


Abstract
Kainate receptors are a subtype of ionotropic glutamate receptors, permeable to cations and thus expected to have an excitatory depolarizing action on neurons. However, kainate receptor activation inhibits γ-aminobutyric acid release in the hippocampus through activation of protein kinase C in a pertussis toxin-dependent manner, suggesting a coupling of kainate receptors to G proteins. Thus, we directly investigated the G protein coupling of kainate receptors in the rat hippocampus by using a selective kainate receptor agonist, [3H](2S,4R)-4-methylglutamate ([3H]MGA). [3H]MGA bound to a single site to hippocampal membranes with a K D value of 32 nM and a B max value of 1024 fmol/mg protein. This binding likely represents kainate receptors because it was displaced by domoate (K i = 4 nM), kainate (K i = 11 nM), and 6-cyano-7-nitroquinoxaline-2,3-dione (K i = 1.4 μM), but not by α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate (K i > 10 μM), (RS)-α-methyl-4-phosphonophenylglycine (K i > 10 μM), or (±)-1-aminocyclopentane-trans-1,3-dicarboxylic acid (K i > 10 μM). Guanylylimidodiphosphate (30 μM), which uncouples all G protein-coupled receptors, shifted to the right the saturation curve of [3H]MGA (K D = 133 nM). This effect was mimicked by pretreatment of hippocampal membranes with modifiers of Gi/Go proteins [30 μMN-ethylmaleimide (K D = 98 nM) or 25 μg/ml pertussis toxin (K D = 95 nM)] but not by a modifier of Gs proteins [50 μg/ml cholera toxin (K D = 32 nM)]. Treatment of solubilized hippocampal membranes with pertussis toxin (25 μg/ml) decreased [3H]MGA affinity (K D = 105–113 nM), which was recovered by reconstitution of these pretreated solubilized hippocampal membranes with Gi/Go proteins (K D = 41–76 nM). These results indicate that hippocampal kainate receptors are coupled to Gi/Go proteins.
In the central nervous system, fast, excitatory transmission is mostly mediated by glutamate acting on postsynaptic ionotropic glutamate receptors of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate subtypes (Hollmann and Heinemann, 1994). A third subtype of ionotropic glutamate receptors, kainate receptors, has been identified by both pharmacological and molecular genetic approaches (Hollmann and Heinemann, 1994; Bettler and Mulle, 1995), but their role is just starting to be revealed. It is still a matter of debate whether kainate receptor activation participates in fast synaptic transmission and/or in presynaptic modulation of neurotransmitter release (Lerma et al., 1997; Malva et al., 1998). One likely role for kainate receptors in the hippocampus is the modulation of GABAergic transmission (Clarke et al., 1997; Rodrı́guez-Moreno et al., 1997; Cossart et al., 1998;Frerking et al., 1998), which is essential for the control and coordination of hippocampal excitability (Buckmaster and Soltesz, 1996) and may be the basis of the profound epileptogenic and cytotoxic effects of kainate in the hippocampus (Coyle, 1983). This effect of kainate on hippocampal GABAergic transmission might be due to a combined postsynaptic modulation of spontaneous firing (Cossart et al., 1998; Frerking et al., 1998) and to a presynaptic inhibition of γ-aminobutyric acid (GABA) release (Cunha et al., 1997;Rodrı́guez-Moreno et al., 1997). This latter effect is paradoxical in view of the expected excitatory depolarizing action of kainate receptors, which are mostly permeable to cations (Bettler and Mulle, 1995). The observation that the mechanism through which kainate receptors inhibit GABA release in the hippocampus is independent of ion channel current and is sensitive to pertussis toxin and to inhibitors of protein kinase C (Rodrı́guez-Moreno and Lerma, 1998) raises the question of whether kainate receptors might also behave as metabotropic receptors.
Taking advantage of the recent introduction of a selective kainate receptor agonist, (2S,4R)-4-methylglutamate (MGA;Gu et al., 1995; Toms et al., 1997; Zhou et al., 1997), we now report that kainate receptors in the hippocampus are coupled to Gi/Go proteins, thus providing a molecular basis for the inhibitory metabotropic action of kainate receptor activation in the hippocampus (Rodriguéz-Moreno et al., 1998).
Materials and Methods
Drugs and Solutions.
[3H]MGA, MGA, (±)-1-aminocyclopentane-trans-1,3-dicarboxylic acid (t-ACPD), and (RS)-α-methyl-4-phosphonophenylglycine (MPPG) were purchased from Tocris Cookson (Bristol, UK). Kainate,N-ethylmaleimide, guanylylimidodiphosphate [Gpp(NH)p], adenylylimidodiphosphate, and cholera toxin were purchased from Sigma Chemical Co. (St. Louis, MO). Domoic acid, AMPA, and 6-cyano-2,3-dihydroxy-7-nitroquinoxaline (CNQX) were obtained from Research Biochemicals Inc. (Natick, MA), and pertussis toxin and G proteins from bovine brain were obtained from Calbiochem (La Jolla, CA).
Preparation of Membranes and Pretreatment with G Protein Modifiers.
Membranes were prepared as described previously (Cunha et al., 1999). Briefly, two to four male Wistar rats (160–180 g) were sacrificed by decapitation after halothane anesthesia. The hippocampi were dissected out at 4°C and homogenized in 10 ml of sucrose solution (0.32 M) containing 50 mM Tris · HCl, 2 mM EGTA, and 1 mM dithiothreitol (pH 7.6). The homogenates were centrifuged at 3000g for 10 min at 4°C, and the supernatants were transferred to new tubes and centrifuged at 20,000g for 20 min at 4°C. The pellets were then resuspended in the incubation solution containing 50 mM Tris · HCl and 0.5 mM MgCl2 (pH 7.4) and maintained at 4°C (incubation buffer). In some experiments, hippocampal synaptosomes were first prepared by sucrose/Percoll isopicnic centrifugations (Cunha et al., 1997), and membranes were prepared after sonication of the synaptosomes.
When the effect of Gpp(NH)p was tested, the membranes were pretreated for 20 min at 37°C with Gpp(NH)p (30 μM) in incubation buffer and then cooled at 4°C and used for binding assays, with Gpp(NH)p (30 μM) being present during the incubation binding reaction. When the effect of N-ethylmaleimide was tested, the membranes were pretreated for 20 min at 37°C with N-ethylmaleimide (30 μM) in incubation buffer. Reactions were stopped by the addition of 2.5 mM dithiothreitol, followed by centrifugation at 20,000gfor 10 min at 4°C. The pelleted N-ethylmaleimide-treated membranes were resuspended in the incubation buffer at 4°C and used for binding assays. Bacterial toxins were activated in 50 mM Tris · HCl (pH 7.4) and 5 mM MgCl2 for 15 min at 37°C just before use. Bacterial toxin-catalyzed G protein modification was basically performed as described previously (Cunha et al., 1999). Membranes were pretreated for 45 min at 37°C with either pertussis toxin (25 μg/ml) or cholera toxin (50 μg/ml) in the following buffer: 50 mM Tris · HCl with 2 mM MgCl2 (pH 7.4), 10 mM thymidine, 1 mM ATP, 0.5 mM GTP, 1 mM NAD, and 10 mM dithiothreitol. The reactions were stopped by the addition of 10 ml of the incubation buffer followed by centrifugation at 20,000g for 10 min at 4°C. The pelleted bacterial toxin-treated membranes were resuspended in the incubation buffer at 4°C and used for binding assays.
Reconstitution of [3H]MGA Binding with Gi/Go Proteins in Pertussis Toxin-Pretreated Membranes.
Hippocampal membranes were prepared as described above and resuspended in a reaction buffer containing 50 mM Tris · HCl with 0.5 mM MgCl2 (pH 7.4), 10 mM thymidine, 1 mM ATP, 0.5 mM GTP, 10 mM dithiothreitol, and 2.5 mM 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate (CHAPS). Knockout of native Gi/Goproteins and later reconstitution with exogenously added Gi/Go proteins of solubilized hippocampal membranes were performed as described previously (Cunha et al., 1999). Basically, pertussis toxin (25 μg/ml), preactivated as described above, and 1 mM NAD+ were then incubated with membranes for 45 min at 37°C. The reaction was terminated by the addition of 10 ml of ice-cold reaction buffer, followed by pelleting of the membranes by centrifugation at 20,000g for 10 min at 4°C. As a control, hippocampal membranes were subject to the same procedure but without the addition of NAD. Gi/Go proteins were resuspended in a solution containing 50 mM HEPES (pH 7.6), 1 mM EDTA, 1 mM dithiothreitol, 20% glycerol, 0.05% phosphatidylcholine, and 12.5 mM 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate and added to membranes (0.980–1.105 mg of protein) at a final concentration of 600 nM. After a 15-min incubation at 4°C, the membranes reconstituted with G proteins were diluted 20-fold with the incubation buffer at 4°C. Saturation curves of [3H]MGA binding were then performed in parallel in pertussis toxin-treated membranes without or with NAD and without or with G proteins.
Binding Assays.
Binding of [3H]MGA was for 90 min at 4°C with 190 to 370 μg of membrane protein in a final volume of 300 μl in the incubation solution containing 50 mM Tris · HCl and 0.5 mM MgCl2 (pH 7.4), essentially as described previously for [3H]kainate (e.g., London and Coyle, 1979) or for [3H]MGA (Toms et al., 1997). Under these conditions, an apparent equilibrium of [3H]MGA binding (30 nM) was reached after 45 min of incubation and binding remained virtually unchanged up to 150 min (data not shown). Specific binding was determined by subtraction of the nonspecific binding that was measured in the presence of 100 μM kainate. The binding reactions were stopped by vacuum filtration through Whatman GF/C glass-fiber filters, followed by washing of the filters and reaction tubes with 8 ml of incubation solution, maintained at 4°C. The filters then were placed in scintillation vials with 5 ml of scintillation liquid (Scintran Cocktail T; Wallac, Turku, Finland). Radioactivity bound to the filters was determined after 12 h with an efficiency of 55 to 60% for 2 min. Saturation curves were performed in triplicate with 10 different [3H]MGA concentrations ranging from 0.3 to 120 nM. Competition curves were performed in duplicate with 30 nM [3H]MGA and eight different concentrations of competitors ranging from 1 nM to 10 μM. The amount of membrane protein was determined according to the methods of Peterson (1977). The data were initially processed in Microsoft Excel software (Microsoft Corp., Redmond, WA) to determine the average specific binding and then fitted by nonlinear regression with the Raphson-Newton method, performed with the GraphPAD Software (San Diego, CA) InPlot software package. When performing [3H]MGA competition experiments, the IC50 values were converted into K i values on nonlinear fitting of the semilogarithmic curves derived from the competition curves, with the K D value for MGA derived from saturation experiments. An F test (P > .05) was used to determine whether the curves were fitted best by one or two independent binding sites. The values are presented as mean ± S.E.M. of n experiments, exceptK D values, which are presented as mean (95% confidence interval).
Results and Discussion
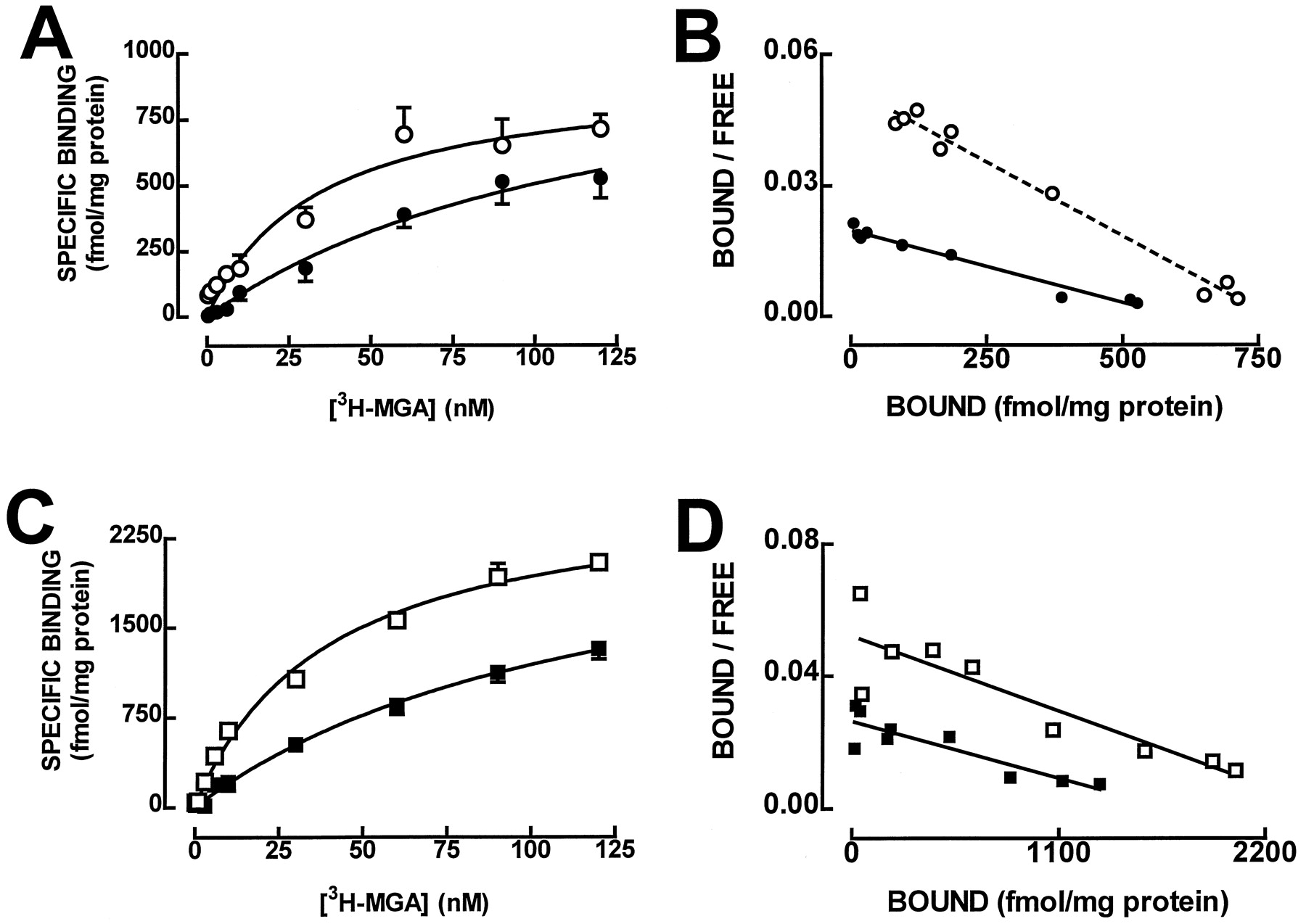
Saturation curves of [3H]MGA binding were performed in hippocampal membranes (Fig.1A). The fitting of the saturation isotherm showed a single binding site for [3H]MGA (F > 0.05 for two binding sites in each individual experiment) with the average binding parameters presented in Table 1. Notably, the apparent affinity of MGA found in the present study is similar to that originally reported (Gu et al., 1995) and is slightly lower than the K D value for [3H]MGA binding found in rabbit membranes (Toms et al., 1997). The inability to detect two affinity binding sites for [3H]MGA (Toms et al., 1997) may be related to the presence of divalent cations in the incubation solution that affects [3H]kainate binding (e.g., Honoréet al., 1986). The density of [3H]MGA-binding sites in hippocampal membranes is similar to that of [3H]kainate-binding sites (London and Coyle, 1979).

Average saturation curves (A and C) and corresponding Scatchard plots (B and D) of [3H]MGA binding to whole membranes (A and B) and to synaptosomal membranes (C and D) of the rat hippocampus in the absence (○, ■) or presence (●, ▪) of guanylylimidodiphosphate (30 μM). The ordinates in A and C represent specific binding of [3H]MGA on subtraction of the nonspecific binding, determined in the presence of 100 μM kainate, from total binding. The curves in A and C were generated from the average binding parameters obtained on fitting by nonlinear regression assuming a single binding site. Results are mean ± S.E.M. of four experiments performed in triplicate. The S.E.M. values are not presented in the Scatchard plot for the sake of simplicity.
Effect of G protein modifiers on the binding parameters of the kainate receptor agonist [3H]MGA to rat hippocampal membranes
We then compared the density of [3H]MGA binding in whole hippocampal membranes with [3H]MGA binding in membranes prepared from hippocampal synaptosomes, because it has been proposed that kainate receptors might be predominantly presynaptically located (Coyle, 1983; Malva et al., 1998). As illustrated in Fig. 1B, [3H]MGA also bound to a single binding site in hippocampal synaptosomal membranes (F > 0.05 for two binding sites in each individual experiment) with a K D value of 40 nM (28–52 nM; n = 4), which was similar (P > .05) to that found in whole hippocampal membranes. However, the density of [3H]MGA binding, measured as B max, was significantly (P < .05) higher in synaptosomal (2692 ± 41 fmol/mg protein; n = 4) than in whole membranes from the hippocampus (1024 ± 39 fmol/mg protein;n = 15). This confirms previous data obtained with [3H]kainate (Foster et al., 1981) and provides a biochemical confirmation of previous neurochemical (for a review, seeMalva et al., 1998), neurotoxic (Represa et al., 1997), and anatomic studies (Foster et al., 1981), emphasizing the predominant presynaptic location of kainate receptors in the hippocampus. This does not exclude the simultaneous presence of functional postsynaptic kainate receptors in the hippocampus (Castillo et al., 1997; Clarke et al., 1997; Lerma et al., 1997; Cossart et al., 1998; Frerking et al., 1998; Vignes et al., 1998).
One of the major characteristics of metabotropic G protein-coupled receptors is the sensitivity of agonist binding to the state of the receptor (i.e., coupled or uncoupled to G proteins), and this equilibrium can be shifted toward uncoupled receptors by increasing the concentration of GTP (Stiles et al., 1984). We now observed that the stable GTP analog Gpp(NH)p (30 μM) shifted to the right the saturation curve of [3H]MGA binding (Fig.1). As presented in Table 1, the effect of Gpp(NH)p was mostly reflected by an increase in K D, with no significant change in B max. In contrast, adenylylimidodiphosphate (30 μM) was devoid of effect on [3H]MGA binding because neither theK D value (35–59 versus 42–48 nM) nor theB max value (921–1089 versus 879–1089 fmol/mg protein) was modified by adenylylimidodiphosphate (30 μM). The shift in K D caused by Gpp(NH)p, quantified by the GTP shift [K D in the presence of Gpp(NH)p/K D in control conditions], which is a measure of the coupling of receptor to G proteins (Stiles et al., 1984), is significantly (P < .05) larger than unity (Table 1). This strongly suggests that kainate receptors in the mammalian brain can be coupled to G proteins, as has previously been suggested for nonmammalian kainate-binding sites, which have a shorter amino acid sequence than their mammalian counterparts and do not exhibit ion channel activity (for a review, see Henley, 1994).
Other glutamate receptors, such as metabotropic glutamate receptors (Hollmann and Heinemann, 1994) or AMPA receptors (Wang et al., 1997), also couple to G proteins. Despite previous evidences that MGA is a selective ligand for kainate versus nonkainate glutamate receptors in different systems (Gu et al., 1995; Toms et al., 1997; Zhou et al., 1997), we decided to exclude the possibility that the observed GTP shift of [3H]MGA binding could be attributed to binding of [3H]MGA to nonkainate glutamate receptors in rat hippocampal membranes. Figure2 shows that two kainate receptor agonists, kainate and domoate, completely displaced [3H]MGA (30 nM) binding withK i values of 11 and 4 nM (n= 3). A non-N-methyl-d-aspartate ionotropic glutamate receptor antagonist, CNQX, also displaced [3H]MGA (30 nM) binding, with aK i value of 1.4 μM (n = 3). In contrast, AMPA was unable to displace [3H]MGA (30 nM) binding within the concentration range tested (n = 3). This profile of displacement of [3H]MGA binding (domoate ≥ kainate ≫ CNQX ≫ AMPA) is fully compatible with binding of [3H]MGA to kainate but not to AMPA receptors in hippocampal membranes (Bettler and Mulle, 1995). Finally, the observation that a selective group I metabotropic glutamate receptor agonist (t-ACPD) and a nonselective group II/group III metabotropic glutamate receptor antagonist (MPPG) were virtually ineffective displacers of [3H]MGA (30 nM) binding (Fig. 2) excludes the measurable contribution of group I, II, and III metabotropic glutamate receptors for [3H]MGA binding to hippocampal membranes.

Inhibition of [3H]MGA (25 nM) binding to hippocampal membranes by the kainate receptor agonists domoate (▪) and kainate (●), by AMPA (▴), by the mixed AMPA/kainate receptors antagonist CNQX (▵), by the group I/II metabotropic agonistt-ACPD (○), and by the group II/III metabotropic antagonist MPPG (■). The ordinates represent specific binding of [3H]MGA obtained on subtraction of the nonspecific binding, determined in the presence of 100 μM kainate, from total binding. The binding of [3H]MGA (30 nM) in the absence of competitors (345 ± 17 fmol/mg protein) corresponds to 100%. The curves were generated from the average binding parameters obtained on fitting by nonlinear regression assuming a single binding site. The Hill slopes were lower than unity (0.69–0.77), indicating that [3H]MGA-binding sites might not be homogeneous. Results are mean ± S.E.M. of three experiments performed in duplicate.
To further investigate whether a particular group of G proteins might be coupled to kainate receptors, we tested the effect of pretreatment of membranes with modifiers of G proteins (Yamane and Fung, 1993) on the binding of [3H]MGA to whole hippocampal membranes. As presented in Table 1, the sulfhydryl alkylating agentN-ethylmaleimide (30 μM), which uncouples Gi and Go proteins by interacting with the carboxyl-terminal cysteine residue found in the α subunit of Gi/Goproteins (Yamane and Fung, 1993), increased theK D value of [3H]MGA binding, with no significant change inB max. Another modifier of Gi/Go proteins, pertussis toxin (25 μg/ml), which ADP-ribosylates a cysteine residue of the α subunit of Gi/Go proteins, blocking their interaction with the receptors (Yamane and Fung, 1993), also increased the K D value of [3H]MGA binding, with no significant change inB max (Table 1). However, a modifier of another group of G proteins, cholera toxin (50 μg/ml), which ADP-ribosylates arginine residues of αs, inhibiting their intrinsic GTPase activity and resulting in continuous activation of αs (Yamane and Fung, 1993), virtually did not affect [3H]MGA-binding parameters (Table 1). These results suggest that Gi/Go proteins are likely candidates to directly or indirectly couple to hippocampal kainate receptors. These results also suggest that the effect of Gpp(NH)p is likely due to its ability to activate G proteins rather than to directly interact with hippocampal kainate receptors, as it has been shown to occur in chick cerebellar kainate-binding protein (Paas et al., 1996). This was further confirmed by the ability ofN-ethylmaleimide to occlude the inhibitory effect of Gpp(NH)p on [3H]MGA binding. Thus, inN-ethylmaleimide (30 μM)-treated membranes, theK D value of [3H]MGA binding was 120 to 139 nM in the absence and 95 to 132 nM in the presence of Gpp(NH)p (30 μM).
To demonstrate the coupling of Gi/Go proteins to kainate receptors, we tested the ability of exogenously added Gi/Go proteins to revert the GTP shift of [3H]MGA binding caused by pertussis toxin. As shown in Fig. 3, in membranes treated with pertussis toxin (25 μg/ml) in the presence of NAD+, there was a decrease in [3H]MGA binding compared with nontreated membranes (i.e., treated with pertussis toxin in the absence of NAD+). This was reflected by a change (P < .05) in K D value from 46 nM (95% confidence interval, 27–89 nM) in nontreated solubilized membranes to a K D value of 108 nM (95% confidence interval, 97–119 nM) in the presence of pertussis toxin (25 μg/ml) and NAD+ (n = 3). In contrast, when the membranes pretreated with pertussis toxin and NAD+ were reconstituted with Gi/Go proteins, there was a recovery of [3H]MGA binding to values close to these found in nontreated membranes (Fig. 3), with an averageK D value of 56 nM (95% confidence interval, 17–89 nM; n = 3). Finally, the binding of [3H]MGA in nontreated membranes was not markedly affected (P > 0.05) by the addition of Gi/Go proteins (K D = 57 nM; 95% confidence interval, 36–79 nM; n = 3; Fig. 3).
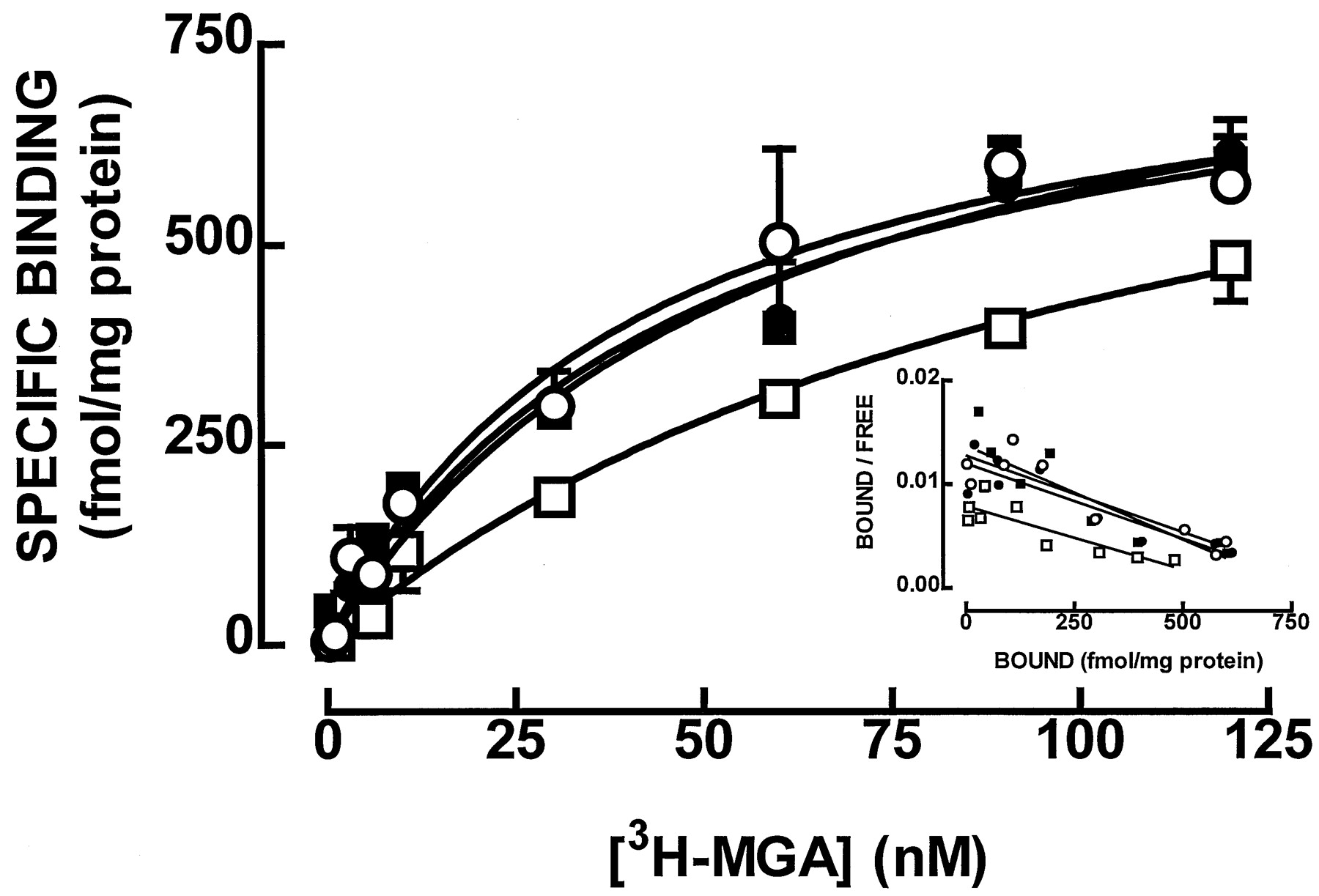
Reconstitution of [3H]MGA binding in pertussis toxin-treated rat hippocampal membranes. Rat hippocampal membranes were treated with pertussis toxin (25 μg/ml) in the absence (○, control) and presence (■) of 1 mM NAD+ and then reconstituted with solubilized Gi/Go proteins (600 nM) from bovine brain (●, control; ▪, with NAD+). The mixture was diluted 20-fold, and [3H]MGA binding was performed. The ordinates represent the specific binding of [3H]MGA obtained on subtraction of the nonspecific binding, determined in the presence of 100 μM kainate, from total binding. Inset, the corresponding Scatchard plot for each of the average saturation isotherms. The values are mean ± S.E.M. of three experiments. The S.E.M. values are not presented in the Scatchard plot for the sake of simplicity.
These results indicate that kainate receptors are coupled to G proteins of the Gi/Go group in rat hippocampal membranes and substantiate the proposal that the presynaptic inhibition of GABA release by kainate receptor activation in the hippocampus (Cunha et al., 1997; Rodrı́guez-Moreno et al., 1997) involves a metabotropic transduction system (Rodrı́guez-Moreno and Lerma, 1998). This G protein coupling of kainate receptors is also a candidate molecular basis for the inhibition of synaptic transmission in the hippocampus (Vignes et al., 1998) or for the hyperpolarization of CA1 neurons caused by a kainate receptor (GLuR5) agonist (Clarke et al., 1997), with both effects being difficult to reconciliate with the activation of ionotropic receptors permeable to cations. However, kainate receptors have a topology typical of ionotropic receptors (Bettler and Mulle, 1995), making it difficult to conceive a direct interaction with G proteins. Alternatively, this interaction might be indirect because kainate receptors are clustered into macromolecular signaling complexes by SAP proteins (Garcia et al., 1998), with some of these proteins, like SAP-97, being presynaptic (Müller et al., 1995). This association of kainate receptors with the SAP-90/PSD-95 family controls the rate of receptor desensitization (Garcia et al., 1998) but might also allow indirect coupling of kainate receptors with G proteins because PDZ domains interact with different regulatory proteins, namely with Ras GTPase activities (Kim et al., 1998). However, it remains to be demonstrated whether the interaction of kainate receptors with SAP proteins allows coupling to G proteins or whether kainate receptors couple directly to G proteins.
Acknowledgments
We acknowledge Drs. L. V. Lopes, J. Coelho, and A. R. Costenla for valuable assistance in the filtration assays. R. A. C. is indebted to Professor Moniz Pereira (Fac. Pharmacy of Lisbon) for scintillation counting facilities.
Footnotes
- Received February 2, 1999.
- Accepted April 21, 1999.
-
Send reprint requests to: Dr. R. A.Cunha, Laboratory of Neurosciences, Faculty of Medicine, University of Lisbon, Avenida Prof. Egas Moniz, 1649-035 Lisbon, Portugal. E-mail:racunha{at}neurociencias.pt
-
This work was supported by Fundação para a Ciência e Tecnologia (Praxis/2/2.1/SAU/1348/95).
Abbreviations
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- GABA
- γ-aminobutyric acid
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate
- CNQX
- 6-cyano-2,3-dihydroxy-7-nitroquinoxaline
- Gpp(NH)p
- guanylylimidotriphosphate
- MGA
- (2S,4R)-4-methylglutamate
- MPPG
- (RS)-α-methyl-4-phosphonophenylglycine
- t-ACPD
- (±)-1-aminocyclopentane-trans-1,3-dicarboxylic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}