


Abstract
Retigabine [N-(2-amino-4-[fluorobenzylamino]-phenyl) carbamic acid; D-23129] is a novel anticonvulsant, unrelated to currently available antiepileptic agents, with activity in a broad range of seizure models. In the present study, we sought to determine whether retigabine could enhance current through M-like currents in PC12 cells and KCNQ2/Q3 K+ channels expressed in Chinese hamster ovary cells (CHO-KCNQ2/Q3). In differentiated PC12 cells, retigabine enhanced a linopirdine-sensitive current. The effect of retigabine was associated with a slowing of M-like tail current deactivation in these cells. Retigabine (0.1 to 10 μM) induced a potassium current and hyperpolarized CHO cells expressing KCNQ2/Q3 cells but not in wild-type cells. Retigabine-induced currents in CHO-KCNQ2/Q3 cells were inhibited by 60.6 ± 11% (n = 4) by the KCNQ2/Q3 blocker, linopirdine (10 μM), and 82.7 ± 5.4% (n = 4) by BaCl2 (10 mM). The mechanism by which retigabine enhanced KCNQ2/Q3 currents involved large, drug-induced, leftward shifts in the voltage dependence of channel activation (−33.1 ± 2.6 mV,n = 4, by 10 μM retigabine). Retigabine shifted the voltage dependence of channel activation with an EC50value of 1.6 ± 0.3 μM (slope factor was 1.2 ± 0.1,n = 4 to 5 cells per concentration). Retigabine (0.1 to 10 μM) also slowed the rate of channel deactivation, predominantly by increasing the contribution of a slowly deactivating tail current component. Our findings identify KCNQ2/Q3 channels as a molecular target for retigabine and suggest that activation of KCNQ2/Q3 channels may be responsible for at least some of the anticonvulsant activity of this agent.
Retigabine [N-(2-amino-4-[fluorobenzylamino]-phenyl) carbamic acid; D-23129] is a novel anticonvulsant, unrelated to currently available antiepileptic agents, with activity in a broad range of seizure models. Retigabine prevents epileptiform activity induced by 4-aminopyridine, bicuculline, low Mg2+ and low Ca2+ in hippocampal slices (Armand et al., 1999,Dost and Rundfeldt, 2000), and seizures induced by pentylenetetrazol, maximal electroshock, kainate, penicillin, picrotoxin, andN-methyl-d-aspartate in rodents (Nikel et al., 1993a,b; Rostock et al., 1996). In addition, retigabine is also effective against audiogenic seizures in DBA/2J mice, against seizures in epilepsy-prone rats, and against seizures in an amygdala-kindling model (Dailey et al., 1995; Rostock et al., 1996; Tober et al., 1996).
Retigabine seems to exert its anticonvulsant action via multiple mechanisms. Evidence exists to suggest that retigabine enhances γ-aminobutyric acid (GABA)-ergic transmission in the central nervous system. Thus, retigabine has been shown to increase the synthesis of GABA in rat hippocampal slices and to enhance GABA-induced chloride currents in cultured rat cortical neurons (Kapetanovic et al., 1995;Rundfeldt et al., 1995). Retigabine may also possess weak sodium and calcium channel blocking activity (Rundfeldt et al., 1995). More recently, retigabine has been shown to induce membrane hyperpolarization in neurones in rat hippocampal-entorhinal cortex slices (Hetka et al., 2000) and to exert potassium channel opening activity in neuronal cells (Rundfeldt, 1997, 1999). In differentiated NG108–15, hNT, and PC12 cells, retigabine increased a barium and weakly tetraethylammonium (TEA)-sensitive, 4-aminopyridine-insensitive potassium conductance. Similar results were obtained in isolated mouse cortical neurons. The ability to open potassium channels in neuronal cells sets retigabine apart from currently available anticonvulsant agents, such as phenytoin, carbamazepine, and valproate (Rundfeldt, 1997). By virtue of this novel mechanism of action, therefore, retigabine may be particularly useful in the treatment of drug-resistant human epilepsies.
The molecular nature of the potassium channel opened by retigabine is unknown. Although the retigabine-induced current is blocked by barium and possesses an apparently linear current-voltage (I-V) relationship over a wide voltage range, it does not seem to be carried by channels such as rTASK, TWIK-1, or a variety of inward rectifiers (Rundfeldt, 1997, 1999). In the present study, we sought to determine whether retigabine could enhance current through KCNQ2/Q3 potassium channels. KCNQ2 and KCNQ3 mRNA is abundant in regions associated with epileptiform activity, such as the hippocampus, thalamus, and cortex (Biervert et al., 1998; Schroeder et al., 1998; Yang et al., 1998). Furthermore, mutations in KCNQ2/Q3 channels were recently identified in a rare form of neonatal epilepsy (Biervert et al., 1998, Charlier et al., 1998; Singh et al., 1998; Lerche et al., 1999). These findings suggest an important role for KCNQ2/Q3 channels in the control of neuronal excitability. KCNQ2/Q3 channels are thought to underlie the M-current, a noninactivating, slowly deactivating potassium current with similar pharmacology (i.e., barium- and weakly TEA-sensitive, 4-aminopyridine-insensitive) and distribution (i.e., present in NG108–15, PC12, and cortical cells) to the retigabine-induced current (Watson and Pittman, 1988; Villarroel 1996; Noda et al., 1998; Wang et al., 1998; Yang et al., 1998; Selyanko et al., 1999). We show that retigabine potently enhances KCNQ2/Q3 currents by inducing profound leftward shifts in the voltage dependence of channel activation. These findings identify KCNQ2/Q3 channels as one of the molecular targets for retigabine and open the way for the design and development of optimized second-generation agents as novel antiepileptic drugs.
Materials and Methods
Plasmid Constructs, Transfection Procedure, and Generation of Stable Cell Lines.
h-KCNQ2 was kindly provided by Dr. T. J. Jensch [Zentrum fur Molekulare Neurobiologie (ZMNH), University of Hamburg, Hamburg, Germany]. An h-KCNQ3 expression vector was constructed using a combination of fragments amplified from a human hippocampus library (extending from a NotI site near the 5′ end through the stop codon) and synthetic oligonucleotides (extending from the start codon to the NotI site). The synthetic oligonucleotides were designed to encode a protein identical with KCNQ3, but silent substitutions were made to reduce GC content for easier cloning. The entire KCNQ3 construct was subcloned into the pOX expression vector using ClaI/XbaI sites introduced on either side of the start and stop codons, respectively. The construct was sequenced in its entirety to make sure that no mutations were introduced during the amplification and cloning process. To make a KCNQ2-KCNQ3 tandem construct, the open reading frame of h-KCNQ3 was linked to the h-KCNQ2 open reading frame by a nucleic acid sequence encoding the polypeptide GSQQQQQQQQQQ. The resulting tandem construct encoded a protein in which the C terminus of KCNQ3 was linked to the N terminus of KCNQ2.
Chinese hamster ovary (CHO-K1) cells were transfected with either h-KCNQ2, h-KCNQ3, or the h-KCNQ2/Q3 tandem construct in pcDNA3.1 (InVitrogen, San Diego, CA) using lipofectamine reagent, according to the manufacturers instructions. Cells stably expressing the KCNQ2 or KCNQ2/Q3 constructs were identified by their resistance to G-418 (400 μg/ml). Clones were screened for expression using the whole-cell, voltage-clamp technique.
Cell Culture.
CHO-K1 cells stably transfected with either KCNQ2 or the KCNQ2/Q3 tandem construct (CHO-KCNQ2/Q3), or transiently transfected with KCNQ3, were maintained in Ham's F-12 medium supplemented with 10% heat-inactivated fetal bovine serum and 400 μg/ml G418 sulfate in an incubator at 37°C with a humidified atmosphere of 5% CO2. PC12 cells (a kind gift from Dr Aaron Kitzmiller, University of North Carolina, Chapel Hill, NC) were maintained in 45% Ham's F-12 medium, 45% Dulbecco's modified Eagle's medium-high glucose, 10% heat-inactivated fetal bovine serum, 50 I.U./ml penicillin, and 50 μg/ml streptomycin. For electrophysiological studies, cells were removed from the culture flask by brief trypsinization and replated at low density onto glass cover slips. PC12 cells on cover slips were differentiated by treatment with nerve growth factor (7–50 ng/ml) for 48 to 96 h before study.
Drugs.
Retigabine was synthesized in the laboratories of ICAgen Inc. (Durham, NC). A 1 mM stock solution of retigabine in distilled water was prepared on the day of the experiment and diluted to concentrations in the range 0.1 to 10 μM in extracellular solution (composition as described below). Linopirdine (Sigma Research Biochemicals International, Natick, MA) was stored at −20°C as a 10 mM stock solution in dimethyl sulfoxide and was diluted to 10 μM in extracellular solution (composition as described below) on the day of the experiment. The final dimethyl sulfoxide concentration was 0.1%, which had no effect on membrane currents in CHO-KCNQ2/Q3 or PC12 cells. BaCl2 was prepared as a 1 M stock in distilled water and diluted to a final concentration of 10 mM in extracellular solution (composition as described below). Nerve growth factor (NGF-7S) was purchased form Sigma Chemical Company (St. Louis, MO).
Electrophysiological Recording.
Cover slips containing PC12 cells or CHO cells expressing KCNQ2, KCNQ3, or KCNQ2/Q3 were placed in a bath on the stage of an inverted microscope and perfused (approximately 1 ml/min) with extracellular solution of the following composition: 138 mM NaCl, 2 mM CaCl2, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4, with NaOH. Pipettes were filled with an intracellular solution of the following composition: 140 mM KCl, 2 mM MgCl2, 10 mM EGTA, 10 mM HEPES, 5 mM K2ATP, pH 7.3 to 7.4 with KOH, and had a resistance of 1 to 2 MΩ. The osmolarity of the extracellular and intracellular solutions was 300 mmol/kg and 306 mmol/kg, respectively. All recordings were made at room temperature (22–24°C) using an AXOPATCH 200B amplifier and PCLAMP6.1 software (Axon Instruments, Burlingame, CA).
KCNQ2/Q3 currents and membrane currents in PC12 cells were measured using the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). Uncompensated series resistance was typically 2 to 5 MΩ and >90% series resistance compensation was routinely achieved. As a result, voltage errors were negligible. Current records were acquired at 2 to 10 KHz and filtered at 1 to 2 KHz.
Retigabine-induced currents were measured as increases in outward current at a holding potential of −30 (PC12 cells) or −40 mV (CHO-KCNQ2/Q3 cells). KCNQ2/Q3 channel activation is submaximal at this potential, thereby providing a window for drug-induced current. Multiple concentrations of retigabine were tested on each cell, with washout in between each drug application.
Steady-state current amplitude was measured at the end of a series of 3-s depolarizing steps (−100 to +30 mV in 10 mV increments from a holding potential of −80 mV). Whole cell conductance (G) was calculated according to the equation G =I/(V − Ek), where I is the steady-state current, V is the step potential, andEk is the reversal potential for potassium, which was calculated to be −82.9 mV. Activation curves were generated by plotting normalized conductance against the step potential and were fitted with a Boltzman distribution according to the equation:Y = A/{1 + exp[V h− V m)/k]}, where A is the amplitude of the relationship, V h is the voltage for half-activation, V m is the test potential, and k is the slope factor.
To describe the time course of current deactivation, tail currents were elicited by repolarizing to a series of potentials between −60 and −100 mV in 5-mV steps, after a 3-s depolarization to −30 mV (for PC-12 cells) or +30 mV (for CHO-KCNQ2/Q3 cells). Some experiments were conducted in an extracellular solution containing 40 mM K+ to enhance inward tail currents in the −60 to −100 mV voltage range. Tail currents in PC-12 cells were best fit with a single exponential function according to the equation:I = A × [1 − exp(−t/τt)], where I is current,A is amplitude, t is time, and τ is the time constant. Tail current decay in CHO-KCNQ2/Q3 cells was best fit with a double exponential function, according to the equation:I = A fast × [1 − exp(−t/τfast)] +A slow × [1 − exp(−t/τslow)].
To determine the KCNQ2/Q3 current reversal potential, tail currents were elicited by repolarizing the membrane to potentials in the range −100 to −20 mV in 5-mV increments after a 3-s voltage step to 30 mV. Measurements of tail currents were made in the standard extracellular solution (composition as above) and in extracellular solution containing 40 mM KCl. Tail currents were fitted with a double exponential function (as described above) and the instantaneous tail current amplitudes were calculated by extrapolation to the time at which the membrane was repolarized. Reversal potentials were calculated from linear fits of instantaneous tail current amplitude against repolarization potential and were corrected for the liquid junction potential (calculated to be 6 mV).
Semilog plots of drug concentration against effect were fit with a logistic function according to the equation: Response = {A 1 − A 2/[(1 + x/x 0)p]} +A 2, where A 1 is the initial response, A 2 is the final response,x 0 is the mid-point (i.e., EC50), and p is power (slope factor). Multiple concentrations of drug were tested per cell, and EC50 and slope values were calculated. Mean ± S.E.M. values for EC50 and slope factors were calculated by averaging data obtained from individual cells. The resting membrane potentials of KCNQ2/Q3 and PC12 cells were measured using the current-clamp configuration of the patch-clamp technique (Hamill et al., 1981).
Statistics.
All data are expressed as mean ± S.E.M. for n ≥ 4 observations. Statistical significance was determined using a suitable (paired, hetero-, or homoscedastic) two-tailed t test. P values <.05 were considered significant.
Results
Retigabine has previously been shown to induce a current in neuronal cells (such as NG108-15, hNT, and PC12 cells) with some similarities to an M-current [i.e., similar distribution and pharmacology (Watson and Pittman, 1988; Villarroel 1996; Noda et al., 1998; Wang et al., 1998; Yang et al., 1998; Selyanko et al., 1999)]. Therefore, as a first step toward elucidating the molecular nature of the potassium channel opened by retigabine, we sought to determine whether retigabine could enhance M-like currents in differentiated PC12 cells.
Retigabine Enhances a Linopirdine-Sensitive Current in Differentiated PC12 Cells
A characteristic feature of M-currents is their sensitivity to block by linopirdine at concentrations that are largely without effect on other membrane currents and cloned ion channels (Aiken et al., 1995;Lamas et al., 1997; Schnee and Brown, 1998; Wang et al., 1998). Therefore, we determined whether retigabine-induced currents in PC12 cells were linopirdine-sensitive. Figure1A shows a representative voltage-clamp recording from a NGF- differentiated PC12 cell. In the example shown, a small outward current was apparent at a holding potential of −30 mV. Retigabine (10 μM) induced a reproducible increase in outward current that was completely reversible upon removal of the drug. Superfusion of the cell with 10 μM linopirdine decreased both the holding current and the retigabine-induced current. Similar observations were made from a total of seven cells, in which the average inhibition of the retigabine-induced current by 10 μM linopirdine was 63.9 ± 2.9%.

Retigabine enhances a linopirdine-sensitive, slowly deactivating current in differentiated PC12 cells. A representative voltage-clamp recording from a NGF-differentiated PC12 cell is shown in A. The recording shows that retigabine (R, 10 μM, open horizontal bar) induced a reproducible increase in outward holding current at −30 mV, which was partially inhibited by superfusion of the cell with 10 μM linopirdine (solid horizontal bar). On average, linopirdine (10 μM) inhibited the retigabine-induced current by 63.9 ± 2.9% (n = 7). To study the effects of retigabine on M-like tail currents, PC12 cells were voltage clamped at −30 mV and then repolarized. The voltage clamp records in B and C show the results of typical experiments in which PC12 cells were repolarized to either −50 mV (B) or −100 mV (C). The figures show that superfusion of cells with 10 μM retigabine (R) increased the amplitude of the tail current and slowed the rate of tail current deactivation relative to control (C). The effect of retigabine was fully reversible (W = wash).
Retigabine Enhances a Slowly Deactivating Tail Current in Differentiated PC12 Cells
Although linopirdine seems to be a relatively selective blocker of the M-current at the concentration used in the present study (Aiken et al., 1995; Lamas et al., 1997; Schnee and Brown, 1998; Wang et al., 1998), it is possible that retigabine may enhance a previously uncharacterized linopirdine-sensitive current in PC12 cells. Therefore, we sought additional evidence for M-current involvement in the actions of retigabine. In a separate series of experiments, we studied the effects of retigabine on M-like tail currents in PC12 cells. Cells were voltage clamped at −30 mV to activate M-like currents and then repolarized to either −50 mV (Fig. 1B) or −100 mV (Fig. 1C) to induce deactivating tail currents. As shown in Fig. 1B, repolarization of PC12 cells to −50 mV induced a very slowly decaying outward tail current, whereas Fig. 1C shows that repolarization to −100 mV induced a somewhat faster deactivating inward tail current. Current deactivation rates (τ, ms) were measured by fitting tail currents with a single exponential function and were 146 ± 19 ms (n = 6) and 23.4 ± 2.4 ms (n = 8) at −50 mV and −100 mV, respectively. Superfusion of cells with 10 μM retigabine increased both the amplitude of the outward holding current at −30 mV and the amplitude of the tail currents induced by repolarization. In addition, retigabine slowed the rate of tail current deactivation. This was particularly evident at −50 mV, where tail currents showed little deactivation over 500 ms (Fig. 1B). Slowing of deactivation was also apparent at −100 mV; however, where deactivation rates were slowed from 23.4 ± 2.4 ms (n = 8) in the absence of retigabine to 70.3 ± 6.0 ms (n = 8,P < .05, paired two-tailed t test, Fig. 1C) in the presence of retigabine. Tail current amplitudes at −100 mV were increased from 38.2 ± 5.4 pA in control recordings to 109.1 ± 17.9 pA after superfusion of retigabine (n = 8,P < .05, paired two-tailed t test, Fig.1C).
Our findings in differentiated PC12 cells are suggestive of an effect of retigabine on native M-like currents. Because KCNQ2/KCNQ3 heteromultimers may underlie M-currents in some neuronal cells (Wang et al., 1998, Selyanko et al., 1999), we investigated whether retigabine could enhance heterologously expressed h-KCNQ2/Q3 heteromultimeric channels.
Stable Expression of KCNQ2/Q3 Heteromeric Channels in a Mammalian Cell Line
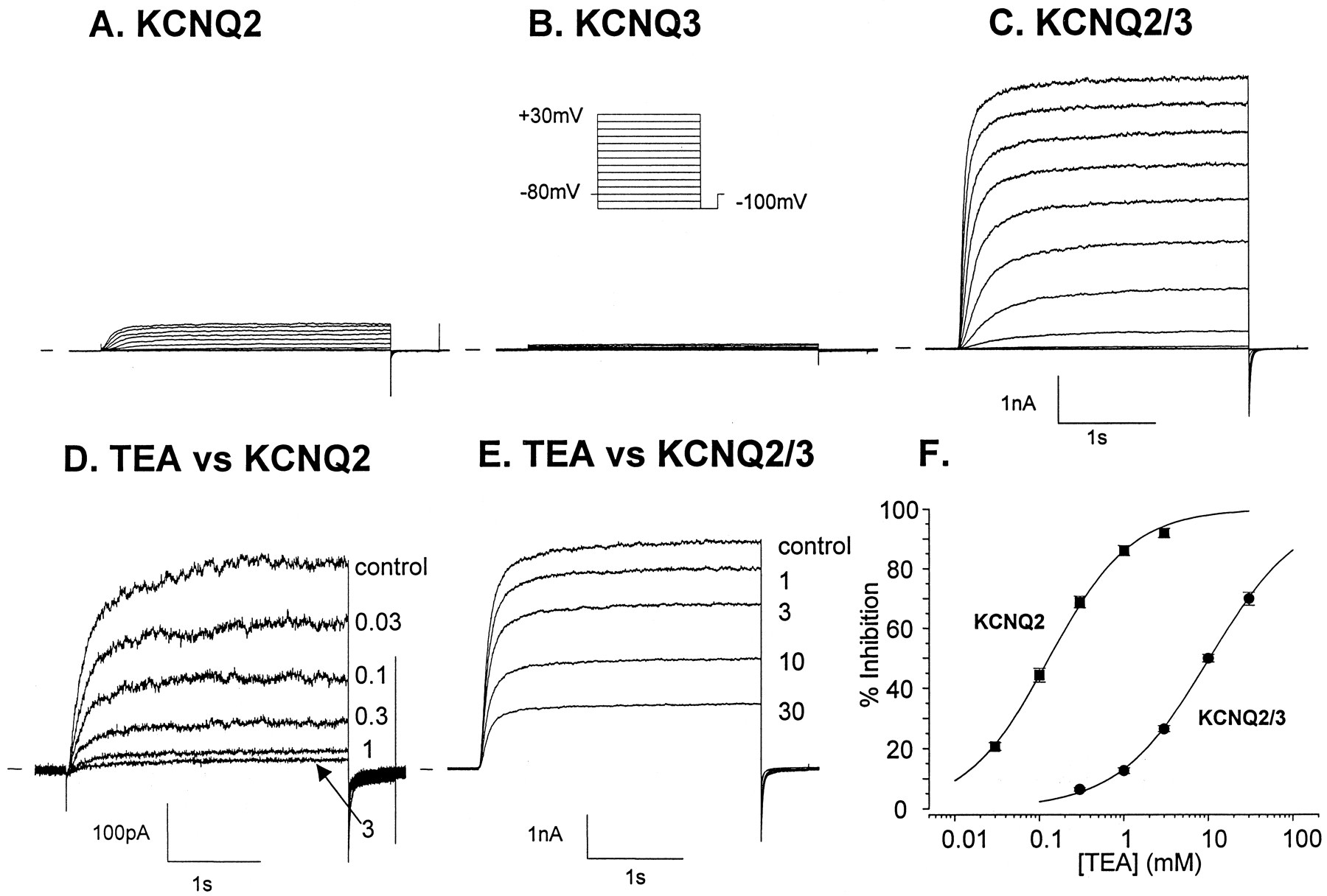
To test the effects of retigabine on KCNQ2/KCNQ3 heteromultimeric channels, we generated a cell line stably expressing an h-KCNQ2/Q3 tandem construct (see Materials and Methods). We chose this approach (rather than cotransfection of KCNQ2 and KCNQ3) in an attempt to fix the stoichiometry of the potassium channel complex, thereby potentially reducing variability in our experimental data. As shown in Fig. 2C, robust currents (i.e., >2 nA at +20 mV) were routinely recorded from cells expressing the tandem construct (CHO-KCNQ2/Q3). KCNQ2/Q3 currents were much larger than currents recorded from CHO-K1 cells expressing h-KCNQ2 [which were invariably less than 1 nA at +20 mV (Fig. 2A)] or h-KCNQ3 [which failed to express (Fig. 2B)]. The TEA sensitivity of currents recorded from cells expressing the h-KCNQ2/Q3 tandem construct was similar to that recorded previously from Xenopus laevis oocytes coinjected with KCNQ2 and KCNQ3 (Wang et al., 1998; Yang et al., 1998); i.e., KCNQ2/Q3 currents were only weakly TEA sensitive (IC50 = 10.5 ± 0.9 mM, slope = 0.81 0.02, n = 4; Fig. 2, E and F). In contrast, currents recorded from cells expressing KCNQ2 alone were potently inhibited by TEA, with an IC50 value of 0.13 ± 0.01 mM (slope = 0.92 ± 0.04, n = 5; Fig. 2, D and F). These data on current magnitude and TEA sensitivity provide evidence to suggest that expression of the h-KCNQ2/Q3 tandem construct gave rise to a uniform population of heteromultimeric KCNQ2/Q3 channels. Currents recorded from cells expressing the tandem construct were also inhibited, in a concentration-dependent manner, by linopirdine (IC50 = 3.5 ± 0.1 μM, slope 1.26 ± 0.03, n = 6 to 7 cells per concentration; Fig. 3, A and B). Linopirdine has been shown previously to inhibit M-currents with similar potency (Wang et al., 1999).

Outward currents in Chinese hamster ovary (CHO) cells stably transfected with h-KCNQ2/Q3 tandem construct. Typical voltage-clamp recordings showing families of outward currents recorded from CHO cells transfected with h-KCNQ2 (A), h-KCNQ3 (B), and an h-KCNQ2/Q3 tandem construct (C) are shown. Membrane currents were elicited by a series of depolarizing steps (−100 to +30 mV in 10 mV increments; see inset) from a holding potential of −80 mV. KCNQ2/Q3 currents were much larger than currents recorded from CHO-K1 cells expressing h-KCNQ2 or h-KCNQ3. KCNQ2 currents in CHO cells were highly TEA-sensitive, as indicated by the representative voltage-clamp recordings in D, whereas currents in cells expressing the tandem construct were only weakly TEA-sensitive (E). Percentage inhibition of KCNQ2 or KCNQ2/Q3 by TEA was plotted against TEA concentration in F. The TEA IC50 for block of KCNQ2 currents was 0.13 ± 0.01 mM (slope = 0.92 ± 0.04, n = 5) and 10.5 ± 0.9 mM (slope = 0.81± 0.02, n = 4) for block of KCNQ2/Q3 currents. Symbols represent the mean ± S.E.M. of four to five cells.

h-KCNQ2/Q3 currents in Chinese hamster ovary (CHO) cells are linopirdine-sensitive. Representative h-KCNQ2/Q3 currents (elicited by depolarization to +30 mV from a holding potential of −80 mV) recorded from a CHO cell, in the absence or presence of linopirdine (lino, 1 to 30 μM) are shown in A. Mean percentage inhibition of KCNQ2/Q3 current was calculated and plotted against the linopirdine concentration in B. The linopirdine IC50 value was 3.5 ± 0.1 μM, slope 1.26 ± 0.03 (n = 6 to 7 cells per concentration). The symbols represent the mean ± S.E.M. of four to five cells.
Retigabine Induces a Potassium Current and Hyperpolarizes CHO-KCNQ2/Q3 Cells but Not WT CHO Cells
CHO-KCNQ2/Q3 cells were voltage clamped at −40 mV. At this potential, an outward current of variable amplitude was observed in the majority of cells (consistent with some steady-state activation of the KCNQ2/Q3 channel at this voltage; see Fig. 6C). In these cells, retigabine (10 μM) induced a large increase in outward current (Fig.4A, inset, upper trace). In current-clamp configuration, the resting membrane potential of CHO-KCNQ2/Q3 cells was typically between −40 and −50 mV and application of 10 μM retigabine hyperpolarized the membrane potential by as much as 20 mV (Fig. 4B, inset, upper trace). Both effects were readily reversible on removal of the drug. Retigabine-induced increases in current or membrane potential were never observed in wild-type CHO-K1 cells (Fig.4, A and B, insets, lower traces). Representative examples of concentration-response curves, generated by plotting increases in outward current or membrane potential against retigabine concentration and fitting the data with a logistic function (see Materials and Methods) are shown in Fig. 4, A (outward current) and B (hyperpolarization). For the examples shown, a half-maximal increase in outward current was observed at 0.3 μM retigabine (slope factor = 2.3) and a half-maximal membrane hyperpolarization was observed at 0.4 μM retigabine (slope factor = 1.3). Data from four independent experiments were averaged to generate retigabine EC50 values of 0.34 ± 0.05 μM and 0.78 ± 0.17 μM for induction of outward current and hyperpolarization, respectively. The mean (±S.E.M.) slope value was 1.7 ± 0.2 for the relationship between outward current and retigabine concentration and 1.1 ± 0.08 for the relationship between membrane potential and retigabine concentration.

Retigabine shifts the voltage dependence of KCNQ2/Q3 activation. A and B show families of outward currents (elicited by voltage steps from −100 to +30 mV, in 10 mV increments from a holding potential of −80 mV, see inset) recorded from a CHO-KCNQ2Q3 cell in the absence (A) or presence (B) of 10 μM retigabine. A comparison of the voltage-clamp records shown in A and B indicates that KCNQ2/Q3 currents are increased at all voltages in the presence of 10 μM retigabine. The increase in KCNQ2/Q3 current amplitude was most marked at voltages considered threshold in the absence of retigabine. Activation curves for KCNQ2/Q3 currents in CHO cells were generated from steady-state outward currents (see Materials and Methods). Representative activation curves generated in the absence of retigabine (▴), in the presence of 0.1 μM (▾), 0.3 μM (♦), 1 μM (+), 3 μM (▪), and 10 μM (●) retigabine, and after washout of retigabine (×) are shown in C. Half-maximal channel activation in the absence of retigabine occurred at −31.1 mV. Retigabine induced a concentration-dependent leftward shift of the KCNQ2/Q3 channel activation curve, with half-maximal channel activation occurring at −61.1 mV in the presence of 10 μM retigabine. The magnitude of the retigabine-induced shifts in the mid-point of the activation curve were calculated and plotted against retigabine concentration in D. From these data, the retigabine EC50was calculated to be 1.6 ± 0.3 μM and the slope factor was 1.2 ± 0.13. The symbols represent the mean ± S.E.M. of 4 to 5 cells per retigabine concentration.

Retigabine enhances holding current at −40 mV and hyperpolarizes CHO-KCNQ2/Q3 cells. Retigabine (R, 10 μM) induced large increases in outward current in CHO-KCNQ2/Q3 cells voltage clamped at −40 mV (A, inset, upper trace), but not in wild-type CHO cells (A, inset, lower trace). Retigabine-induced increases in holding current were concentration-related. A representative example of a retigabine concentration-response curve, generated by plotting increases in outward current against retigabine concentration, and fitting the data with a logistic function, is shown in A. The average retigabine EC50 value calculated in this way was 0.34 ± 0.05 μM (slope factor = 1.7 ± 0.2,n = 4). Retigabine (R, 10 μM) also induced large membrane hyperpolarizations in CHO-KCNQ2/Q3 cells (B, inset, upper trace) but not in wild-type CHO cells (B, inset, lower trace). A representative example of a concentration-response curve, generated by plotting membrane hyperpolarization against retigabine concentration is shown in B. The average EC50 value was 0.78 ± 0.17 μM (slope factor = 1.1 ± 0.08, n = 4).
Retigabine-Induced Currents in CHO-KCNQ2/Q3 Cells Are Linopirdine- and Barium-Sensitive
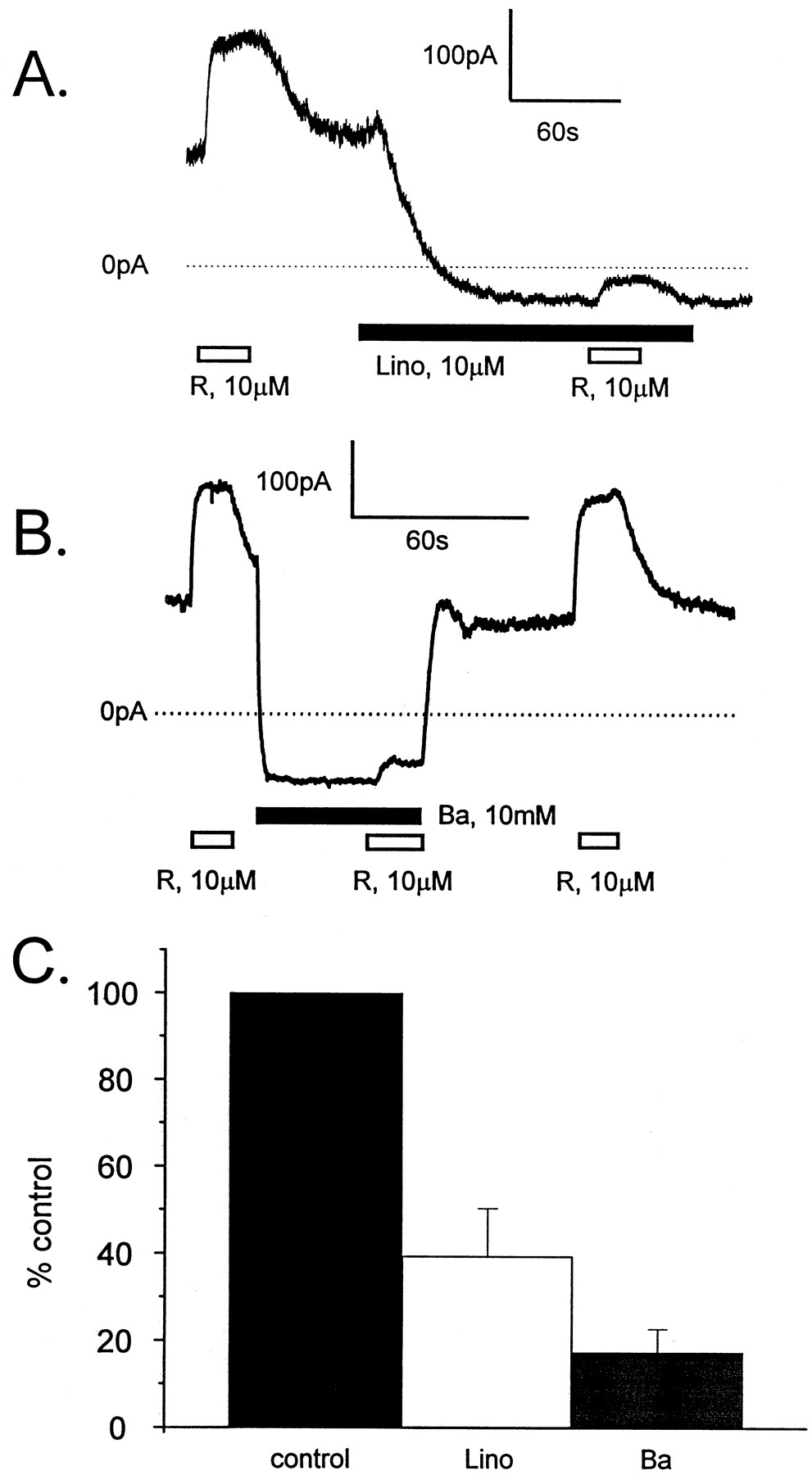
The finding that retigabine increased outward current and hyperpolarized the resting membrane potential in CHO-KCNQ2/Q3 cells but not in wild-type CHO-K1 cells strongly suggests that retigabine enhances activation of KCNQ2/Q3 channels. However, it remains possible that retigabine enhances a background current that becomes selectively up-regulated in the stably transfected cells. For this reason, we determined whether the retigabine-induced current was sensitive to the KCNQ2/Q3 blockers, linopirdine, and barium. Figure5A shows a representative recording from a CHO-KCNQ2/Q3 cell voltage-clamped at −40 mV and superfused with 10 μM retigabine before and after exposure to 10 μM linopirdine. The figure shows that retigabine produced a reversible enhancement of the outward holding current at −40 mV. Superfusion of the cell with 10 μM linopirdine produced a slow decrease in the holding current and markedly inhibited the ability of retigabine to induce current. The effects of linopirdine were not fully reversible over the time course of the experiment. BaCl2 (10 mM) also decreased the holding current and the retigabine-induced current in CHO-KCNQ2/Q3 cells; unlike linopirdine, the effects of BaCl2(10 mM) were rapid and fully reversible (Fig. 5B). Observations similar to those illustrated in Fig. 5, A and B, were made from four cells for each blocker, with the retigabine-induced current being inhibited 60.6 ± 11% by 10 μM linopirdine and 82.7 ± 5.4% by 10 mM BaCl2 (Fig. 5C).

Retigabine-induced currents in CHO-KCNQ2/Q3 cells are linopirdine- and barium-sensitive. Representative current recordings from CHO-KCNQ2/Q3 cells voltage-clamped at −40 mV are shown in A and B. In A, the cell was superfused with 10 μM retigabine (R, open horizontal bar) before and during exposure to 10 μM linopirdine (lino, solid horizontal bar). In B, the cell was superfused with 10 μM retigabine (R, open horizontal bar) before, during, and after exposure to 10 mM BaCl2 (Ba, solid horizontal bar). These figures show that both linopirdine and BaCl2 reduced holding current and markedly inhibited the ability of retigabine to increase current. Only the effects of BaCl2 were reversible. Observations similar to those illustrated in A and B were made from four cells for each blocker, and the average relative amplitudes of the retigabine-induced currents in the presence of linopirdine and BaCl2 are plotted in the bar graph (C). On average, the retigabine-induced currents were reduced to 39.4 ± 11% (open bar) and 17.3 ± 5.4% (gray bar) of control (solid bar) by 10 μM linopirdine and 10 mM BaCl2, respectively.
Retigabine Shifts the Voltage-Dependence of KCNQ2/Q3 Activation and Enhances Slow Deactivation without Altering Channel K+Selectivity
Activation.
To understand the mechanism by which KCNQ2/Q3 currents were increased, we studied the effects of retigabine on the time- and voltage-dependent activation of KCNQ2/Q3 currents. CHO-KCNQ2/Q3 cells were held at −80 mV, stepped to voltages in the range −100 mV to +30 mV for 3 s and then repolarized to −100 mV for 0.5 s (Fig. 6, center inset). Inspection of the voltage clamp records shown in Fig. 6A reveals that, in the absence of retigabine, voltage steps above −50 mV induced a slowly activating, noninactivating outward current, with subsequent repolarizations to −100 mV inducing deactivating inward tail currents. Superfusion of the same cell with 10 μM retigabine dramatically changed both the time and voltage dependence of channel activation. A voltage step to −100 mV induced a small, slowly decaying inward current, whereas steps positive to −80 mV induced outward currents with both instantaneous and time-dependent components. At any given voltage, the amplitude of the outward current was larger in the presence of retigabine. The extent to which KCNQ2/Q3 currents were increased by retigabine was dependent on the voltage. Relatively large increases in outward current were seen at voltages considered threshold in the absence of retigabine [for example, compare current amplitudes in the presence and absence of retigabine at −40 mV (Fig. 6, A and B, arrows)]. In contrast, relatively smaller increases in current were seen at more depolarized potentials. Deactivating inward tail currents were apparent after repolarization from all voltages positive to −90 mV in the presence of 10 μM retigabine.
Theoretically, the observed effects of retigabine could be accounted for, at least in part, by a hyperpolarizing shift in the voltage dependence of channel activation. We therefore constructed activation curves for KCNQ2/Q3, in the absence and presence of retigabine (0.1 to 10 μM; see Materials and Methods). Activation curves generated in a representative experiment are shown in Fig. 6C. In this experiment, half-maximal channel activation in the absence of retigabine occurred at a voltage of −31.1 mV. Retigabine induced a concentration-dependent leftward shift of the KCNQ2/Q3 channel activation curve, with half-maximal channel activation occurring at −32.8, −39.1, −46.3, −51.9, and −61.1 mV in the presence of 0.1, 0.3, 1, 3, and 10 μM retigabine, respectively. The magnitude of the retigabine-induced shifts in the midpoint of the activation curve were calculated and plotted against retigabine concentration, to generate a concentration-response relationship, as illustrated in Fig. 6D. On average, the retigabine EC50 value was 1.6 ± 0.3 μM and the slope factor was 1.2 ± 0.13 (n = 5). The slopes of the activation curves seemed relatively unaffected by retigabine. In the absence of retigabine, the slope of the KCNQ2/Q3 activation curve was 9.2 ± 0.3. Slope values were 9.5 ± 0.4 (P > .05), 8.8 ± 0.5 (P > .05), 7.3 ± 0.7 (P < .05), 10.4 ± 1.8 (P > .05), 8.2 ± 1.4 (P > .05) in the presence of 0.1, 0.3, 1, 3, and 10 μM retigabine, respectively.
Deactivation.
To examine the kinetics of tail current deactivation, KCNQ2/Q3 currents were elicited by 3-s depolarizations to 30 mV and tail currents were obtained by repolarization to a series of potentials in the range −100 to −60 mV in 5-mV increments. Recordings were made in an extracellular solution containing elevated (40 mM) K+, to enhance inward tail currents in the −60 mV to −100 mV voltage range. Tail current deactivation was fitted with a double exponential function, with the fast component accounting for 55.1 ± 10.2% (n = 5) at −60 mV but 99.4 ± 0.4% (n = 5) at −100 mV (Fig.7A). Figure 7B shows that τfast values were voltage-dependent, decreasing from 57.8 ± 7.2 ms (n = 5) at −60 mV to 24.7 ± 2.1 ms (n = 5) at −100 mV (P < .05). In the presence of retigabine (10 μM), tail current deactivation was dominated by the slow component at all voltages (Fig. 7C). This dominant effect prevented meaningful analysis of the effects of retigabine on the voltage dependence of fast deactivation.

Retigabine enhances slow KCNQ2/Q3 channel deactivation. Deactivating tail currents were elicited by repolarization to voltages from −60 to −100 mV in 5 mV increments, after a 3-s depolarization to +30 mV. Recordings were made in extracellular solution containing elevated (40 mM) K+, to enhance inward tail currents in the −60 to −100 mV voltage range. Tail current decay was fitted with a double exponential function and the relative contribution of the fast and slow components for each voltage, in the absence (●) and presence (■) of 10 μM retigabine, plotted in A. In the presence of retigabine (10 μM), tail current deactivation was dominated by the slow component at all voltages. τfast Values were plotted against voltage in B, which shows that τfast values were voltage-dependent. The concentration dependence of the retigabine-induced enhancement of slow deactivation was determined in the experiments summarized in C. KCNQ2/Q3 currents were elicited as described above and tail currents were obtained by repolarization to −100 mV in normal extracellular solution. Retigabine significantly increased the contribution of the slow component of tail current deactivation at concentrations of 0.3 μM or greater. τslow Values are plotted against retigabine concentration in D, which shows that τslowvalues tended to increase with increasing retigabine concentrations. Symbols represent the mean ± S.E.M. of four to five cells. ∗, represents P < .05, retigabine treated versus control, two-tailed unpaired t test.
Because the predominant effect of retigabine was to increase the contribution of the slow component of tail current deactivation, we examined the concentration dependence of this effect in a separate series of experiments. KCNQ2/Q3 currents were elicited with 3-s depolarizations to 30 mV and tail currents were obtained by repolarization to −100 mV. Recordings were made in normal extracellular solution. In the absence of retigabine, tail current decay was well-fit with a double exponential function, with a fast time constant (τfast) of 25.2 ± 2 ms (n = 5) and a slow time constant (τslow) of 125.2 ± 8.8 ms (n = 5). In the absence of retigabine, slow deactivation accounted for only 22.9 ± 4.3% of the tail current amplitude. The contribution of the slow component of tail current deactivation increased to 38.0 ± 7.0%, 49.5 ± 4.0%, 64.1 ± 4.5%, 74.2 ± 1.0%, and 75.1 ± 3.5% in the presence of 0.1, 0.3, 1,3, and 10 μM retigabine, respectively (n = 4 to 5 per concentration; Fig. 7C). The contribution of the slow component was significantly greater than control in the presence of all but the lowest concentration of retigabine. In addition, retigabine significantly increased τslow at the highest concentration tested (P > .05, two-tailed t test; Fig. 7D).
Ionic Selectivity.
To determine whether the ionic selectivity of KCNQ2/Q3 currents was altered by retigabine, we measured the reversal potential of voltage-activated currents in CHO-KCNQ2/Q3 cells, in normal and elevated extracellular K+ and in the presence and absence of 10 μM retigabine. As illustrated in the examples shown in Fig. 8, A to D (and inset), KCNQ2/Q3 currents were elicited by a 3-s voltage step to 30 mV and tail currents were obtained by repolarization to a series of potentials in the range −100 to −20 mV in 5-mV increments. Reversal potentials were calculated from linear fits of instantaneous tail currents (see Materials and Methods) against repolarization potential. The KCNQ2/Q3 reversal potential, in the absence of retigabine, was calculated to be −78.4 ± 1.3 mV (n = 10) in normal extracellular K+ (5.4 mM), and −29.3 ± 0.8 mV (n = 6) in elevated extracellular K+ (40 mM; Fig. 8E). These values were similar to calculated EK values (−82.9 in normal K+ and −32.4 in 40 mM K+), indicating that KCNQ2/Q3 currents were carried mainly by K+. In the presence of 10 μM retigabine, reversal potentials were −75.3 ± 1.3 mV (n = 7) in normal K+ and −28.0 ± 1.1 mV (n = 5) in 40 mM K+ (Fig. 8E). These values were not significantly different (P > .05) from those determined in the absence of retigabine. These findings indicate that the enhancement of KCNQ2/Q3 current by retigabine was not associated with any change in the ionic selectivity of the channel. Furthermore, this observation suggests that retigabine does not enhance any non-K+ currents in the CHO-KCNQ2Q3 cell line.

KCNQ2/Q3 reversal potential in the absence and presence of retigabine. KCNQ2/Q3 currents were elicited by 3-s voltage steps to 30 mV and tail currents were obtained by repolarization to a series of potentials from −100 to −20 mV in 5-mV increments. Tail currents were elicited in the absence (A, B) or presence (C, D) of 10 μM retigabine (R), in extracellular solution containing either 5.4 mM K+ (A, C) or 40 mM K+ (B, D). Reversal potentials were calculated from linear fits of instantaneous tail current amplitude against repolarization potential, and are plotted against extracellular K+ concentration in E. Reversal potentials measured in the absence of retigabine are denoted by the open squares, whereas reversal potentials measured in the presence of 10 mM retigabine are denoted by closed squares. Reversal potentials were similar to calculated EK values (−82.9 in normal K+ and −32.4 in 40 mM K+) under all conditions.
Discussion
Retigabine is a novel antiepileptic drug whose mechanism of action involves potassium channel opening activity in neuronal cells (Rundfeldt, 1997, 1999). However, the molecular nature of the potassium channel opened by retigabine has not been previously identified. In the present study, we found that retigabine enhanced an outward current in PC12 cells, confirming the findings of Rundfeldt (1999). The current activated by retigabine shared some similarities with an M-like current. For example, retigabine-induced currents in PC12 cells are inhibited by the M-current blocking drugs, linopirdine (present study), and Ba2+ (Rundfelt, 1999). Also, the induction of outward currents in PC12 cells by retigabine was associated with slowed M-like current deactivation. These findings suggest that retigabine activates native M-like currents in PC12 cells.
Because KCNQ2/KCNQ3 heteromultimers may underlie M-currents in some neuronal cells (Wang et al., 1998, Selyanko et al., 1999), we investigated whether retigabine could enhance heterologously expressed h-KCNQ2/Q3 heteromultimeric channels. In the present study, we show that retigabine markedly enhances KCNQ2/Q3 currents. Enhanced KCNQ2/Q3 activation occurs largely as a result of profound retigabine-induced leftward shifts in the voltage dependence of channel activation. In addition, retigabine also enhances slow channel deactivation. A recent report has suggested that erg1 channels may also contribute to M-currents in some cells (Selyanko et al., 1999). In preliminary studies, retigabine did not enhance h-erg1 expressed in COS-7 cells (Yu and Wickenden, unpublished observations).
The underlying mechanism by which retigabine modifies the gating of KCNQ2/Q3 channels is not completely clear from the present study. However, the findings that retigabine induces strong hyperpolarizing shifts in activation and enhances slow deactivation suggest that retigabine may either destabilize a closed conformation or stabilize an open conformation of the channel. Interestingly, a recent gating model of the M-current in rat sympathetic neurons [to which KCNQ2/Q3 channels are thought to contribute (Wang et al., 1998)] proposes the involvement of at least three closed states with long (CL), medium (CM), and short (CS) closed times and two open states (OS and OL;Selyanko and Brown, 1999). These authors have demonstrated that the CL state can be effectively removed by patch excision into calcium free solutions, which is associated with gating modifications that are qualitatively similar to those caused by retigabine. Clearly, an analysis of the effects of retigabine on single KCNQ2/Q3 channel behavior would provide important insights into the mechanism of action of this compound.
Previous studies have shown that retigabine-induced currents in PC12 cells possess an apparently linear I-V relationship over a wide voltage range (Rundfeldt, 1999). Furthermore, we found that the kinetics of KCNQ2/Q3 deactivation (biexponential) differ from the kinetics of deactivation of the retigabine sensitive current in PC12 cells (monoexponential). These findings may suggest KCNQ2/Q3-based currents are not the only target for retigabine in PC12 cells. Indeed, it has been suggested that retigabine enhances a background current, possibly carried by an inward rectifier or a two-pore channel (Rundfeldt, 1997,1999). However, it is possible that the apparently linear I-V relationship can be accounted for by large retigabine-induced leftward shifts in the voltage dependence of KCNQ2/Q3 activation, coupled with a drug-induced slow deactivation. Furthermore, differences in the voltage-clamp protocols, NGF treatment, and/or the small amplitude of the deactivating tail currents in PC12 cells may account for the apparent kinetic differences in current deactivation.
In summary, retigabine is a novel anticonvulsant, unrelated to currently available antiepileptic agents, with activity in a broad range of seizure models. Although retigabine possesses multiple mechanisms of action (Kapetanovic et al., 1995; Rundfeldt et al., 1995;Rundfeldt, 1997, 1999), it is tempting to speculate that activation of KCNQ2/Q3 channels may be responsible for at least some of the anticonvulsant activity of this agent. Indeed, our findings, coupled with the recent genetic evidence linking KCNQ2/Q3 channel mutations with benign familial neonatal epilepsy (Biervert et al., 1998, Charlier et al., 1998; Singh et al., 1998; Lerche et al., 1999), strongly suggest that KCNQ2/Q3 channels may represent novel targets for antiepileptic drugs.
Acknowledgments
We thank Rick Payne and Louise Heath for help with cell culture, Jayne Bachman for help with preparation of plasmid constructs, Dr. Joe Lennox for synthesis of retigabine, and Dr. Doug Krafte for helpful comments during the preparation of the manuscript.
Footnotes
- Received December 3, 1999.
- Accepted June 5, 2000.
-
Send reprint requests to: Dr. Alan D. Wickenden, ICAgen Inc., Suite 460, 4222 Emperor Boulevard, Durham, NC 27702. E-mail:awickenden{at}icagen.com
Abbreviations
- GABA
- γ-aminobutyric acid
- TEA
- tetraethylammonium
- I-V
- current-voltage
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}