


Abstract
Nicotinic receptors containing α7 subunits are ligand-gated ion channels widely distributed in the nervous system; they influence a diverse array of events because of their high relative calcium permeability. We show here that nicotine-induced whole-cell responses generated by such receptors can be dramatically potentiated in a rapidly reversible manner by some but not all albumins. The potentiation involves increases both in potency and efficacy with no obvious differences in rise and fall times of the response. The potentiation is not reduced by removing absorbed components; it is abolished by proteolysis, suggesting that the albumin protein backbone is essential. The fact that some albumins are ineffective indicates that minor differences in amino acid sequence may be critical. Experiments with open channel blockers indicate that the potentiation involves increased responses from active receptors rather than recruitment of receptors from a previously silent pool. Single channel recordings reveal that the potentiation correlates with increased single channel opening probability, reflected in increased frequency of channel opening and increased mean channel open time. The potentiation can be exploited to overcome blockade by noncompetitive inhibitors such as β-amyloid peptide. The results raise the possibility that endogenous compounds use the site to modulate receptor function in vivo, and suggest that the receptors may represent useful targets for therapeutic intervention in cases where they have been implicated in neuropathologies such as Alzheimer's disease.
The ability to modulate the function of ionotropic receptors in situ offers a potentially powerful method for therapeutic intervention. By altering the response of a receptor to endogenous agonists, the modulator can change the level of signaling without necessarily changing the temporal or spatial pattern of the signaling. One of the most successful examples of therapeutic intervention based on this strategy is the use of benzodiazepines to enhance the response of GABAA receptors and augment inhibitory activity in the central nervous system (Macdonald and Olsen, 1994).
Nicotinic acetylcholine receptors (nAChRs) are widely expressed in the nervous system and have been implicated in a variety of behaviors and neuropathologies. One of the most abundant is a species composed of α7 subunits (α7-nAChRs) that has a high relative permeability to calcium, exceeding that of NMDA receptors. Because of this and because of their diverse locations, α7-nAChRs can influence a wide range of cellular functions, including enhancement of transmitter release (McGehee et al., 1995; Gray et al., 1996), generation of synaptic currents, activation of second messenger cascades, and regulation of neurite extension, apoptosis, and neuron survival (for reviews, seeBroide and Leslie, 1999; Margiotta and Pugh, 2003). Recent evidence suggests that α7-nAChRs are also involved in cognitive events (Levin et al., 1999) and may be specifically blocked in Alzheimer's disease (Liu et al., 2001; Pettit et al., 2001).
Most therapeutic strategies targeting neuronal nAChRs have made use of compounds designed to act as agonists or antagonists. Examples include compounds to treat nicotine addiction, generate nociception, and ameliorate Alzheimer's disease (for reviews, see Francis et al., 1999;Dani et al., 2001). Modulation of receptor function may offer an alternative strategy, however, as suggested by the finding that plant alkaloids can potentiate the response of α4/β2-containing nAChRs (Schrattenholz et al., 1996). Evidence that α7-nAChRs may also be subject to potentiation comes from reports that the antihelmintic ivermectin (Krause et al., 1998), the neuropeptide PACAP (Pardi and Margiotta, 1999), and the hormone prostaglandin E2 (Du and Role, 2001) can each increase the whole-cell response generated by the receptors.
We now report a dramatic potentiation of α7-nAChR responses by specific albumins. The effect seems to depend on the peptide sequence of the albumin rather than on an absorbed component, and it includes both an increase in the affinity of the receptor for agonist and an increase in the maximum response the receptors generate. The potentiation is mediated by an increase in channel open time rather than recruitment of previously inactive receptors. The results suggest that it may be possible to exploit existing sites on α7-nAChRs to increase substantially the response they produce in vivo.
Materials and Methods
Cell Cultures.
Chick cultures were prepared from embryonic day 8 (E8) ciliary ganglion neurons and maintained for 4 to 7 days as described previously (Nishi and Berg, 1981) before analysis. Freshly dissociated ciliary ganglion neurons were obtained from E13 to 14 embryos and were examined 1 to 5 h later as described previously (Liu and Berg, 1999b). Rat hippocampal cultures were prepared by dissociating E18/19 hippocampi and maintaining the cells in culture for 8 to 18 days as described previously (Liu et al., 2001).
Electrophysiology.
All experiments were performed at room temperature (20–23°C). Both perforated-patch and conventional whole-cell patch-clamp recordings were performed as described previously (Liu and Berg, 1999b, and references therein). The composition of the bathing solution and of the pipette solutions for perforated-patch and conventional patch-clamp recordings was as described previously (Liu and Berg, 1999b). Agonist (nicotine) with and without the indicated albumins was rapidly applied using a multibarrel rapid-exchange system that achieved fluid exchange in ≤3 ms (Zhang et al., 1994). A relatively large recording electrode was used (1–1.5 MΩ resistance with 80% series resistance compensation). The rapid application and low-resistance recording were required to observe the full extent of potentiation, given the rapid desensitization of the response. When compounds were to be applied for several minutes or more [e.g., albumins for several minutes, α-bungarotoxin (αBgt) for an hour], they were also added to the bath for the indicated times.
The procedures for single channel data acquisition and analysis have been described in detail previously (McNerney et al., 2000). Briefly, E14 ciliary ganglion neurons were dissociated and bathed in a standard recording solution (RS), containing 145.0 mM NaCl, 5.3 mM KCl, 5.4 mM CaCl2, 0.8 mM MgSO4, 5.6 mM glucose, and 5.0 mM HEPES acid pH 7.4, or in RS containing 75 μM bovine serum albumin (BSA) and termed RS+BSA. Outside-out membrane patches were excised from the neurons, held at −100 mV, and exposed to 0.5 μM nicotine dissolved in RS or RS+BSA by gentle pressure microperfusion (2–5 psi). The resulting currents were digitized at 50 kHz and filtered to a final cutoff frequency (fc) of 6.8 kHz (−3 dB), allowing resolution of channel openings with durations >98 μs [2 times the filter rise time (tr = 0.3321/fc); Colquhoun and Sigworth, 1995]. Single nAChR channel events were detected by visual inspection of transitions from the baseline current and manually selected such that only those events exceeding preset thresholds for amplitude (2 times noise) and duration (98 μs) were subsequently analyzed. For each recording, single channel currents were first compiled into amplitude histograms (0.2-pA bins) and fit to multiple Gaussian functions using a Simplex least-squares algorithm that minimizes the difference between actual and fit values (pSTAT; Axon Instruments, Foster City, CA). The sum of four Gaussian functions best fit the amplitude distributions, as determined by eye, and in seven cases, by comparing F statistic values obtained from fits conducted with four versus five or three functions. In each of these cases, the fits using four Gaussian components were significantly improved over those using three components (p < 0.005) and not significantly different from those using five components (p > 0.1). Because the individual functions overlapped somewhat, the range of each current class was determined by calculating the critical amplitudes (Ac ) between peaks in the distribution that minimized misclassified events from adjacent ranges, as described previously (Colquhoun and Sigworth, 1995).
To analyze BSA effects on nAChRs, the single channel conductance, steady-state open probability, open frequency, and open duration were compared for each current class, x. Single channel conductances (γx) were determined using reversal potential values determined previously from patches where full current-voltage plots were obtained (McNerney et al., 2000). The steady-state opening probability for each class (Popen,x) was assessed using %Popen,x = 100[Σt,x/(TLx )], where Σt,x was obtained from the summed open durations of channel class x. Trepresents the total record time (ca. 40 s in all cases), andLx represents the number of channels of that current class in the patch. Lx was estimated from the number of x value current levels visualized in a record and was usually 1 or 2. This method provides an upper limit estimate of %Popen,x becauseLx is likely to be an underestimate of the actual number of channels in the patch (Colquhoun and Hawkes, 1995). The open frequency for each class (Fopen,x) was determined usingFopen,x =Nx/(TLx ), where Nx represents the total number of events in that conductance class. Average channel open times (Topen,x) were estimated from the average durations of events in each class usingTopen,x = (Σt,x/Nx ) − tmin wheretmin is the minimum resolvable interval (100 μs). In patches where Nx exceeded 50, logarithmically binned histograms were constructed for the open durations of accepted events in each conductance class (Sigworth and Sine, 1987; Colquhoun and Sigworth, 1995) and fitted by maximum likelihood methods using Intrv5 [Interval Analysis 3.12 (1994), generously provided by Dr. Barry S. Pallotta, University of North Carolina, Chapel Hill, NC]. Such histograms revealed heterogeneous open duration distributions for the 25- and 40-pS events requiring up to three time constants [brief (τx,b ≤ 0.20 ms), intermediate (0.20 < τx,i ≤ 1.0 ms), and long (τx,l > 1.0 ms)] to optimize the fits. Because a detailed kinetic analysis was not possible for each patch, however, and long-duration events were relatively rare, %Popen,x,Fopen,x, andTopen,x were calculated using an arbitrary cutoff of 200 μs to separate brief from longer 25- and 40-pS events. All kinetic parameters are expressed as mean ± S.E.M., and the statistical significance (p < 0.05) of comparisons determined using Student's unpaired, two-tailedt test, conducted assuming equal or unequal variances where applicable.
Biochemical Procedures.
BSA was extracted with the organic solvents chloroform, methanol, or acetonitrile. BSA (1 g) was suspended in 50 ml of solvent, shaken vigorously for 2 h, centrifuged to collect the precipitate, and dried by a stream of nitrogen. The BSA was then resuspended in recording solution and tested. Dialysis of denatured BSA was done by dissolving BSA in 8 M urea to yield 10 mg/ml, adjusting the pH to 8.0 with sodium hydroxide, and then adding iodoacetic acid to a final concentration of 20 mM and incubating the solution at room temperature for 30 min to alkylate-free sulfhydryls. The BSA solution was then dialyzed against 500 ml of 8 M urea for 15 h followed by dialysis against 4 × 2 liters of recording solution for 24 h and then tested for activity. BSA at 10 mg/ml in recording solution was also treated with 10 mM dithiothreitol for 30 min at room temperature, alkylated with 20 mM iodoacetic acid, dialyzed for 2 h against recording solution, and then tested.
Protease treatments of BSA were conducted by reducing and alkylating BSA disulfides with dithiothreitol and iodoacetic acid as described above, and then incubating with 0.25 ml of α-chymotrypsin-agarose orN-tosyl-l-phenylalanine chloromethyl ketone-trypsin-agarose for 15 h at 37°C with shaking. The proteases were removed by centrifugation, and the BSA solutions were diluted with recording solution and tested. Reduction and alkylation were required to fully inactivate the BSA activity by the proteases. Pepsin digestion of BSA was conducted by dissolving BSA in 1% acetic acid to yield 10 mg/ml, adding 0.2 ml of pepsin-agarose, incubating 15 h at 37°C with shaking, removing the pepsin-agarose by centrifugation, lyophilizing the peptide solution, and resuspending in recording solution for testing. Controls for the pepsin digestion included either omission of the pepsin-agarose or conducting the experiment as described but adjusting the pH of the BSA solution to 7.0 before adding the pepsin-agarose.
Limited pepsin digestion of BSA and purification of peptide fragments were conducted as described previously (Feldhoff and Peters, 1975). The carboxyl terminal (amino acids 308–583) and amino terminal (amino acids 1–307) halves of BSA obtained from a 20-min pepsin digestion in the presence of octanoic acid were purified by a combination of ion exchange and size exclusion chromatography. Additional smaller peptides were purified by size-exclusion and reverse-phase chromatography. These included peptides corresponding to amino acids 308 to 386 and 506 to 583. Peptide identities were confirmed by mass determinations by electrospray ionization mass spectrometry.
Materials.
White Leghorn chick embryos were obtained locally and maintained at 37°C in a humidified incubator. αBgt was purchased from Biotoxins (St. Cloud, FL). β-Amyloid peptide1–42 (Aβ) was obtained from Calbiochem (La Jolla, CA) and prepared as described previously (Liu et al., 2001).N-tosyl-l-phenylalanine chloromethyl ketone-trypsin-agarose and pepsin agarose were purchased from Pierce Chemical (Rockford, IL). All other drugs, including ivermectin, BSA, other albumins, and α-chymotrypsin-agarose were purchased from Sigma-Aldrich (St. Louis, MO). GenBank accession numbers for albumin sequences analyzed were as follows: bovine, ABBOS; horse, ABHOS; sheep, ABSHS; dog, CAA76841; rabbit, P49065; cat, JC4660; pig, P08835; human, ABHUS; rat, P02770; mouse, CAA09617; and chicken, ABCHS.
Results
BSA-Mediated Enhancement of α7-nAChR Responses.
Typically α7-nAChRs produce rapidly activating and rapidly desensitizing currents (Zorumski et al., 1992; Alkondon and Albuquerque, 1993; Zhang et al., 1994), although some variants can produce a slowly desensitizing response (Cuevas and Berg, 1998; Yu and Role, 1998). Diagnostically, α7-nAChR responses can be blocked by nanomolar concentrations of either αBgt (Couturier et al., 1990) or methyllycaconitine (Alkondon et al., 1992).
Chick ciliary ganglion neurons in cell culture express substantial numbers of α7-nAChRs as measured with125I-αBgt binding and exhibit spontaneous excitatory synaptic potentials generated in part by α7-nAChR activation (Chen et al., 2001). Whole-cell patch-clamp recording from E8 ciliary ganglion neurons grown 1 week in culture, however, failed to detect significant nicotine-induced responses that could be attributed to α7-nAChRs (Fig. 1A). Unexpectedly, the inclusion of BSA in the perfusion solution allowed a large, rapidly activating and rapidly desensitizing nicotinic response to be seen. The response was not seen after treatment with αBgt (Fig. 1B), indicating that it was generated by α7-nAChRs (Fig. 1C).

BSA-mediated appearance of αBgt-sensitive nicotinic responses from ciliary ganglion neurons in cell culture. E8 ciliary ganglion neurons were maintained in dissociated cell culture for 5 days and then tested for whole-cell currents induced by 20 μM nicotine (Nic, horizontal line) in the absence (left) and presence (right) of 75 μM BSA (BSA, horizontal open bar) before (A) and after (B) treatment with 100 nM αBgt for 1 h. C, compiled results (n = 8–12 neurons/condition). Only in the presence of BSA can a rapidly activating, rapidly desensitizing αBgt-sensitive current characteristic of α7-nAChR responses be discerned. Holding potential, −60 mV.
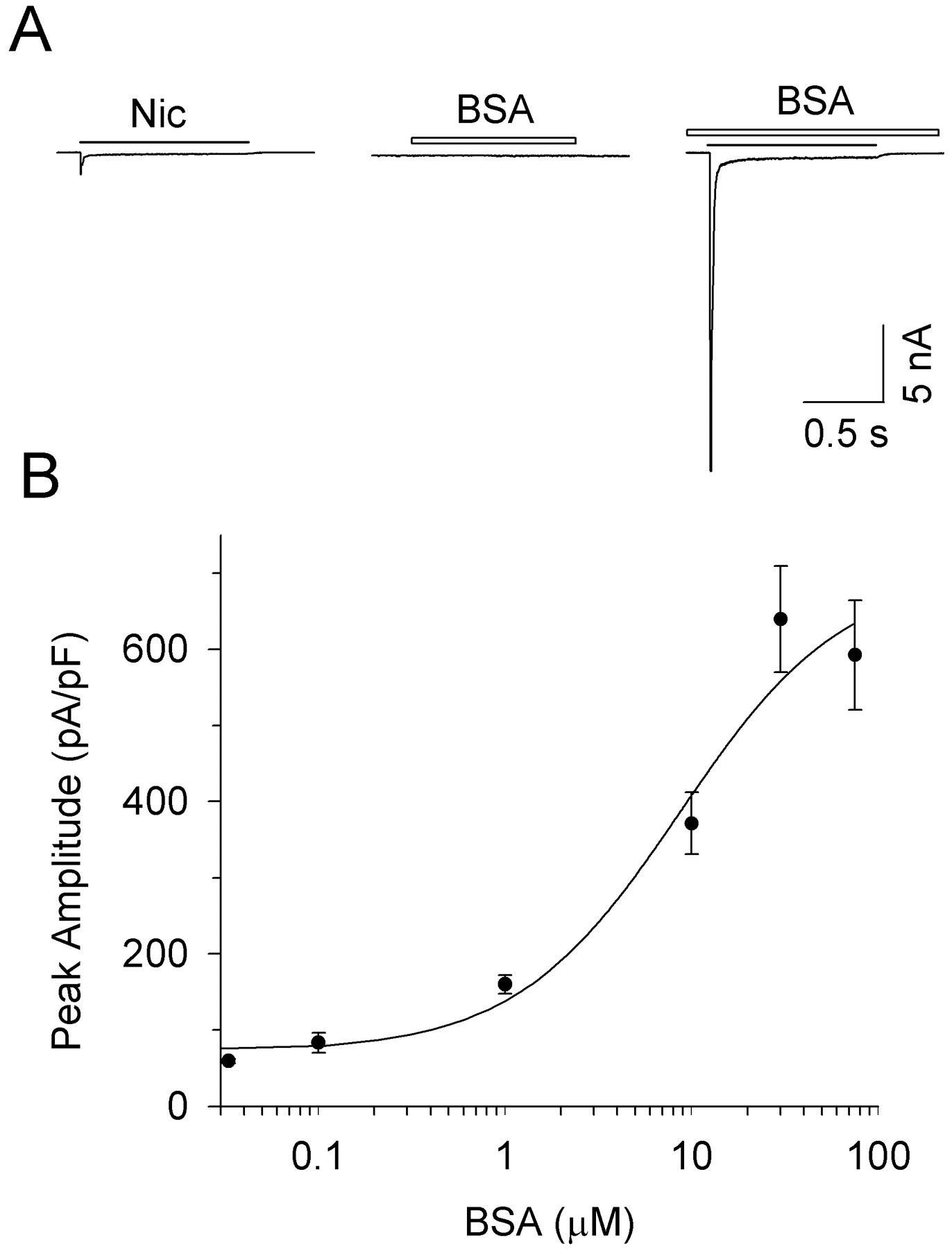
In contrast to ciliary ganglion neurons maintained in culture, freshly dissociated E13 neurons display a substantial α7-nAChR response when challenged with nicotine. Nonetheless, BSA had an enormously potentiating effect on the response. The effect was so great that the α7-nAChR currents could not be accurately measured when recorded in neurons voltage-clamped at −60 mV. At a holding potential of −20 mV, 75 μM BSA increased the peak nicotine-induced whole-cell current by an order of magnitude (Fig. 2A), and the increased response was blocked by αBgt as expected for α7-nAChRs (data not shown). The temporal profile of the response, including both rise and fall times, were similar to those of α7-nAChRs in untreated cells. Thus, at −20 mV, rise times (10–90% of peak) for control and BSA-treated neurons were 4.8 ± 0.3 and 5.4 ± 0.4 ms, respectively, whereas decay time constants for the large, rapidly decaying component of the response were 21.3 ± 2.8 and 15.4 ± 1.4 ms, respectively (mean ± S.E.M.; n = 10). The similarity in decay time suggests that the BSA-mediated potentiation of the α7-nAChR response is unlikely to result from a dramatic reduction in the rate of receptor desensitization.

BSA potentiation of α7-nAChR responses from freshly dissociated E13 ciliary ganglion neurons. A, whole-cell responses elicited from a neuron voltage-clamped at −20 mV and challenged with 20 μM nicotine (Nic, horizontal line) alone (left), 75 μM BSA (BSA, horizontal open bar) alone (middle), or both nicotine and BSA together (right) with the BSA application in this case being initiated before the nicotine. B, dose-response curve for BSA potentiation of the peak whole-cell nicotine-induced currents. Values represent the mean ± S.E.M. of 7 to 14 neurons for each concentration; the curve represents the best least-squares fit of the data.
Generating a dose-response curve for BSA potentiation of the α7-nAChR response indicated an EC50 value of about 10 μM (Fig. 2B). The BSA effect was completely reversible: less than 20 s was required for maximal potentiation and less than 20 s for return to control levels (Fig. 3, A and B). Reapplication of BSA reproduced the potentiation (data not shown). The potentiation seemed to include both an effect on agonist affinity and an effect on the maximal whole-cell response. Thus, in the presence of BSA, the dose-response curve for nicotine was shifted slightly to lower agonist concentrations but also reached significantly greater maximum values (Fig. 4). Interpreting changes in the dose-response curve for α7-nAChRs, however, is complicated by the fact that it is influenced by receptor desensitization, which can limit response amplitude and varies with agonist concentration (Zhang et al., 1994).

Time course of BSA potentiation and reversal. Mean peak responses were elicited from E13 ciliary ganglion neurons by 20 μM nicotine with the indicated exposure to 10 μM BSA. A multibarrel rapid applicator was used, permitting fluid exchange within 3 ms. A, time course of potentiation. B, time course of recovery after BSA removal. Values represent the mean ± S.E.M. of results from 8 to 12 neurons for each point; individual neurons were used for only a single determination.

Effects of BSA on the dose-response curve for nicotine. Neurons were tested for the peak whole-cell response induced by the indicated concentrations of nicotine before (●) and after (○) application of 10 μM BSA. Values represent the mean ± S.E.M. of results from 7 to 14 neurons for each condition; the curves represent the best least-squares fit of the data. BSA increased the affinity of the receptor for agonist as reflected in the lower EC50 value and increased the maximum whole-cell response elicited from the neurons.
Specificity of the Potentiation.
The potentiation preferentially affected α7-nAChRs and was produced by albumins obtained from several, but not all, species. Little potentiation was seen in either the non-α7 nicotinic response or the GABAA response in E13 ciliary ganglion neurons treated with BSA (Fig. 5A). BSA had no significant effect on either the AMPA- or NMDA-induced responses from rat hippocampal neurons, although it did potentiate α7-nAChR responses from the neurons as it did in chick ciliary ganglion neurons (Fig. 5B). Over half of the albumins tested on chick neurons produced substantial potentiation, whereas four, including chick and rat albumin, had no effect (Fig. 5C). Rat albumin also failed to potentiate significantly the rat hippocampal α7-nAChR response: peak values induced by 1 mM ACh in the presence of 10 μM rat albumin were 1.3 ± 0.2-fold that of control responses (mean ± S.E.M.;n = 8 cells). The results suggest that “competent” albumins either share a critical amino acid sequence or have an absorbed component necessary to produce the potentiation.

Receptor type and albumin specificity. A, effects of 10 μM BSA on E13 chick ciliary ganglion α7-nAChR, non-α7 nAChR, and GABAA receptor responses were assessed by measuring the whole-cell response either to 20 μM nicotine, or to nicotine after treatment with 100 nM αBgt, or to 20 μM GABA, respectively, and normalizing the results to control responses from the same neurons before BSA application. Individual neurons were tested for a single kind of receptor response ± BSA. B, effects of 10 μM BSA on the AMPA, NMDA, and α7-nAChR responses of rat hippocampal neurons in cell culture, elicited with 100 μM AMPA, 100 μM NMDA (+ 1 μM glycine in the absence of Mg2+), and 1 mM acetylcholine (in the presence of 0.5 μM atropine), respectively, and analyzed as in A for ciliary ganglion neurons. BSA significantly potentiated both ciliary ganglion and hippocampal α7-nAChR responses while having only a small effect on ciliary ganglion non-α7 nAChR and GABAAreceptor responses, and no significant effect on hippocampal AMPA or NMDA receptor responses. C, mean peak nicotine-induced response of E13 ciliary ganglion neurons was measured after treatment with 10 μM albumin from the indicated species, and the results were normalized to control responses from the same neurons. Seven albumins significantly potentiated the response, whereas four did not. Values represent the mean ± S.E.M. from 10 to 16 neurons per condition.
Mechanism of the Potentiation.
It is known that albumins can act as scavengers to absorb lipids and other components from blood (Peters, 1985). One family of candidates is ivermectin-like components that might have been present in the feed of some but not all animal species supplying the albumins; ivermectin has been reported to produce significant potentiation of heterologously expressed α7-nAChRs (Krause et al., 1998). Treating E13 ciliary ganglion neurons with 10 to 30 μM ivermectin for 2 to 5 min, however, had no effect on the peak α7-nAChR response (data not shown). Moreover, extracting BSA with a variety of organic solvents, including chloroform, methanol, and acetonitrile, to remove absorbed components, had no effect on the ability of the BSA to potentiate α7-nAChR responses (Fig.6A). Nor was the potentiation diminished when the BSA was partially denatured with either urea or dithiothreitol in the presence of iodoacetamide and then dialyzed to remove absorbed components (Fig. 6A). In contrast, extensive proteolysis to destroy the BSA protein backbone eliminated the potentiation. Pepsin, chymotrypsin, and trypsin each reduced the potentiating ability of BSA to background levels, whereas negative controls, e.g., pepsin incubation at a nonpermissive pH, had no effect (Fig. 6B). The results are most consistent with the BSA itself generating the potentiation, and using specific domains of the protein not common to albumins from all species.

Treatments of BSA to identify required features for potentiation. A, aliquots of BSA were extracted with the organic solvents methanol, chloroform, or acetonitrile (ACN), or reversibly denatured by dialysis against urea and iodoacetamide (urea/IAA), or reduced by treatment with DTT and then IAA (DTT/IAA) before dialysis into perfusion buffer and application to E13 ciliary ganglion neurons for testing. B, aliquots of BSA were proteolyzed with pepsin, chymotrypsin, or trypsin, and then, after enzyme removal or inactivation either by inhibitor or by pH adjustment, the samples were dialyzed or lyophilized and resuspended as described underMaterials and Methods and tested. (Pepsin is inactive at pH 7.) Values have been normalized to control responses and represent the mean ± S.E.M. for 6 to 14 neurons for each condition. Only protease treatment abolished the capacity of BSA to potentiate nicotine-induced peak responses.
Limited proteolytic cleavage of the BSA using pepsin as described previously (Feldhoff and Peters, 1975) yielded a 30-kDa carboxy terminal half of the protein that remained competent. The fragment was isolated by using a combination of size exclusion and ion exchange chromatography. At 10 μM, the fragment produced 560 ± 80% (mean ± S.E.M.; n = 9) potentiation of the peak nicotine-induced response. This was indistinguishable from potentiation by intact BSA (p > 0.5). Further proteolysis, followed by high-performance liquid chromatography fractionation to separate individual fragments, failed to identify active components. Either the proteases used (pepsin or trypsin) destroyed the active site, or protein conformation, rather than sequence alone, was important for the potentiation.
The BSA-mediated potentiation was not voltage-dependent. The ratio of peak nicotine-induced responses obtained at +40 mV to those seen at −20 mV in the same neurons under control conditions was 9.2 ± 1.2% (n = 5). The ratio seen in the presence of 10 μM BSA was identical: 9.2 ± 1.3% (n = 6).
The BSA effect was not mediated via common second messenger pathways such as those depending on intracellular calcium or G protein-coupled receptors. Dialyzing the interior of the neuron from the patch pipette either with BAPTA to chelate calcium or with a stable GDP analog had no effect on the BSA-induced potentiation (Fig.7). Neither did the compounds nor a stable GTP analog have any effect on control responses in the absence of BSA. Blockers of protein kinases A, C, and G also seemed to be without effect on the potentiation (Fig. 7). The results suggest a direct interaction between albumin and α7-nAChRs or possibly one mediated by surface components on the neurons; intracellular signaling cascades are not likely to be required.

Lack of second messenger involvement in BSA potentiation of α7-nAChR responses. E13 ciliary ganglion neurons were internally perfused 3 min via the patch pipette with 10 mM BAPTA to chelate calcium, 0.5 mM guanosine 5′-O-(2-thio)diphosphate (GDPβS) or guanosine 5′-O-(3-thio)triphosphate (GTPγS) to alter G protein-coupled pathways, and either 0.1 mM Rp-cAMP, H-7, or H-8 to inhibit predominantly protein kinases A, C, and G, respectively. The cells were then tested for nicotinic responses in the presence or absence of BSA, as indicated. Values have been normalized to control responses and represent the mean ± S.E.M. for 7 to 11 neurons for each condition. None of the treatments altered the ability of BSA to potentiate the response.
Properties of α7-nAChRs Affected by the Potentiation.
It has been suggested that only a small fraction of the α7-nAChRs present on chick ciliary ganglion neurons may be functionally available (McNerney et al., 2000). This assessment may require revision because it relied on αBgt binding to quantify α7-nAChRs; the stoichiometry of αBgt binding may be greater than previously thought, if the results obtained with soluble acetylcholine binding protein (Brejc et al., 2001) can be extended to α7-nAChRs. Nonetheless, it was important to consider the possibility that the potentiation of the whole-cell α7-nAChR response by BSA represented a recruitment of receptors from a previously “silent” pool to a pool that was now functionally available for nicotine-induced activation. This was tested by using an open channel blocker. We reasoned that if the potentiation represented silent receptors being recruited de novo by the BSA treatment, then previous exposure to agonist in the presence of an open channel blocker (while the receptors were in silent mode) would have no effect on the ability of BSA subsequently to produce a large nicotine-induced response. The protocol would require removing agonist and unbound open channel blocker before adding the BSA so that no blockade of newly recruited receptors would occur. Alternatively, if the open channel blocker was able to attenuate the BSA effect as it did control responses even though the blocker was only available before the BSA incubation, the results would suggest that the same receptor population produced both the control and potentiated whole-cell responses.
Chlorisondamine has been commonly used as an open channel blocker of nAChRs (Amador and Dani, 1995; Hicks et al., 2000). At 20 μM chlorisondamine quickly produced a dramatic reduction in the peak whole-cell nicotinic response (Fig. 8A), which is dominated by α7-nAChRs (Liu and Berg, 1999b). It also almost completely eliminated the slower non-α7 nAChR response. Agonist was applied for 1 s at 30-s intervals to allow full recovery from receptor desensitization between trials, and perforated patch-clamp recording was used to minimize rundown of the response during the experiment (Liu and Berg, 1999a). After even a few applications of agonist, the chlorisondamine reduced control responses by about 80% (Fig. 8B). The reduction was consistent with an open channel blockade because it did not occur if the chlorisondamine was applied continuously during the 29-s intervals between nicotine applications but excluded during the 1-s nicotine applications themselves (Fig. 8B).

Open channel blockers demonstrate that the BSA effect on α7-nAChRs depends on potentiation of previously competent receptors. A, nicotine (20 μM; horizontal line) was used to elicit responses from the same E13 ciliary ganglion neurons before (left), during (middle), and after (right) application of the open channel blocker chlorisondamine (20 μM, Chlor, horizontal open bar). BSA (horizontal filled bar) was applied during the nicotine tests only after removal of unbound chlorisondamine. Holding potential, −60 mV. B, combined results from neurons tested with nicotine at 30-s intervals before and during chlorisondamine treatment (20 μM; horizontal open bar), and after removal of chlorisondamine and application of BSA (horizontal filled bars). ○, 10 μM BSA; ▵, 75 μM BSA; ●, negative control in which chlorisondamine was applied only between but not during nicotine applications (i.e., chlorisondamine was present 29 out of every 30 s for each of the five trial periods). Results have been normalized to the values obtained from the same neuron at the outset and then averaged among neurons to obtain the mean ± S.E.M. (n = 6–7 except for the 0–4-min points for the BSA trials before BSA application where the results were pooled so that n = 13). The response seen in BSA after removal of the chlorisondamine is that expected for BSA-mediated potentiation of the residual response after open channel blockade; it is much less than expected for activation of a previously silent (and therefore chlorisondamine-resistant) α7-nAChRs.
After inducing open channel blockade with chlorisondamine, we removed unbound material and tested the nicotinic response subsequently in the presence of BSA. At 10 μM BSA potentiated the residual response about 3-fold (Fig. 8B). At 75 μM it produced a 6-fold potentiation (Fig.8B). Similar results were obtained when the experiment was performed with 100 μM nicotine as agonist rather than the usual 20 μM (5.1 ± 0.5-fold potentiation by 75 μM BSA; n = 5 neurons). The results are entirely consistent with the potentiation being exerted on the residual response, according to the dose-response curve of BSA (Fig. 2). Had the potentiation represented BSA recruitment of a previously inactive population that was not affected by the chlorisondamine treatment, the response in the presence of 10 μM BSA, for example, should have been 30-fold greater than the residual response, rather than the 3-fold observed. This was clearly not the case.
Because BSA did not seem to increase the number of functional α7-nAChRs, we conducted recordings from excised outside-out neuron patches to determine which functional single channel properties might be altered to account for the potentiation. Previous studies on chick ciliary ganglion neurons identified single nAChR channel events of 25, 40, 60, and 80 pS activated by 0.5 μM nicotine in excised outside-out neuron patches (Margiotta and Gurantz, 1989; McNerney et al., 2000). The 60- and 80-pS events were brief (<0.2 ms) and were blocked by αBgt, indicating that they probably resulted from activation of α7-nAChRs. The 25- and 40-pS events were of longer duration and not blocked by αBgt, suggesting they were produced by the non-α7 nAChRs on the neurons. Recent studies have further distinguished brief 25- and 40-pS events (<0.2 ms) that can be blocked by αBgt, marking them as additional candidates for α7-nAChR responses (J. F. Margiotta and Q. Nai, unpublished observations). BSA did not detectably change the amplitudes of the single channel events induced by 0.5 μM nicotine (Fig. 9, A and B), indicating no change in single channel conductance. In contrast, BSA treatment significantly increased the steady-state opening probability (%Popen) of both the 60- and 80-pS events with about 5-fold increases evident for both after a 10-min exposure to 75 μM BSA (Fig. 9C; see legend for %Popen values). This effect was accompanied by approximately 2- and 3-fold increases in the average open duration (Topen) and frequency of opening (Fopen), respectively (Fig.9D). The increase in Topen seems to result from a larger fraction of longer duration openings in patches from BSA-treated neurons, as shown for the 60-pS class of events (Fig.9, E and F). Similar results were obtained for the 80-pS events (data not shown). No single channel events were seen during comparable recording times in the absence of nicotine.

Single nAChR channel events modulated by BSA. Outside-out patches excised from 10 control and 10 BSA-treated (75 μM, 10 min) ciliary ganglion neurons were held at −100 mV and perfused with 0.5 μM nicotine. A, amplitude histogram and example records (inset) for single nAChR channel current events recorded in a patch from a control neuron. Modal values for the four current amplitude ranges (1–4) are indicated, corresponding to single channel conductances of 25.1, 39.8, 63.8, and 82.2 pS. In 10 such control patches, mean single channel conductances were 25.6 ± 0.7, 41.9 ± 0.5, 62.7 ± 0.7, and 82.3 ± 1.0 pS for channel classes 1–4, respectively. B, amplitude histogram and example records (inset) for single channel current events recorded in a patch from a BSA-treated neuron. Modal values for the four current amplitude ranges (1–4) corresponded to single channel conductances of 26.2, 42.6, 60.6, and 83.6 pS. In 10 patches from BSA-treated neurons mean single channel conductances were 26.8 ± 0.4, 42.0 ± 0.4, 61.9 ± 0.5, and 83.8 ± 0.6 pS for the four channel classes, respectively; the values were not significantly different from control neurons (p > 0.1). Note, however, that the 60- and 80-pS events were more evident and made a larger contribution to amplitude histograms in patches from BSA-treated neurons. Calibrations in insets indicate 4 pA and 2 ms. Histograms were compiled from a total of 450 accepted events in A and 694 in B. C, BSA treatment resulted in 5-fold higher %Popen values for 60- and 80-pS channel events, all of which have characteristically brief open durations. The results are presented as a percent of controls; actual values were %Popen,60 = 0.008 ± 0.002 and %Popen,80 = 0.005 ± 0.001 for control patches and %Popen,60 = 0.036 ± 0.009 and %Popen,80 = 0.027 ± 0.009 for BSA-treated patches. The effect of BSA was specific because it failed to detectably change %Popen(p > 0.05) for any of the other event categories. In control patches these values were %Popen,25[infi]b = 0.026 ± 0.008, %Popen,25[infi]l = 0.015 ± 0.004, %Popen,40[infi]b = 0.012 ± 0.003, and %Popen,40[infi]l = 0.191 ± 0.068. D, ability of BSA to increase %Popenfor the 60- and 80-pS events was accompanied by an increase in their opening frequency and average open duration (Fopen and Topen; see Materials and Methods for details). Actual values were Fopen,60 = 0.564 ± 0.148 Hz,Topen,60 = 47 ± 5 μs,Fopen,80 = 0.281 ± 0.087 Hz, andTopen,80 = 84 ± 11 μs for control patches. For BSA-treated patchesFopen,60 = 1.820 ± 0.446 Hz,Topen,60 = 103 ± 22 μs,Fopen,80 = 1.066 ± 0.313 Hz, andTopen,80 = 164 ± 35 μs. Asterisks in C and D indicate p < 0.05, by Student's t test. E and F, consistent with the change in Topen, BSA treatments caused the appearance of αBgt-sensitive channels having longer open durations. The 60-pS channel open durations were logarithmically binned (tmin = 100 μs) and fitted using a maximum likelihood method (see Materials and Methods) in two control and three BSA-treated patches. For the control patch depicted in E, a single component (solid line, τ60,[infi]b = 60 μs; 60 events) adequately fit the open duration distribution. For the BSA-treated patch depicted in F, two components (solid lines, sum is topmost) were required for adequate fitting (τ60,[infi]b = 68 μs and τ60,[infi]i = 284 μs; 193 events). The 80-pS channel showed similar changes (data not shown).
The ability of BSA to increase %Popenwas specific for αBgt-sensitive events in the sense that the treatment had no significant effect on the long duration 25- and 40-pS events attributable to non-α7 nAChRs in the same patches (Fig. 9C). The fact that BSA also had no significant effect, however, on the short duration, αBgt-sensitive 25- and 40-pS events (Fig. 9C), suggests that these latter events may be produced by a distinct subclass of α7-nAChRs or a different class of αBgt-sensitive receptors. About 5% of the αBgt binding sites in ciliary ganglia are associated with receptors lacking any known gene product (Pugh et al., 1995); their cellular source and functional responses remain unknown, but they may contribute to some of the single channel events observed. Overall, the single channel results are consistent with the whole-cell recordings presented above, and suggest that the BSA-dependent potentiation of α7-nAChR responses is caused by increases in %Popen,Fopen, andTopen for two prominent classes of channel events.
BSA-Mediated Compensation for Inhibition of α7-nAChRs by Aβ.
Aβ specifically and potently inhibits α7-nAChR function by a noncompetitive mechanism, suggesting the possibility that the receptors represent an important early molecular victim of Alzheimer's disease (Liu et al., 2001; Pettit et al., 2001). The ability to potentiate α7-nAChR function could provide a mechanism for partially overcoming such inhibition and helping to alleviate symptoms of the disease involving the receptors. We evaluated this by examining the inhibition of α7-nAChRs by Aβ in the presence and absence of BSA. Aβ at 100 nM produced a substantial inhibition of the whole-cell α7-nAChR response in E13 ciliary ganglion neurons as reported previously. Coapplication of BSA at a concentration near the EC50 value for potentiation returned the response amplitude to near control levels even in the continued presence of Aβ (Fig. 10). A higher concentration of 75 μM BSA produced a much larger response (Fig. 10), consistent with the concentration dependence of the BSA effect on control responses (Fig. 2). Thus, BSA-mediated potentiation can, in effect, compensate for inhibition by Aβ even though the latter was previously shown to be noncompetitive with respect to agonist (Liu et al., 2001).

BSA-mediated potentiation of α7-nAChR responses can compensate for inhibition of the receptors by Aβ. A, whole-cell nicotine-induced responses were recorded from E13 ciliary ganglion neurons before incubation with 100 nM Aβ (left), during incubation with the peptide (middle), and after continued incubation with the peptide plus the addition of 10 μM BSA (right). Nicotine, horizontal line; Aβ, horizontal open bar; Aβ + BSA, horizontal filled bar. Holding potential, −60 mV. B, combined results normalized for the response seen in the same neurons before application of Aβ (n = 7). Potentiation of the residual response by 10 μM BSA completely compensated for the Aβ blockade; potentiation by 75 μM BSA produced an even greater response.
Discussion
The results demonstrate that native α7-nAChRs can be significantly potentiated, and that the potentiation can produce up to an order of magnitude increase in the whole-cell response. The potentiation includes an increase in agonist affinity and an increase in the maximum amplitude of the response. The mechanistic basis for the potentiation seems to be an increase in the steady-state channel opening probability produced both by an increased frequency of channel opening and an increased mean channel open time. No change is seen in single current amplitude, and no evidence suggests the potentiation depends on the recruitment of a previously silent pool of receptors. No change is seen in the temporal profile of the whole-cell response that could account for the potentiation.
The ability of certain albumins to produce the potentiation seems to depend on information contained in the protein backbone rather than on absorbed components. Neither extensive extraction with organic solvents nor unfolding followed by exhaustive dialysis diminished the ability of BSA to induce the potentiation. Proteolysis, on the other hand, completely abolished it. The finding that the carboxyl half of BSA was sufficient to produce the potentiation focused attention on the amino acid sequences therein. A comparison of the active albumins with those of the inactive ones indicated six distinct sites at which the two groups diverged by at least one amino acid. Examining the three-dimensional X-ray crystallographic structure of human serum albumin indicated that five of the six sites were likely to reside on the surface of the protein where they could interact with other molecules (Curry et al., 1998). One or more of these, possibly in a conformation-dependent manner, may be responsible for causing the potentiation of α7-nAChR responses.
The properties of the potentiation are consistent with the albumins acting as allosteric modulators of the α7-nAChR. The modulation may result from a direct interaction between the albumin and the receptor. This is suggested by the finding that the potentiation was rapid, reversible, and reproducible, and did not depend on any of a variety of common second messenger systems. Moreover, the albumin was effective at sustaining the potentiation when applied to excised outside-out patches. We cannot exclude the possibility, however, that the effect is indirect, e.g., that the albumin reversibly binds a negative modulator tethered in the vicinity of the receptor. The potentiation was most pronounced for α7-nAChRs, although a small potentiation could be seen for certain other ionotropic receptors.
A previous study examined albumin effects on nicotinic receptors and concluded that non-α7 nAChR responses were potentiated in ciliary ganglion neurons (Gurantz et al., 1993). The techniques used then would not have revealed the rapidly decaying α7-nAChR responses normally displayed by the neurons. The present results find little, if any, albumin effect on ciliary ganglion non-α7 nAChRs and suggest instead that the potentiated response seen previously actually arose from enhanced α7-nAChR currents persisting sufficiently long as to override the much smaller non-α7 responses. This possibility was not tested at the time because α7-nAChRs had not yet been shown to represent functional ligand-gated ion channels. In other respects the present results are consistent with the previous study, including the lack of changes seen in non-α7 nAChR single channel properties, the small enhancement of GABAA receptor responses by BSA, and the fact that some albumins are effective, whereas others are not. An additional point worth comment was a finding in the previous study that cAMP produced a small enhancement of the whole-cell nicotinic response and that the enhancement was nonadditive with that produced by BSA (Gurantz et al., 1993). We found no requirement for second messengers in the potentiation of α7-nAChR responses by BSA, although a cAMP-dependent process can increase to some extent the α7-nAChR response (Pardi and Margiotta, 1999). The simplest explanation is that the BSA effect on α7-nAChRs is independent from cAMP pathways and either obscures their effects in this case or converges on a common target.
Three components that increase α7-nAChR responses have been reported previously. Ivermectin was found to produce a large enhancement of the nicotinic responses generated by heterologously expressed α7-nAChRs if supplied in advance of agonist (Krause et al., 1998). No effect of ivermectin was seen in the present experiments, however, with native α7-nAChRs on chick ciliary ganglion neurons. Either the source of ivermectin and associated minor components is critical for the effect, or the source of α7-nAChRs determines the outcome. Another known modulator of α7-nAChRs is prostaglandin E2, which increases the opening probability and open duration of 23-pS single channel events attributed to the receptors on chick sympathetic neurons (Du and Role, 2001). Whole-cell currents generated by such receptors have relatively slow kinetics of activation and desensitization, different from the α7-nAChRs described here. The prostaglandin E2 effect was thought to be indirect, possibly relying on calcium as a second messenger (Du and Role, 2001). A third modulator is PACAP, a neuropeptide endogenous in the ciliary ganglion. PACAP acts through adenylate cyclase and protein kinase A to enhance the responses of both α7- and non-α7 nAChRs on the neurons (Pardi and Margiotta, 1999). Neither prostaglandin E2 nor PACAP, however, produced the dramatic potentiation of α7-nAChR responses seen here with BSA.
The significance of the present findings is 3-fold. First, they indicate that the response of α7-nAChRs can be dramatically potentiated by increasing the steady-state opening probability of the receptors. The previously reported low %Popen values associated with both the 60- and 80-pS channel events of ciliary ganglion α7-nAChRs (McNerney et al., 2000) are not, therefore, inherently rigid features of the receptors but rather are subject to allosteric modulation. The site of interaction and the mechanism accounting for the potentiation will be subjects of considerable interest for future analysis.
The second important aspect of the findings is the recognition that a site exists on native α7-nAChRs capable of dramatic modulation. This finding raises the possibility that endogenous compounds, possibly neuropeptides but conceivably compounds of completely different composition, exploit the site in vivo during normal physiological function. Blood levels of albumin are typically about 40 mg/ml but can be 200-fold less in cerebral spinal fluid (Curry et al., 1998). Although even this reduced level could have some effect, we think albumins are not likely to be the normal ligand acting at such sites. One reason for thinking so is that the cognate albumin (e.g., chicken albumin and chick α7-nAChRs) was ineffective. Identification of endogenous compounds exerting the modulation and elucidation of the physiological significance could have profound implications for nicotinic signaling in the nervous system.
One candidate modulator is lynx1, an endogenous prototoxin that has recently been shown to complex with α7-nAChRs when coexpressed in transfected cells (Ibanez-Tallon et al., 2002). Lynx1 has multiple effects on the response of α4β2-nAChRs, increasing the proportion of large current events but also increasing the EC50 value for agonist and the rate and extent of receptor desensitization (Ibanez-Tallon et al., 2002). The overall effect of lynx1 on α4β2-nAChRs is very different from the dramatic potentiation of α7-nAChRs seen here with select albumins. How lynx1 or related family members might affect α7-nAChR function is not yet known.
Last, the demonstration that α7-nAChR function can be potentiated in a way that compensates for receptor blockade by the Aβ peptide, encourages speculation that the receptor may be a useful target for therapeutic strategies aiming at augmenting nicotinic signaling. The hope would be that knowledge about the modulatory site would permit the design of nonpeptide molecules capable of accessing and modulating the receptors in situ. This could be germane to neuropathologies such as Alzheimer's disease (Liu et al., 2001; Pettit et al., 2001) and neurodisorders such as schizophrenia (Leonard et al., 2000).
Acknowledgments
We thank Dr. Russell F. Doolittle (University of California, San Diego) for advice on albumin structure and protein biochemistry.
Footnotes
-
This study was supported by Tobacco-Related Disease Research Program Grants 9RT-0058 (to W.G.C.) and 9RT-0221, and by National Institutes of Health Grants NS12601, NS35469 (to D.K.B.), and NS24417 (to J.F.M.). Q.-S.L. is an American Heart Association Postdoctoral Fellow.
-
W.G.C. and Q.-S.L. contributed equally to this work.
- Abbreviations:
- nAChR
- nicotinic acetylcholine receptor
- NMDA
- N-methyl-d-aspartate
- PACAP
- pituitary adenylyl cyclase-activating protein
- E
- embryonic day
- αBgt
- α-bungarotoxin
- RS
- standard recording solution
- BSA
- bovine serum albumin
- Aβ
- β-amyloid peptide1–42
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- Received September 6, 2002.
- Accepted October 21, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}