


Abstract
One of the important targets of dopamine D4 receptors in prefrontal cortex (PFC) is the multifunctional Ca2+/calmodulindependent protein kinase II (CaMKII). In the present study, we investigated the effect of D4 receptor activation on subcellular localization of CaMKII. We found that activation of D4 receptors, but not D2 receptors, induced a rapid translocation of α-CaMKII from cytosol to postsynaptic sites in cultured PFC neurons. Activated CaMKII (Thr286 phospho-CaMKII) was also redistributed to postsynaptic sites after D4 receptor stimulation. The translocation was blocked by inhibiting the phospholipase C/inositol 1,4,5-trisphosphate receptor/Ca2+ signaling. Point mutation of the calmodulin binding site (Ala302), but not the autophosphorylation site (Thr286), of α-CaMKII prevented the D4-induced CaMKII translocation. Moreover, D4 receptors failed to induce CaMKII translocation in the presence of an actin stabilizer, and D4 activation reduced the binding of CaMKII to F-actin. Concomitant with the synaptic accumulation of α-CaMKII in response to D4 receptor activation, a D4-induced increase in the CaMKII phosphorylation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor glutamate receptor 1 (GluR1) subunits and the amplitude of AMPA receptor-mediated excitatory postsynaptic currents was also observed. Thus, our results show that D4 receptor activation induces the synaptic translocation of CaMKII through a mechanism involving Ca2+/calmodulin and F-actin, which facilitates the regulation of synaptic targets of CaMKII, such as AMPA receptors.
Dopaminergic inputs to prefrontal cortex (PFC) are believed to play important roles in many physiological functions, such as working memory formation (Brozoski et al., 1979; Goldman-Rakic, 1995; Marie and Defer, 2003). Dysfunction of the dopaminergic system in PFC is considered to be a significant contributor to the pathophysiology of a variety of disorders, including schizophrenia (Grace, 1991; Goldman-Rakic, 1994; Andreasen et al., 1997; Lewis and Lieberman, 2000; Carlsson et al., 2001). The dopamine D4 receptor, a member of the D2 family of receptors, is highly expressed in PFC (Mrzljak et al., 1996; Wedzony et al., 2000). Several lines of evidence have suggested the involvement of D4 receptors in normal PFC functioning and neuropsychiatric disorders (Rubinstein et al., 1997; Dulawa et al., 1999; Oak et al., 2000). The expression of D4 receptors is elevated in PFC of patients with schizophrenia (Seeman et al., 1993). Some antipsychotic drugs have high affinities to D4 receptors (Van Tol et al., 1991; Kapur and Remington, 2001). D4 receptor antagonists are effective in ameliorating cognitive deficits caused by the psychotomimetic drug phencyclidine (Jentsch et al., 1997, 1999). However, the cellular mechanisms underlying these D4 actions have yet to be elucidated.
Our recent studies have shown that one of the important targets of dopamine D4 receptors in PFC is CaMKII (Wang et al., 2003; Gu and Yan, 2004). In PFC neurons with low activity, D4 receptor stimulation increases CaMKII activity through a PLC/IP3R-dependent pathway, but not the classic Gi/o and PKA signaling pathway, whereas D2 receptors fail to increase CaMKII activity (Gu and Yan, 2004). Thus, activation of CaMKII can be one of the features to distinguish D4 receptor signaling from D2 receptor signaling.
CaMKII is a multifunctional kinase highly enriched in neurons. It is activated by elevated intracellular Ca2+, which triggers calmodulin binding to CaMKII at Ala302. CaMKII is autophosphorylated at Thr286 when the enzyme is activated, leading to the appearance of a sustained, Ca2+-independent activity (Miller and Kennedy, 1986). Many signals can elevate intracellular Ca2+ through different pathways, including extracellular Ca2+ influx through voltage-gated Ca2+ channels and NMDA receptor channels, or Ca2+ release from intracellular stores through activation of Gq-coupled receptors. Upon activation, CaMKII phosphorylates dozens of substrates throughout the whole cell (Braun and Schulman, 1995). Like other kinases with broad substrate selectivity, CaMKII achieves the efficacy and specificity of signal transduction via compartmentalized localization (Kennedy, 2000; Hudmon and Schulman, 2002). CaMKII located in postsynaptic sites has been found to play a crucial role in synaptic plasticity, which is integral for learning and memory, through the regulation of several postsynaptic proteins, such as postsynaptic AMPA receptors and NMDA receptors (Silva et al., 1992; Malenka and Nicoll, 1999; Frankland et al., 2001; Soderling et al., 2001).
Because targeting CaMKII to certain subcellular compartments is crucial for the efficacy and specificity of signals in response to various stimuli, we examined whether D4 receptor activation in PFC neurons could change the subcellular localization of CaMKII. We found that D4 receptor stimulation induced a synaptic translocation of CaMKII, and this event was paralleled by a significant increase in the CaMKII phosphorylation of AMPA receptors and the postsynaptic AMPA response after D4 receptor activation.
Materials and Methods
Primary Neuronal Culture. Rat PFC cultures were prepared as described previously (Wang et al., 2003). In brief, PFC was dissected from embryonic day 18 rat embryos and cells were dissociated using trypsin and trituration through a Pasteur pipette. The neurons were plated on coverslips coated with poly-l-lysine in Dulbecco's modified Eagle's medium with 10% fetal calf serum at a density of 100,000 cells/cm2. When neurons attached to the coverslip within 24 h, the medium was changed to Neurobasal medium with B27 supplement (Invitrogen, Carlsbad, CA). Neurons were maintained in the same kind of media for 2 to 3 weeks. Cultured neurons were treated with various agents for the durations as indicated in texts before fixation and immunostaining.
Immunocytochemistry. After treatment, cultured PFC neurons were fixed in 4% paraformaldehyde in PBS for 20 min and were permeabilized with 0.2% Triton X-100 for 5 min. After 1 h of incubation with 5% bovine serum albumin to block nonspecific staining, the cells were incubated with the polyclonal α-CaMKII antibody (1:100; Upstate Biotechnology, Lake Placid, NY) and monoclonal 95-kDa postsynaptic density protein (PSD-95) antibody (1:100; Affinity Bioreagents, Golden, CO) at 4°C overnight. In some experiments, the cells were incubated with the polyclonal Thr286-p-α-CaMKII antibody (1:100; Santa Cruz Biotechnology, Santa Cruz) and monoclonal PSD-95 antibody at 4°C overnight. Otherwise, cells were incubated with the polyclonal β-CaMKII antibody (1:100; Zymed Laboratories Inc., South San Francisco, CA). After washing, the cells were incubated with a fluorescein- and rhodamine-conjugated secondary antibody (1:200; Sigma, St. Louis, MO) for 60 min at room temperature. After three washes in PBS, the coverslips were mounted on slides with VECTASHIELD mounting media (Vector Laboratories, Inc., Burlingame, CA). Fluorescent images were obtained using a 100× objective with a cooled charge-coupled device camera mounted on a Nikon microscope, and analyzed with the ImageJ software (http://rsb.info.nih.gov/ij/). All specimens were imaged under identical conditions and analyzed using identical parameters. To define CaMKII, p-CaMKII, and PSD-95 clusters, a single threshold was chosen manually and corresponded to two times the average intensity of fluorescence in the dendritic shaft. Any CaMKII or p-CaMKII cluster that overlapped with a PSD-95 cluster was defined as synaptic. Three to four independent experiments for each of the treatments were performed. On each coverslip, four to six neurons were chosen and quantified. For each neuron, three to four neurites (50 μm each) were measured. Quantitative analyses were conducted blindly (without knowledge of experimental treatment).
Receptor ligands PD168077 maleate, L-745870 trihydrochloride (Tocris, Ballwin, MO), quinpirole, dopamine, SCH23390, sulpiride, α-methyl-5-hydroxytryptamine (α-Me-5HT), carbachol (Sigma), as well as second messenger reagents cpt-cAMP, PKI14–22, U73122, genistein, Bis-indolyl-maleimide I (Bis1), KN-62, KN-93, 2-aminoethoxydiphenylborane (2APB) (Calbiochem, San Diego, CA), and actin/microtubule agents phalloidin-oleate and paclitaxel (Taxol; Sigma) were made up as concentrated stocks and stored at -20°C. The final dimethyl sulfoxide concentration in all applied solutions was no more than 0.1%. Stocks were thawed and diluted immediately before use.
Western Blot and Coimmunoprecipitation. After treatment with different agents, equal amounts of protein from PFC culture homogenates (in 1% SDS lysis buffer) were separated on 7.5% acrylamide gels and transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat dry milk for 1 h at room temperature. Then the blots were incubated with the anti-pS831-GluR1 (1:500; Upstate Biotechnology) or anti-GluR1 (1:1000; Upstate Biotechnology) for 1 h at room temperature. After being rinsed, the blots were incubated with horseradish peroxidase-conjugated anti-rabbit antibodies (1:1000; Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) for 1 h at room temperature. After three washes, the blots were exposed to the enhanced chemiluminescence substrate. Quantification was obtained from densitometric measurements of immunoreactive bands on films using NIH Image software (http://rsb.info.nih.gov/nih-image).
For coimmunoprecipitation experiments, PFC slices (400 μm) from 3-week-old rats were used. Slices were prepared as described previously (Gu et al., 2003; Gu and Yan, 2004). After treatment with indicated agents, each slice was collected and homogenized in 1 ml of lysis buffer (1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, 50 mM NaPO4, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 1 mg/ml leupeptin). Lysates were centrifuged at 4°C at 16,000g for 30 min. Supernatant fractions were incubated with an anti-actin antibody (Santa Cruz Biotechnology) for 1 h at 4°C, followed by incubation with 50 μl of protein A/G plus agarose (Santa Cruz Biotechnology) for 1 h at 4°C. Immunoprecipitates were washed three times with lysis buffer containing 0.2 M NaCl, boiled in 2× SDS loading buffer for 5 min, and separated on 7.5% SDS-polyacrylamide gels.
CaMKII α Subunit Cloning and Site Mutation. CaMKII α subunit cDNA (GenBank accession number NM012920) was amplified by reverse transcription-PCR from rat brain total RNA. The primers used for PCR were 5′-gcgaattctgccaggatggctaccatcacc-3′ and 5′-gcggatccctggcctggtccttcaatgg-3′. The PCR product was then cloned to PCR2.1-TOPO vector (Invitrogen) and further subcloned to pEGFP-C1 vector (Clontech, Mountain View, CA). Point mutants were made with a site mutation kit (Stratagene, La Jolla, CA) using the primers 5′-catgcacagacaggaggccgtggactgcc-3′ (for Thr286 to Ala mutation) and 5′-ggaaactgaagggacgcatcctcaccac tatgc-3′ (for Ala302 to Arg mutation). Transfection of GFP-fused CaMKII constructs to primary PFC cultures was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Neurons were used 2 days after transfection.
Synaptic Current Recording in Cultures. The recording of AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs) in PFC cultures was similar to that described previously (Cai et al., 2002). The internal solution consisted of 130 mM Cs-methanesulfonate, 10 mM CsCl, 4 mM NaCl, 10 mM HEPES, 1 mM MgCl2, 5 mM EGTA, 2.2 mM QX-314, 12 mM phosphocreatine, 5 mM MgATP, 0.5 mM Na2GTP, 0.1 mM leupeptin, pH 7.2 to 7.3, 265 to 270 mOsM. The external solution consisted of 127 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 12 mM glucose, 10 mM HEPES, 0.001 mM TTX, 0.005 mM bicuculline, pH 7.3 to 7.4, 300 to 305 mosM. d-2-Amino-5-phosphonovalerate (20 μM) was added to block the NMDA receptor-mediated component of mEPSCs. The membrane potential was held at -70 mV. The AMPA receptor mEPSCs in the absence or presence of agonists were compared. Before recording, cultured neurons were treated with TTX (1 μM) for 24 h to lower the basal activity. Synaptic currents were analyzed with Mini Analysis Program (Synaptosoft, Leonia, NJ). Statistical comparisons of the amplitude and frequency of synaptic currents (mean ± S.E.M.) were made using the Kolmogorov-Smirnov (K-S) test.
Results
Induction of CaMKII Postsynaptic Translocation by D4 Receptors in Cultured PFC Neurons. We assessed the effect of D4 receptor activation on the subcellular localization of CaMKII in cultured PFC neurons using immunocytochemical approaches. As shown in Fig. 1A, in the untreated cells (control), CaMKII was mainly distributed uniformly throughout dendritic shafts, similar to what was found in cultured hippocampal neurons (Fong et al., 2002). In the cells treated with PD168077, a selective D4 receptor agonist (Glase et al., 1997; Wang et al., 2003), CaMKII exhibited enrichment in punctate structures along dendritic processes. Counting CaMKII puncta along dendrites (Fig. 1B) showed that the translocation occurred at 2 min (18.3 ± 3.9 clusters/100 μm versus 7.2 ± 1.5 clusters/100 μm in untreated neurons; p < 0.05, ANOVA), peaked at 5 min (43.5 ± 7.5 clusters/100 μm, p < 0.001, ANOVA), lasted for approximately 30 min (17.1 ± 3.8 clusters/100 μm, p < 0.05, ANOVA), and recovered at approximately 60 min (8.9 ± 2.2 clusters/100 μm).

PD168077 induced α-CaMKII clustering along dendrites of cultured PFC neurons in a time-dependent manner. A, immunocytochemical images of α-CaMKII in cultured PFC pyramidal neurons treated with or without PD168077 (20 μM) for different durations. Diffused distribution of CaMKII staining was shown in untreated neurons. CaMKII clusters were formed 2 min after PD168077 application, peaked at 5 min, and returned to basal level at 60 min. Enlarged versions of the boxed regions of dendrites are shown beneath each of the images. B, quantitative analysis of α-CaMKII clusters along dendrites under different treatment. *, p < 0.001 versus control; **, p < 0.05 versus control, ANOVA.
To further investigate the localization of CaMKII puncta after PD168077 treatment, we costained CaMKII with the postsynaptic scaffolding protein PSD-95 (a marker for postsynaptic synapses). In contrast to the translocation of CaMKII, the distribution of PSD-95 was not altered by PD168077 treatment (Fig. 2A). As illustrated on the merged pictures, the punctate distribution of CaMKII after PD168077 treatment showed a marked colocalization with PSD-95, suggesting that CaMKII was translocated to postsynaptic sites in response to D4 receptor activation. Quantification (Fig. 2B) revealed a significant increase in synaptic CaMKII clusters along dendrites after activating D4 receptors (control, 5.3 ± 1.5 clusters/100 μm; PD168077, 35.5 ± 6.4 clusters/100 μm; p < 0.001, ANOVA; n = 24 neurons per group from six cultures), whereas the density of synaptic PSD-95 clusters along dendrites was unchanged by D4 receptor activation (control, 52.9 ± 8.5 clusters/100 μm; PD168077, 54.8 ± 8.7 clusters/100 μm; n = 10 neurons per group from three cultures).

PD168077-induced CaMKII clusters were largely colocalized with PSD-95 in cultured PFC neurons, and autophosphorylated CaMKII (pT286-CaMKII) was also translocated to synapses by PD168077 treatment. A, images of cultured PFC pyramidal neurons coimmunostained with antibodies against α-CaMKII (green) and PSD-95 (red). Before staining, cultured neurons were either untreated (top) or treated with PD168077 (bottom, 20 μM, 5 min). The merged pictures showed great colocalization of CaMKII clusters and PSD-95 after PD168077 treatment. B, quantitative analysis of CaMKII clusters, PSD-95 clusters and colocalized CaMKII and PSD-95 clusters along dendrites with or without PD168077 treatment. *, p < 0.001 versus control, ANOVA. C, images of cultured PFC pyramidal neurons coimmunostained with antibodies against pT286-CaMKII (green) and PSD-95 (red). Similar to total CaMKII, pT286-CaMKII was clustered after application of PD168077 (5 min). The clusters of pT286-CaMKII were also colocalized with PSD-95. D, quantitative analysis of p-CaMKII clusters and colocalized p-CaMKII and PSD-95 clusters along dendrites with or without PD168077 treatment. *, p < 0.001 versus control, ANOVA.

D4 receptor, but not D2 receptor, activation induced CaMKII synaptic translocation. A, immunocytochemical images of α-CaMKII in cultured PFC pyramidal neurons treated with PD168077 (20 μM), PD168077 plus the D4 receptor antagonist L-745870 (20 μM), quinpirole (20 μM), or dopamine (50 μM) in the presence of the D1/D5 antagonist SCH23390 (10 μM) and D2/D3 antagonist sulpiride (10 μM). B, quantitative analysis of CaMKII synaptic clusters along dendrites under different treatment. *, p < 0.001 versus control, ANOVA.
We then examined whether D4 receptor activation triggered the synaptic targeting of activated CaMKII (Thr286-phosphorylated) in cultured PFC neurons. As shown in Fig. 2, C and D, in the untreated cell, p-CaMKII was distributed almost evenly on dendritic arbors. In the PD168077-treated cell, p-CaMKII exhibited a significantly increased clustering at synaptic sites, as indicated by the enhanced colocalization with PSD-95 (synaptic p-CaMKII clusters/100-μm dendrite: 6.2 ± 1.7 in controls, 37.3 ± 6.7 in PD168077-treated neurons; p < 0.001, ANOVA; n = 22 neurons per group from four cultures). These results suggest that activated CaMKII, like CaMKII, was redistributed to postsynaptic sites by D4 receptor activation.
Mechanisms for the D4-Induced Synaptic Translocation of CaMKII in PFC Cultures. If the PD168077-induced CaMKII translocation to postsynaptic sites was indeed mediated by D4 receptors, selective D4 receptor antagonists should prevent the action of PD168077. Consistent with this, application of L745870 (20 μM), a selective D4 receptor antagonist (Kulagowski et al., 1996; Patel et al., 1997), strongly inhibited the synaptic translocation of CaMKII induced by PD168077 (Fig. 3, synaptic CaMKII clusters/100-μm dendrite: 5.2 ± 1.7 in untreated neurons; 35.7 ± 6.2 in PD168077-treated neurons; 6.1 ± 1.6 in L745870+ PD168077-treated neurons; n = 18 neurons per group from four cultures). In addition, we found that quinpirole, a dopamine D2 receptor agonist, failed to induce CaMKII translocation (4.9 ± 1.9 synaptic CaMKII clusters/100-μm dendrite, n = 20 neurons from four cultures), indicating that the effect on CaMKII translocation is specific for D4 receptor activation.
We further tested whether dopamine can mimic the effect of D4 agonist. PFC cultures were treated with SCH23390 (10 μM) and sulpiride (10 μM) for 15 min to block D1/D5 and D2/D3 receptors, followed by a 5-min treatment with dopamine (50 μM). SCH23390 and sulpiride treatment alone did not induce CaMKII clustering (5.6 ± 3.8 clusters/100-μm dendrite, n = 18 neurons from three cultures). However, in the presence of these D1/D5 and D2/D3 antagonists, dopamine induced significant CaMKII synaptic translocation (26.8 ± 4.5 synaptic CaMKII clusters/100-μm dendrite, n = 20 neurons from three cultures, p < 0.001, ANOVA), similar to the effect of PD168077.
It has been shown previously that D4 receptor activation increased CaMKII activity through a mechanism involving the stimulation of PLC/IP3R pathway, but not the classic D2 receptor-coupled Gi/o and PKA pathway in PFC slices (Gu and Yan, 2004). We then examined the participation of these signaling molecules in D4-induced CaMKII translocation. As shown in Fig. 4, neither the PKA activator cpt-cAMP (100 μM) nor the PKA inhibitor PKI14–22 (100 nM) prevented the PD168077-induced CaMKII translocation (synaptic CaMKII clusters/100-μm dendrite: 32.7 ± 5.7 in cAMP+PD168077-treated neurons; 33.2 ± 5.8 in PKI+PD168077-treated neurons; n = 18 neurons per group from three cultures), indicating the lack of involvement of the PKA pathway in this event. On the other hand, in the presence of the PLC inhibitor U73122 (1 μM), after PD168077 treatment, CaMKII still exhibited the uniform distribution along dendritic processes (5.8 ± 1.3 clusters/100 μm in U73122+PD168077-treated neurons, n = 16 neurons from three cultures), indicating that the D4-induced CaMKII synaptic translocation is abolished by the inhibition of the PLC pathway.
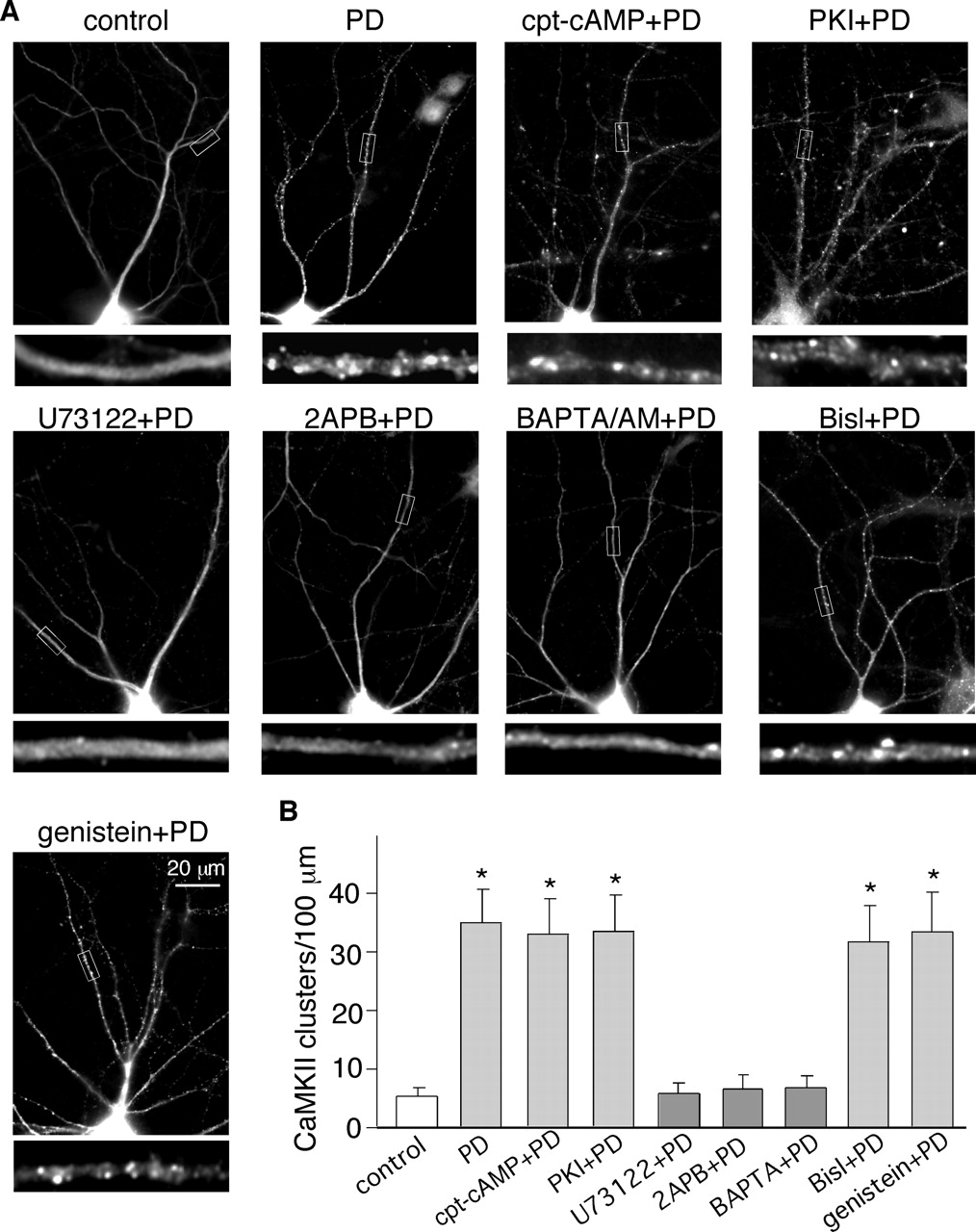
The PD168077-induced CaMKII synaptic translocation was depended on the PLC/IP3R/Ca2+ pathway, but not PKA or PKC. A, immunocytochemical images of α-CaMKII in cultured PFC pyramidal neurons preincubated (20 min) with various agents followed by the PD168077 treatment (20 μM, 5 min). Agents include the PKA activator cpt-cAMP (100 μM), the PKA inhibitor PKI14–22 (100 nM), the PLC inhibitor U73122 (1 μM), the IP3 receptor inhibitor 2APB (15 μM), the membrane permeable Ca2+ chelator BAPTA/AM (50 μM), the PKC inhibitor Bis1 (4 μM), or the tyrosine kinase inhibitor genistein (100 μM). B, quantitative analysis of CaMKII synaptic clusters along dendrites under different treatment. *, p < 0.001 versus control, ANOVA.
Because PLC stimulation leads to the release of intracellular Ca2+ via IP3 receptors and activation of PKC, we further examined the role of these molecules in D4-induced CaMKII synaptic translocation. As shown in Fig. 4, blocking IP3 receptors with 2APB (15 μM) or inhibiting the elevation of intracellular Ca2+ with BAPTA/AM (50 μM) prevented CaMKII from clustering at synaptic sites in response to PD168077 treatment (synaptic CaMKII clusters/100-μm dendrite: 6.5 ± 1.4 in 2APB+PD168077-treated neurons; 6.8 ± 1.2 in BAPTA+PD168077-treated neurons; n = 14 neurons per group from three cultures). In contrast, inhibiting PKC activation with the specific inhibitor Bis1 (4 μM) or inhibiting tyrosine kinases with genistein (100 μM) failed to alter the PD168077-induced CaMKII synaptic targeting (synaptic CaMKII clusters/100-μm dendrite: 31.7 ± 5.3 in Bis1+PD168077-treated neurons; 33.6 ± 6.2 in genistein+ PD168077-treated neurons; n = 18 neurons per group from four cultures). Taken together, these results suggest that the D4 receptor-mediated CaMKII translocation depends on intracellular Ca2+ elevation induced by the PLC/IP3R pathway.
To understand how Ca2+ is potentially involved in the D4 induction of CaMKII translocation, we examined the influence of CaMKII mutants on the translocation. We expressed GFP-fused wild-type α-CaMKII, the autophosphorylation-deficient mutant (T286A), or the calmodulin binding-deficient mutant (A302R) form of α-CaMKII in PFC cultures. As shown in Fig. 5, similar to the endogenous CaMKII, GFP-fused α-CaMKII diffused throughout dendritic shafts in untreated neurons (synaptic CaMKII clusters/100-μm dendrite: 6.4 ± 1.8, 6.8 ± 1.7, and 5.2 ± 1.4 for wild-type, autophosphorylation-deficient, and calmodulin binding-deficient CaMKII, respectively, n = 20 neurons per group from four cultures). PD168077 treatment (5 min) significantly increased CaMKII-GFP puncta along dendrites in neurons transfected with wild-type CaMKII (38.8 ± 6.7 clusters/100 μm, p < 0.001, ANOVA; n = 20 neurons from four cultures) or autophosphorylation-deficient CaMKII (39.4 ± 6.6 clusters/100 μm, p < 0.001, ANOVA; n = 20 neurons from four cultures), but not in neurons transfected with the calmodulin binding-deficient CaMKII (8.3 ± 1.9 clusters/100 μm; n = 20 neurons from four cultures). It suggests that calmodulin binding, but not autophosphorylation, of CaMKII is required to trigger its synaptic translocation in response to D4 receptor activation.
To determine whether calmodulin binding is sufficient to induce CaMKII redistribution, we examined the effect of several other agents on CaMKII translocation, including glutamate (50 μM), muscarinic receptor agonist carbachol (10 μM), 5-HT2 receptor agonist α-Me-5HT (20 μM), and the calcium ionophore A23187 (2 μM), all of which can elevate intracellular Ca2+ and thus promote the calmodulin binding and activation of CaMKII. Indeed, similar to PD168077, all these agents induced CaMKII activation, as indicated by the increased level of Thr286-phosphorylated CaMKII (Fig. 6A and 6B). However, only glutamate induced a significant increase in synaptic CaMKII clustering along dendrites (Fig. 6, C and D, 38.1 ± 5.5 clusters/100 μm, p < 0.001, ANOVA; n = 18 neurons from three cultures), consistent with the previous result (Shen and Meyer, 1999). None of the other three agents showed any effect on CaMKII clustering (Fig. 6, C and D, CaMKII clusters/100-μm dendrite: 6.5 ± 1.5, 6.3 ± 1.3 and 6.0 ± 1.4 for carbachol, α-Me-5HT and A23187, respectively), suggesting that calmodulin binding is not sufficient to induce CaMKII redistribution.
What is the other factor, in addition to Ca2+/calmodulin binding, that is necessary for CaMKII translocation? Previous studies have shown that CaMKII could associate with actin filaments (Ohta et al., 1986) and microtubules (Vallano et al., 1986). We hypothesize that the D4-induced elevation of intracellular Ca2+ and calmodulin binding of CaMKII may affect the association of CaMKII with the cytoskeletal network, therefore facilitating the translocation of CaMKII. To test this, we exposed neuronal cultures to various agents that disturb F-actin or microtubule before activating D4 receptors with PD168077 treatment. As shown in Fig. 7, A and B, the F-actin stabilizer phalloidin-oleate (1 μM) prevented PD168077-induced synaptic CaMKII translocation along dendrites (9.4 ± 2.1 clusters/100 μm, n = 20 neurons from four cultures), whereas the microtubule stabilizer paclitaxel (10 μM) had no effect (33.0 ± 4.9 CaMKII clusters/100-μm dendrite, p < 0.001, ANOVA; n = 20 neurons from four cultures). Neither of these stabilizers induced CaMKII clustering by itself. These results prompted us to hypothesize that the F-actin stabilizer may prevent the reorganization of actin cytoskeleton and the dissociation of CaMKII from actin, thus blocking the D4-induced CaMKII translocation.
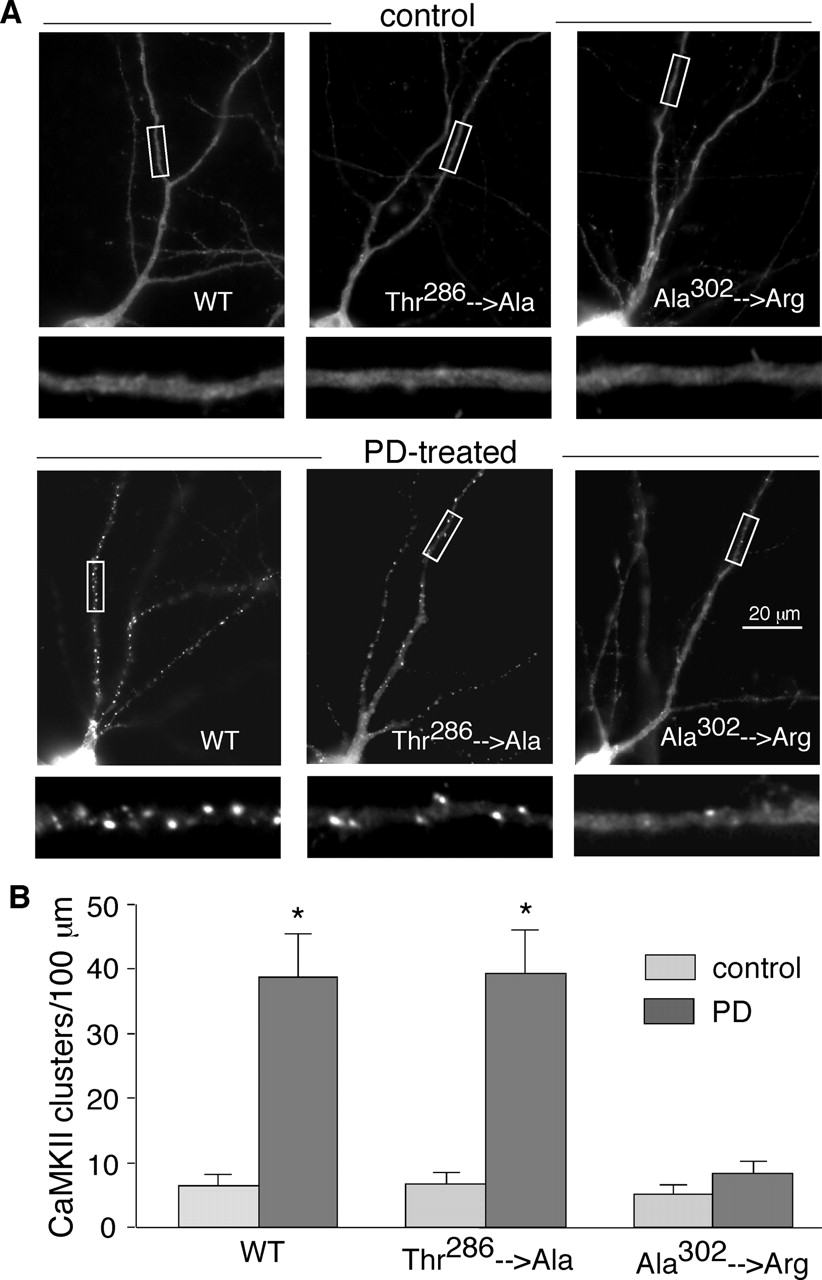
Mutation of the calmodulin binding site (Ala302), but not the autophosphorylation site (Thr286), of CaMKII prevented the PD168077-induced CaMKII synaptic translocation. A, fluorescence images of GFP-CaMKII in cultured PFC pyramidal neurons transfected with wild-type or site mutants of CaMKII. All three kinds of GFP-CaMKII were distributed evenly throughout the dendrites in untreated neurons, similar to the distribution pattern of endogenous CaMKII. Application of PD168077 induced a dramatic synaptic translocation of the wild-type GFP-CaMKII and the autophosphorylation-deficient GFP-CaMKII (Thr286→ Ala), but not the calmodulin binding-deficient GFP-CaMKII (Ala302→ Arg). B, quantitative analysis of GFP-CaMKII synaptic clusters along dendrites in neurons transfected with different CaMKII constructs before (control) and after PD168077 treatment (PD). *, p < 0.001 versus control, ANOVA.
To test this, we examined the effects of various agents on the binding between CaMKII and F-actin. As shown in Fig. 7, C and D, both PD168077 and glutamate caused a significant reduction of CaMKII that bound to F-actin (PD168077, 35 ± 8% of control; Glu, 29 ± 6% of control, n = 6; p < 0.001, ANOVA), whereas carbachol, α-Me-5HT, and A23187 had no effect (carbachol, 97 ± 15% of control; α-Me-5HT, 98 ± 13% of control; A23187, 95 ± 19% of control; n = 6). In the presence of phalloidin, both PD168077 and glutamate lost the capability to reduce the level of actin-binding CaMKII. These results suggest that CaMKII redistribution may require its dissociation from actin cytoskeleton, and the different effects of those agents on the binding of CaMKII to F-actin may underlie their distinct effects on CaMKII translocation.
Because β-CaMKII binds to F-actin directly and α-CaMKII is docked to the actin cytoskeleton by forming hetero-oligomers with β-CaMKII (Shen et al., 1998), we further examined the possible translocation of β-CaMKII in response to D4 receptor activation. As shown in Fig. 8A and 8B, treatment of PFC culture with PD168077 (20 μM, 5 min) induced a significant increase of β-CaMKII clusters along dendrites (control, 6.8 ± 4.5 clusters/100 μm, n = 20 neurons from three cultures; PD168077, 41.3 ± 7.3 clusters/100 μm, n = 24 neurons from three cultures; p < 0.001, ANOVA), similar to the effect of glutamate (50 μM, 5 min) treatment (45.8 ± 7.8 clusters/100 μm, n = 20 neurons from three cultures). In contrast, carbachol (10 μM, 5 min) treatment had no effect on the distribution of β-CaMKII (7.5 ± 5.5 clusters/100 μm, n = 20 neurons from three cultures).

Various agents that could activate CaMKII had different effects on CaMKII translocation. A, immunoblots of phospho-CaMKII and CaMKII in PFC slices incubated with PD168077 (PD, 20 μM), glutamate (50 μM), muscarinic receptor agonist carbachol (10 μM), 5-HT2 receptor agonist α-Me-5HT (20 μM), or the calcium ionophore A23187 (2 μM) for 10 min. Extracts of slices were immunoblotted with an anti-phospho-α-CaMKII antibody. After stripping out signals, membranes were reblotted with an antibody recognizing the total α-CaMKII. B, quantification of p-CaMKII induced by different treatment. *, p < 0.001 versus control, ANOVA. C, immunocytochemical images of α-CaMKII in cultured PFC pyramidal neurons treated for 5 min with various agents, including glutamate (50 μM), carbachol (10 μM), α-Me-5HT (20 μM), and A23187 (2 μM). Among them, only glutamate induced CaMKII translocation. D, quantitative analysis of CaMKII synaptic clusters along dendrites under different treatment. *, p < 0.001 versus control, ANOVA.
Increased CaMKII Phopsphorylation of GluR1 and Miniature EPSCs after D4 Receptor Activation. The synaptically translocated CaMKII after D4 receptor activation can phosphorylate many synaptic substrates. One potential target is the AMPA receptor GluR1 subunit (Soderling and Derkach, 2000). We then investigated whether D4 receptor activation can modulate the CaMKII phosphorylation of GluR1. Previous studies have identified that both CaMKII and PKC phosphorylate GluR1 at the same site Ser831 (Mammen et al., 1997). As shown in Fig. 9, A and B, application of PD168077 (5 min) induced a significant increase in GluR1 phosphorylation at Ser831 (3.4 ± 0.3-fold, n = 8, p < 0.001, ANOVA). The effect was largely blocked by the D4 receptor antagonist L-745870 (1.3 ± 0.2-fold, n = 6) or the CaMKII inhibitor KN-62 (1.4 ± 0.2-fold, n = 6), but not by the PKC inhibitor Bis1 (3.1 ± 0.3-fold, n = 6, p < 0.001, ANOVA). It suggests that D4 receptor activation increases the CaMKII-mediated phosphorylation of GluR1.
To determine whether the increased phosphorylation of GluR1 is caused by synaptically translocated CaMKII, we then examined the effect on GluR1 phosphorylation by other agents that might activate but not translocate CaMKII. As shown in Fig. 9, C and D, the calcium ionophore A23187 (2 μM, 5 min) was unable to increase GluR1 phosphorylation (1.2 ± 0.2-fold, n = 6). Glutamate (50 μM, 5 min) and carbachol (10 μM, 5 min) induced a strong increase in GluR1 phosphorylation (Glu, 3.7 ± 0.5-fold; carbachol, 3.9 ± 0.6-fold, n = 6, p < 0.001, ANOVA), and α-Me-5HT (20 μM, 5 min) had a moderate effect (2.1 ± 0.4-fold, n = 6, p < 0.05, ANOVA). We further found that the glutamate-induced GluR1 phosphorylation was largely blocked by the CaMKII inhibitor KN-62 (1.8 ± 0.3-fold, n = 6, p < 0.05, ANOVA), and partially blocked by the PKC inhibitor Bis1 (2.2 ± 0.4-fold, n = 6, p < 0.05, ANOVA). However, the effect of carbachol or α-Me-5HT on GluR1 phosphorylation was insensitive to CaMKII inhibition but was almost completely blocked by inhibiting PKC, indicating that the GluR1 phosphorylation induced by carbachol or α-Me-5HT probably occurs through activated PKC rather than CaMKII. Taken together, our data show that the agents that can induce CaMKII translocation, such as PD168077 and glutamate, increase CaMKII phosphorylation of GluR1, whereas the agents that can not induce CaMKII translocation, such as A23187, carbachol, and α-Me-5HT, fail to increase CaMKII-dependent phosphorylation of GluR1.

The CaMKII synaptic translocation involved F-actin. A, immunocytochemical images of α-CaMKII in cultured PFC pyramidal neurons preincubated (1 h) with the F-actin stabilizer phalloidin-oleate (1 μM) or the microtubule stabilizer paclitaxel (10 μM), followed by the PD168077 treatment (20 μM, 5 min). Phalloidin, but not paclitaxel, prevented the PD168077-induced CaMKII synaptic translocation. B, quantitative analysis of CaMKII synaptic clusters along dendrites under different treatment. *, p < 0.001 versus control, ANOVA. C, effect of various agents on the interaction of CaMKII with F-actin. Cell lysates from PFC slices were incubated with various agents in the absence or presence of phalloidin-oleate (1 μM), followed by immunoprecipitation with anti-actin antibody and Western blot analysis for CaMKII and actin. PD168077 and glutamate, but not carbachol (10 μM), α-Me-5HT (20 μM), or A23187 (2 μM), disrupted the binding of CaMKII to F-actin. Phalloidin blocked this effect of PD168077 and glutamate. D, bar graphs showing the levels of CaMKII bound to F-actin under different treatment. *, p < 0.001 versus control, ANOVA.
To understand the functional consequence of the D4-induced synaptic translocation of CaMKII, we examined the effect of PD168077 on AMPA receptor-mediated synaptic transmission. mEPSCs are believed to result from the random release of single glutamate packets (quanta), and a significant effect on their amplitude is usually considered good evidence for a modification of postsynaptic AMPA receptor properties. Thus, neuronal cultures were exposed to tetrodotoxin (1 μM) and mEPSCs were recorded in PFC pyramidal neurons. As shown in Fig. 10, A and B, bath application of PD168077 significantly enhanced the mEPSC amplitude (p < 0.01, K-S test), as indicated by a rightward shift of the distribution. In the presence of the CaMKII inhibitor KN-93 (10 μM, 10-min pretreatment), PD168077 failed to enhance the mEPSC amplitude (Fig. 10, C and D). In a sample of neurons we tested, PD168077 increased the mean amplitude of mEPSCs by 18.7 ± 2.4% (n = 7, p < 0.01, K-S test), which was abolished by KN-93 (2.7 ± 0.9%, n = 4). The mEPSC frequency was either slightly increased or not changed by PD168077 (data not shown). These results suggest that AMPA receptors are up-regulated by synaptically translocated CaMKII in response to D4 receptor activation.

PD168077 induced β-CaMKII clustering along dendrites of cultured PFC neurons. A, immunocytochemical images of β-CaMKII in cultured PFC pyramidal neurons treated with or without PD168077 (20 μM, 5 min), glutamate (50 μM, 5 min), or carbachol (10 μM, 5 min). Enlarged versions of the boxed regions of dendrites are shown beneath each of the images. B, quantitative analysis of β-CaMKII clusters along dendrites under different treatment. *, p < 0.001 versus control, ANOVA.
Discussion
Given the broad substrate selectivity of CaMKII, the control of specificity becomes a crucial issue in signal transduction. Subcellular targeting has emerged as an important mechanism by which signaling enzymes achieve precise substrate recognition and enhanced efficacy of signal transduction (Pawson and Scott, 1997; Hudmon and Schulman, 2002). Different pools of CaMKII compartmentalized at membrane, PSD, nucleus, and cytosol are responsible for regulating distinct substrates. A previous study has revealed the CaMKII translocation after NMDA receptor activation (Shen and Meyer, 1999). Little is known about CaMKII translocation in response to other stimuli that can elevate intracellular Ca2+ from internal stores. Our previous studies have found that D4 receptor stimulation changes the enzymatic activity of CaMKII (Wang et al., 2003; Gu and Yan, 2004). In this study, we showed that stimulation of D4 receptors also changed the subcellular localization of CaMKII. After a short treatment with the D4 receptor agonist, both endogenous CaMKII and transfected CaMKII-GFP exhibited rapid redistribution to synapses. Activated CaMKII (Thr286-phosphorylated) was also recruited to synapses after D4 receptor stimulation. The synaptic accumulation of CaMKII provides a mechanism for limiting activation of all cellular CaMKII and phosphorylation of all of its substrates when D4 receptors are activated.

PD168077 increased CaMKII phosphorylation of GluR1 in PFC culture. A, immunoblots of Ser831 phospho-GluR1 (top) and total GluR1 (bottom) in PFC neurons untreated (ctl) or treated with PD168077 (PD, 20 μM) or quinpirole (quin, 20 μM) in the absence or presence of various agents. Agents include the D4 receptor antagonist L-745870 (20 μM), the CaMKII inhibitor KN-62 (10 μM), and the PKC inhibitor Bis-indolyl-maleimide (Bis1, 4 μM). C, immunoblots of Ser831 phospho-GluR1 in PFC neurons treated with various agents in the absence or presence of KN-62 or Bis1. Agents used: A23187 (2 μM), glutamate (Glu, 50 μM), carbachol (10 μM), and α-Me-5HT (20 μM). B and D, bar graphs showing the -fold increase of GluR1 phosphorylation at Ser831 under different treatment. *, p < 0.001 versus ctl; **, p < 0.05 versus ctl; #, p < 0.05 versus Glu alone, ANOVA.

PD168077 increased the mEPSC amplitude in PFC cultures. A and C, representative mEPSC traces obtained from cultured PFC pyramidal neurons treated with PD168077 (PD, 40 μM) in the absence or presence of KN-93 (10 μM). Scale bars, 25 pA, 1 s. B and D, cumulative plots of the distribution of miniature inhibitory postsynaptic current amplitude before and during PD168077 treatment in the absence or presence of KN-93.
CaMKII has multiple interacting partners in the PSD, including actin (Shen et al., 1998) and NMDA receptors (Gardoni et al., 1998; Leonard et al., 1999; Strack et al., 2000). It has been shown that the regulated CaMKII interaction with NR2B subunits provides a mechanism for the glutamate-induced synaptic translocation of CaMKII-GFP (Strack and Colbran, 1998; Bayer et al., 2001). Possible mediators for the D4-induced CaMKII translocation include synaptic activity, actin reorganization, kinase activation, and an alteration in the affinity of protein interactions. We show that the D4-induced CaMKII synaptic translocation depends on the stimulation of PLC pathway, elevation of intracellular Ca2+, but not PKC or tyrosine kinases. Similar to NMDA-induced CaMKII translocation (Shen and Meyer, 1999), mutation of the calmodulin binding site, but not the autophosphorylation site, of CaMKII prevented CaMKII synaptic accumulation after D4 receptor activation, suggesting that Ca2+/calmodulin binding is the driving force for CaMKII translocation under these conditions. However, although calmodulin binding is necessary for the D4-induced CaMKII synaptic translocation, it is not a sufficient factor. Many other agents, such as the agonists for Gq-coupled muscarinic and serotonergic receptors (carbachol and α-Me-5HT) and the calcium ionophore A231087, show no effect on CaMKII distribution, despite their capability to elevate intracellular Ca2+ and promote calmodulin binding to CaMKII. Additional mechanisms must be involved in the D4-induced CaMKII translocation.
CaMKII has been found to bind to actin filaments in a calmodulin-sensitive manner (Ohta et al., 1986). The predominant α-CaMKII is docked to the actin cytoskeleton by forming hetero-oligomers with β-CaMKII, which is bound to F-actin (Shen et al., 1998). It also binds to the actin network through α-actinin, an actin-binding protein (Walikonis et al., 2001; Dhavan et al., 2002). We speculate that the D4-induced Ca2+/calmodulin binding to CaMKII leads to the decreased association of CaMKII with F-actin and increased association of CaMKII with PSD-localized binding partners, thereby facilitating the clustering of CaMKII at PSD. In agreement with this, our data show that an F-actin stabilizer prevents the D4-induced CaMKII translocation, and D4 receptor activation reduces the binding of CaMKII to F-actin. The lack of effect of carbachol, α-Me-5HT, and A231087 on CaMKII association with F-actin provides a possible reason to explain why not all agents that can promote calmodulin binding of CaMKII will induce CaMKII redistribution. Taken together, these results suggest that the dissociation of CaMKII from F-actin is required for the D4-induced synaptic translocation of CaMKII.
By inducing synaptic targeting of CaMKII, D4 receptors could more effectively modulate synaptic efficacy through the specific regulation of CaMKII substrates that are enriched in PSD, such as AMPA receptors (Barria et al., 1997; Hayashi et al., 2000). Indeed, we observed a CaMKII-mediated increase of GluR1 phosphorylation after D4 receptor activation. It is noteworthy that other agents that do not induce CaMKII synaptic translocation fail to increase CaMKII-dependent GluR1 phosphorylation, suggesting that synaptically translocated CaMKII is a more effective regulator of AMPA receptors. The D4-induced potentiation of mEPSC amplitudes is consistent with the finding that CaMKII phosphorylation of GluR1 enhances the AMPA channel conductance (McGlade-McCulloh et al., 1993; Derkach et al., 1999). Therefore, D4 receptors could achieve efficient and specific regulation of synaptic strength through modulating postsynaptic AMPA receptors by PSD-accumulated CaMKII.
This study mechanistically links together D4 receptors and CaMKII, both of which have been implicated in cognitive and emotional processes (Chen et al., 1994; Oak et al., 2000; Hudmon and Schulman, 2002) associated with PFC. In addition to the dynamic regulation of CaMKII activity (Gu and Yan, 2004), D4 receptors cause the synaptic translocation of CaMKII through a mechanism depending on the PLC pathway and intracellular Ca2+ release. This dynamic trafficking of CaMKII enables D4 receptors to facilitate the preferential regulation of CaMKII substrates localized at postsynaptic sites, including AMPA and NMDA receptors. By focusing the range of CaMKII substrates, D4 receptors should be able to achieve an increased “signal-to-noise” ratio in signal transduction. Because “loss of central filtering” caused by unbalanced excitatory and inhibitory activity has been postulated as a mechanism for the sensorimotor gating deficiencies in schizophrenia (McGhie and Chapman, 1961; Braff and Geyer, 1990), the accurate regulation of CaMKII signaling provides a cellular mechanism for the functional roles of D4 receptors in neuropsychiatric disorders.
Footnotes
-
This work was supported by National Institutes of Health grants MH63128, NS48911, and AG21923 and National Alliance for Research on Schizophrenia and Depression (NARSAD) Independent Investigator Award (to Z.Y.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.018853.
-
ABBREVIATIONS: PFC, prefrontal cortex; PLC, phospholipase C; IP3R, inositol 1,4,5-triphosphate receptor; PKA, protein kinase A; CaMKII, Ca2+/calmodulin-dependent protein kinase II; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-d-aspartate; PSD-95, 95-kDa postsynaptic density protein; PD168077, [(4-phenylpiperazinyl)-methyl]benzamide; L-745870, 3-(4-[4-chlorophenyl]piperazin-1-yl)methyl-1H-pyrrolo[2,3b]pyridine; SCH23390, R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine; α-Me-5HT, α-methyl-5-hydroxytryptamine; cpt-cAMP, chlorophenylthio-cAMP; U73122, 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; Bisl, Bis-indolyl-maleimide I; KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine; KN-93, 2-[N-(2-hydroxyethyl)-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine; 2APB, 2-aminoethoxydiphenylborane; PCR, polymerase chain reaction; GFP, green fluorescent protein; mEPSC, miniature excitatory postsynaptic current; QX-314, 2-((2,6-dimethylphenyl)amino)-N,N,N-triethyl-2-oxoethanaminium; TTX, tetrodotoxin; K-S, Kolmogorov-Smirnov; ANOVA, analysis of variance; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; AM, acetoxymethyl ester; A23187, calcimycin.
- Received September 12, 2005.
- Accepted December 19, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}