiments were performed. In the first, cells were pretreated for 15 min with either buffer (control), 10 μm WT, 100 μm LY-294002 or 20 μm PAO; washed free of inhibitor; and then subjected to subcellular fractionation. PI4K activity was then monitored in P1, V1, and S2 (cytosol) fractions obtained from control or inhibitor-pretreated cells. Pretreatment of SH-SY5Y cells with either WT or PAO resulted in a significant loss of PI4K activity from both V1 and S2 fractions, whereas no reduction of enzyme activity was observed for the P1 fraction. In contrast, pretreatment of cells with LY-294002 did not elicit a loss of PI4K activity from any of the subcellular fractions (Table 2). Results from this series of experiments suggest that both WT and PAO induce a persistent inhibition of PI4K activity, whereas that elicited by the addition of LY-294002 is reversible on cell lysis. In the second series of experiments, the ability of WT, LY-294002, or PAO to inhibit PI4K activity when added directly to the subcellular fractions was evaluated. All three inhibitors were found to preferentially inhibit enzyme activity in the V1 and S2 fractions, whereas little or no inhibition of PI4K activity present in the P1 fraction was observed (Table 2). Unexpectedly, the addition of PAO at a 10-fold higher concentration (200 μm) than that added to intact cells was required for inhibition of PI4K activity, a result consistent with a previous study of the effects of PAO on PI4K activity in cell lysates (Wiedemann et al., 1996). One possible explanation for this anomalous result is that PAO is concentrated by SH-SY5Y cells such that the intracellular concentrations of the inhibitor significantly exceed those outside the cell, as has been recently demonstrated for PAO added to chromaffin cells (Wiedemann et al., 1996).
Inhibition of PI4K activity in subcellular fractions of SH-SY5Y cells by WT, LY-294002, PAO, or the monoclonal antibody 4C5G
The ability of WT to inhibit PI4K activity present in the V1 fraction was further examined in a series of dose-inhibition studies (Fig. 9). Maximum inhibition (∼50%) was observed at a concentration of WT of <3 μm. In contrast, an almost complete inhibition of PI4K activity present in the S2 fraction could be observed. The concentrations of WT necessary for 50% of maximum inhibition were ∼200–300 nm for the S2 and V1 fractions, values similar to those observed for inhibition of the endocytosis of mAChRs (Fig. 1B) but distinctly different from those required for inhibition of PI3K (Fig. 9).

WT inhibition of PI4K activity in V1and S2 fractions. SH-SY5Y cells were hypotonically lysed, and subcellular fractions were isolated. PI4K activity present in V1 (•) and S2 (○) fractions was monitored in the presence of WT at the concentrations indicated. Values are expressed as PI4K activity (percent of control) and are from one of three experiments that gave similar results. Dashed line, the dose-inhibition curve for PI3K activity in immunoprecipitates of whole cell lysates (data taken from Fig. 3).
When subcellular fractions of SH-SY5Y cells were preincubated with 4C5G, an inhibitory monoclonal antibody specific for the 56- and 97-kDa, WT-insensitive isoforms of PI4K (Endemann et al., 1991; Wong and Cantley, 1994), a profile of inhibition distinct from that observed for WT, PAO or LY-294002 was obtained. In this case, only pretreatment of the P1 and V1 fractions with 4C5G resulted in a significant inhibition of enzyme activity (Table 2).
Subcellular distribution of isoforms of PI4K.
Four isoforms of PI4K have been identified, two of which [56- and 97-kDa (PI4Kα)] are inhibited by 4C5G and are WT-insensitive, whereas two additional isoforms are WT-sensitive (Endemann et al., 1991; Wong and Cantley, 1994; Downing et al., 1996; Nakagawa et al., 1996b; Balla et al., 1997;Meyers and Cantley, 1997). The latter consist of a 110-kDa form (PI4Kβ) and a 230-kDa form of the enzyme. Immunoblot analysis of either lysates or subcellular fractions with an antibody raised to the carboxyl terminus of PI4Kα (which also recognizes the 230-kDa form of PI4K, a splice variant of PI4Kα) did not indicate the presence of the 230-kDa isoform in SH-SY5Y cells, even though the same isoform was readily detectable in rat brain lysates (data not shown). In contrast, PI4Kβ was readily detected in SH-SY5Y cells and was most prevalent in the V1 and S2fractions, whereas relatively little immunoreactivity associated with PI4Kβ was observed in the P1 fraction (Fig.10).

Western blot analysis of PI4Kβ in subcellular fractions of SH-SY5Y cells. SH-SY5Y cells were hypotonically lysed and subcellular fractions were isolated. Equivalent aliquots (25 μg of protein) of whole-cell lysates or subcellular fractions were electrophoresed through 10% SDS-polyacrylamide gels and transferred to PVDF membranes. Membranes were then immunoblotted for PI4Kβ as described in the text. Top, Western blots representative of the results obtained in three separate experiments. Bottom, Densitometric analysis of the Western blots. Values are expressed as the density in each fraction relative to lysate and results shown are the mean ± standard error for three separate experiments.
Inhibition of PI4Kβ activity present in immunoprecipitates.
If PI4Kβ plays a role in receptor endocytosis, then it should be inhibited not only by WT but also by LY-294002 and PAO. To address this issue, PI4Kβ was immunoprecipitated from hypotonic lysates of SH-SY5Y cells and enzyme activity monitored in the absence or presence of 10 μm WT, 100 μm LY-294002, or 200 μm PAO. The addition of either of these three inhibitors resulted in a marked (>75%) inhibition of PI4Kβ activity (Fig.11). Dose-inhibition studies indicated an IC50 value of ∼200 nm for WT-mediated inhibition of PI4Kβ (see Fig. 11, inset).
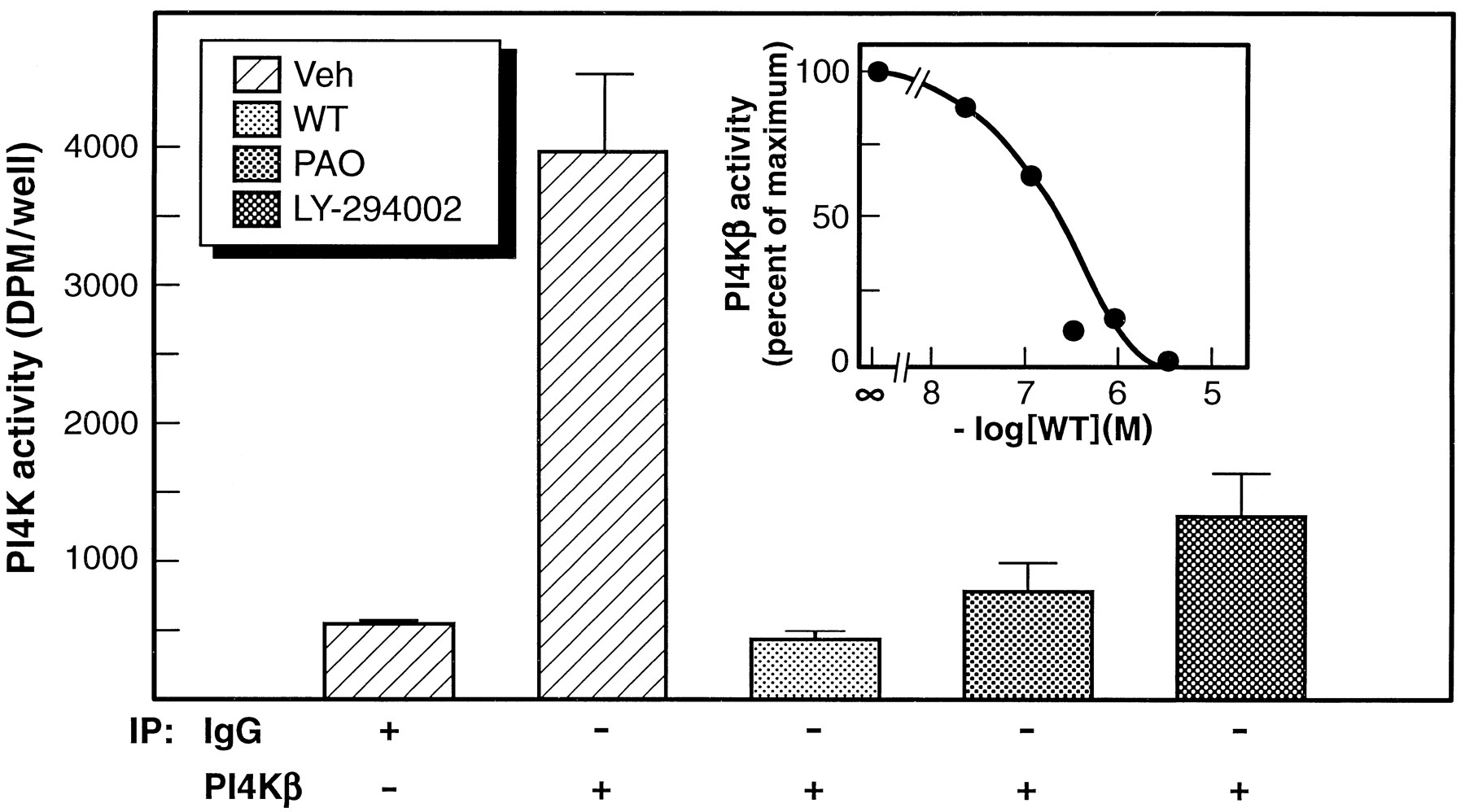
WT, LY-294002, and PAO inhibit PI4K activity in immunoprecipitates of PI4Kβ. Lysates from individual 35-mm cultures of SH-SY5Y cells were immunoprecipitated with control IgG or anti-PI4Kβ as outlined in the text. PI4K activity in the immunoprecipitates was then monitored in the presence of vehicle alone, 10 μm WT, 200 μm PAO, or 100 μm LY-294002. Results shown are the mean ± standard error for three replicates obtained from one experiment. Similar results were obtained in a second experiment. Inset, WT dose-inhibition of PI4Kβ activity in immunoprecipitates of SH-SY5Y cell lysates. Results from two separate experiments are shown. IC50 value was ∼200 nm.
Discussion
The results of the current study strongly suggest that inositol lipid synthesis is a prerequisite for the endocytosis of mAChRs in SH-SY5Y cells because receptor internalization can be blocked by inclusion of WT, LY-294002, or PAO, three chemically distinct agents that prevent PIP synthesis via inhibition of PI4K activity (Downinget al., 1996; Wiedemann et al., 1996; Meyers and Cantley, 1997). Although each of these agents can be viewed as relatively nonspecific, the ability of all three to inhibit both mAChR endocytosis and phosphoinositide synthesis provides a high degree of stringency in establishing a connection between these two parameters. Two additional lines of evidence point to a link between the endocytosis of mAChRs and PIP synthesis. First, similar concentrations of WT were required for half-maximal inhibition of PIP synthesis, PI4K activity, and receptor internalization (∼300 nm). In contrast, >85% inhibition of PI3K occurred at a WT concentration (10 nm) that did not perturb mAChR endocytosis (Fig. 3). Although the presence of an atypical WT-insensitive form of PI3K cannot be excluded (Domin et al., 1997), the current results indicate that the generation of 3-phosphoinositides is not required for mAChR endocytosis in SH-SY5Y cells. In keeping with this conclusion, a role for PI3K in the postendosomal sorting of the platelet-derived growth factor, but not in its internalization, has been proposed (Jolyet al., 1995). Furthermore, WT does not seem to exert its effect on mAChR internalization via its ability to inhibit myosin light-chain kinase, because a known inhibitor of the latter (ML-7) was without effect on receptor endocytosis (Fig. 5). Second, a complete recovery of mAChR endocytosis in SH-SY5Y neuroblastoma could be obtained when PAO-pretreated cells were incubated in the presence of BAL, conditions under which PIP synthesis is also fully restored (Figs.6 and 7). Although the ability of PAO to inhibit PI4K activity was only recently appreciated (Wiedemann et al., 1996), PAO has previously been observed to block the endocytosis of β2-adrenergic receptors in 1321N1 astrocytoma cells (Hertel et al., 1985) and angiotensin receptors in adrenal glomerulosa cells (Hunyady et al., 1991). These results, together with the data obtained in the current study for mAChR internalization in both SH-SY5Y cells and fibroblasts, suggest that PI4K activity, and hence PIP synthesis, are general requirements for the endocytosis of GPCRs. In quiescent SH-SY5Y cells, the addition of either WT, LY-294002, or PAO results in a selective inhibition of the synthesis of PIP. Presumably in the absence of agonist, there is only a very limited conversion of PIP to PIP2, and the selective reduction of PIP labeling observed in the presence of PI4K inhibitors reflects degradation of PIP by a PIP 4-phosphatase. After mAChR activation, the demands for PIP2resynthesis are increased significantly and PIP2stores become rapidly depleted in the presence of the inhibitors (see Fig. 2B). A conclusion to be drawn from these observations is that although in the current study PI4K activity has been the primary target for inhibition, it may transpire that both PIP and PIP2 play a functional role in the endocytosis of mAChRs.
Significant progress has been made in the identification of distinct isoforms of PI4K. Initial biochemical studies indicated the presence of a membrane-bound 56-kDa enzyme that was inhibited by the monoclonal antibody 4C5G and a large-molecular-weight enzyme (∼200 kDa) that was insensitive to 4C5G (Endemann et al., 1991). More recently, cloning studies have revealed the presence of three distinct isozymes of molecular weight 97, 110, and 230kDa (Wong and Cantley, 1994;Nakagawa et al., 1996b; Meyers and Cantley, 1997). The 97-kDa enzyme (termed PI4Kα) exhibits biochemical characteristics similar to those attributed to the 56-kDa enzyme and is WT-insensitive. In contrast, the 110-kDa (92-kDa in rat: Nakagawa et al., 1996a) isoform (termed PI4Kβ) and 230-kDa isoform of PI4K are both WT-sensitive. Given that both 32P-labeling studies of intact SH-SY5Y cells and direct enzyme assay of PI4K in subcellular fractions demonstrate that WT is an effective inhibitor of PIP synthesis in these cells, two candidates for mediation of the inhibitory effects of WT are PI4Kβ and/or the 230-kDa form of PI4K. Of these, only PI4Kβ is readily demonstrable in SH-SY5Y cells. In this context, of particular significance was the observation that the addition of WT (and LY-249002 and PAO) preferentially inhibited PI4K activity in subcellular fractions enriched in endocytic vesicles and cytosol. The subcellular distribution of PI4Kβ in SH-SY5Y cells, as revealed by Western blot analysis, is entirely consistent with the observed profile of inhibition (see Fig. 10) and is in accord with previous studies that have indicated PI4Kβ to be either loosely membrane associated or cytosolic (Wong et al., 1997). Direct evidence that PI4Kβ is inhibited by WT, LY-294002, and PAO was obtained from experiments in which enzyme activity in immunoprecipitates of SH-SY5Y cell lysates could be inhibited >75% by inclusion of each of the three inhibitors. Furthermore, the IC50 value for WT inhibition of PI4Kβ activity in these immunoprecipitates (200 nm) is in close agreement with that previously obtained for inhibition of enzyme activity in Jurkat cell lysates (140 nm: see Meyers and Cantley, 1997). Although the possibility remains that additional, as yet unidentified, isoforms of PI4K are present in cells (Wong et al., 1997), the current data are consistent with the involvement of a WT-sensitive form of PI4K, such as PI4Kβ, in the regulation of mAChR endocytosis in SH-SY5Y cells.
The mechanism or mechanisms by which phosphoinositides might regulate receptor endocytosis remain to be elucidated. However, the possibility that these lipids modulate endocytosis via downstream effects on the cytoskeleton can be ruled out because disruption of the cytoskeleton with either cytochalasin D or colchicine had no effect on receptor internalization. One possibility that merits attention is that high concentrations of phosphoinositides may be localized to sites of vesicle budding, where, due to their highly negative polar headgroups, they alter membrane curvature and thereby promote membrane budding. Alternatively, the inositol lipids may serve to recruit, activate, or modulate other factors necessary for membrane trafficking. For example, these lipids may serve to recruit key proteins to their appropriate intracellular locations and thereby regulate protein/protein interactions that could facilitate endocytic events (see DeCamilliet al., 1996).
In conclusion, the results of the current study demonstrate that the synthesis of phosphoinositides, in particular that of PIP (and possibly, PIP2), is obligatory for mAChR endocytosis in SH-SY5Y cells. The activity of a WT-sensitive isoform of PI4K, such as PI4Kβ, may represent one of the factors involved in the regulation of the phosphoinositide pool required for the occurrence of receptor endocytosis.
Acknowledgments
We thank Ms. Jo Ann Kelsch and Mr. T. Desmond for preparation of the manuscript and Dr. R. R. Neubig for his helpful suggestions.
Footnotes
-
Send reprint requests to: Dr. Stephen K. Fisher, Neuroscience Laboratory, University of Michigan, 1103 E. Huron St., Ann Arbor, MI 48104-1687. E-mail: skfisher{at}umich.edu
-
This work was supported by National Institutes of Health Grants NS23831 (S.K.F.) and MH46252 (S.K.F., A.M.H.). S.D.S. and D.A.L. were supported by National Institutes of Health Training Grant GM07767.
- Abbreviations:
- GPCR
- G protein-coupled receptor
- Oxo-M
- 2-butyn-1-ammonium,N,N,N-trimethyl-4-(2-oxo-1-pyrrolidinyl)iodide
- mAChR
- muscarinic acetylcholine receptor
- PEP
- priming of exocytosis proteins
- PLC
- phospholipase C
- PI
- phosphatidylinositol
- PIP
- phosphatidylinositol-4-phosphate
- PIP2
- phosphatidylinositol-4,5-bisphosphate
- PA
- phosphatidic acid
- PI4K
- phosphatidylinositol-4-kinase
- PI3K phosphatidylinositol-3-kinase
- PAO, phenylarsine oxide
- TCA
- trichloroacetic acid
- BAL
- 2,3-dimercaptopropanol (British anti-Lewisite)
- QNB
- quinuclidinyl benzilate
- NMS
- N-methylscopolamine
- WT
- wortmannin
- Received December 11, 1997.
- Accepted February 4, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}