


Abstract
Zinc has been shown to be present in synaptic vesicles of a subset of glutamatergic boutons and is believed to be coreleased with glutamate at these synapses. A variety of studies have suggested that zinc might play a role in modulation of excitatory transmission, as well as excitotoxicity, by inhibitingN-methyl-d-aspartate (NMDA)-type glutamate receptors. To further investigate the modulatory effects of zinc on NMDA receptors of different subunit compositions, we coexpressed the recombinant subunit NR1 with NR2A and/or NR2B in HEK 293 cells. In whole-cell patch-clamp recordings from these transfected cells, zinc inhibited peak glutamate-evoked current responses in a noncompetitive manner, but there were significant differences between the receptor subtypes in sensitivity to zinc inhibition. For NR1/NR2A, ∼40% of the peak current was inhibited by zinc in a voltage-independent manner with an IC50 value of 5.0 ± 1.6 nm and at a V H value of −60 mV; the remainder was blocked at a second, voltage-dependent site with an IC50 value of 79 ± 18 μm. In contrast, NR1/NR2B currents showed nearly complete inhibition at a voltage-independent site with an IC50 value of 9.5 ± 3.3 μm. Cells cotransfected with NR1, NR2A, and NR2B showed zinc sensitivity intermediate between that characteristic of NR1/NR2A and that of NR1/NR2B. Furthermore, zinc accelerated the macroscopic desensitization of both NR1/NR2A and NR1/NR2B in a dose-dependent manner, apparently independently of glycine-sensitive desensitization and Ca2+-dependent inactivation; maximal effects were to decrease desensitization time constants for NR1/NR2A by ∼75% and for NR1/NR2B by ∼90%. Differential modulation of NR1/NR2A and NR1/NR2B currents by zinc may play a role in regulating NMDA receptor-induced synaptic plasticity and neurotoxicity.
Glutamate is the major excitatory neurotransmitter in the adult central nervous system; it has been shown to play an important role in synaptogenesis, motor control, learning, and memory, as well as a variety of neuropathological conditions such as stroke, epilepsy, and some neurodegenerative diseases (1, 2). Glutamate mediates rapid excitatory neurotransmission via three classes of ionotropic receptors: NMDA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid, and kainate, which are separated on the basis of physiological and pharmacological properties and named for their preferred agonists (3). NMDA receptors are distinguished by their relatively high calcium permeability and voltage-dependent Mg2+ block (3), and receptor function is modulated by a variety of endogenous molecules, including glycine (a coagonist with glutamate), polyamines, protons, oxidizing and reducing agents, and zinc (4-9). Accumulating evidence indicates that NMDA-type glutamate receptors make a critical contribution to mechanisms underlying synaptic plasticity and excitotoxic cell death in a wide variety of neuronal populations (10, 11); thus, endogenous modulators of NMDA receptor function may play a regulatory role in these processes as well.
Adult brain has been shown to contain large amounts of chelatable zinc, which is predominantly localized to glutamatergic terminals in the hippocampal formation, although zinc release has been reported in only the CA3 region (12-14). Release of zinc from synaptic terminals has been shown to be calcium dependent and is increased at higher rates of neuronal firing, giving a peak concentration of ∼300 μm(15, 16). Thus, a role for zinc as an endogenous modulator of glutamatergic transmission has been suggested. In support of this hypothesis, results of several studies in neuronal culture indicate that zinc differentially modulates glutamate receptors, having an inhibitory effect on NMDA receptors while potentiating the response of non-NMDA receptors (5, 17, 18).
Although previous results indicate that zinc inhibits neuronal NMDA receptor-mediated currents (5, 17, 19-21), as well as NMDA receptor-mediated neurotoxicity (22, 23), the reported IC50values vary from 0.5 to 80 μm. Discrepancies in reported values may be due, at least in part, to differences in neuronal NMDA receptor subunit composition. Recent cloning and expression studies indicate that NMDA receptors are likely composed of two NR1 (ζ1) subunits (24), along with combinations of NR2A, NR2B, NR2C, or NR2D (ε1–4), and that the different NR2 subunits confer differences in receptor/channel function, including agonist and antagonist affinity (for reviews, see Refs. 25-27). To investigate whether subunit composition regulates NMDA receptor sensitivity to zinc inhibition, we have recorded whole-cell current responses to rapid agonist application from HEK 293 cells expressing NR1/NR2A, NR1/NR2B, or NR1/NR2A/NR2B, since NR2A and NR2B are the two NR2 subunits known to be widely expressed in cerebral cortex and hippocampus (27). We show that zinc inhibits peak current and alters desensitization of receptors containing either NR2A or NR2B. However, NR1/NR2A exhibits two distinct binding sites for zinc (a voltage-independent, high potency site and a lower potency, voltage-dependent site), whereas NR1/NR2B seems to have only one, largely voltage-independent, relatively low potency binding site for zinc inhibition. Currents recorded from cells transfected with all three subunits generally exhibit characteristics of both NR1/NR2A and NR1/NR2B.
Experimental Procedures
Cell culture and transfections.
HEK 293 cells (CRL 1573; American Type Culture Collection, Rockville, MD) were maintained at 37° and 5% CO2 in minimum essential medium containing Earle’s salts and supplemented with l-glutamine (2 mm), sodium pyruvate (1 mm), penicillin/streptomycin (100 units/ml), and 10% fetal bovine serum. The cells were passaged every 3–4 days and plated at a density of ∼1 × 106/ml at 10–24 hr before transfection. As previously described (28), cells were transfected according to the method of calcium phosphate precipitation with a total of 10 μg of plasmid DNA/10-cm plate. Cells were transfected with a 1:1 ratio of cDNAs encoding NR1a (nomenclature of 29) and NR2 (A and/or B) subunits. Transfected cells were maintained on glass coverslips in medium containing 1 mm (±)-2-amino-5-phosphonopentanoic acid. In some cases, NR1/NR2B-transfected cells were maintained in medium containing 100 μm memantine.
Electrophysiology.
At 20–48 hr after the start of transfection, the cells were transferred to the stage of an inverted microscope (Axiovert 100, Carl Zeiss, Thornburg, NY). Patch-clamp recordings in the whole-cell configuration (30) were made under voltage-clamp (VH = −60 mV) at room temperature. Electrodes were pulled from borosilicate glass (Warner Instruments, Hamden, CT) with the Narishige PP-83 electrode puller (Narishige Scientific Instruments, Tokyo, Japan) and then fire-polished just before recording. Electrodes with resistance of 1–5 MΩ were used.
After establishment of the whole-cell recording mode, the cell was lifted from the recording chamber floor, and agonist was rapidly applied by a piezo-driven θ tube (Hilgenburg, Malsfeld, Germany) positioned within ∼100 μm of the cell. Control and agonist solutions were gravity fed continuously through the two different sides of the θ tube, and the flow rate at the tip was 6–7 cm/sec (from each side). Switching between control and agonist solutions was accomplished by computer-controlled lateral movement of the θ tube via a fast piezo switch (Physik Instruments, Waldbronn, Germany). The 10–90% rise time for exchange of the two solutions was <0.5 msec at the tip of the recording electrode. The rise time for glutamate-evoked NMDA receptor mediated current is glutamate concentration dependent and thus will not accurately reflect solution exchange time over the whole cell. However, the 10–90% rise time for 100 μm glutamate-evoked NR1/NR2A- and NR1/NR2B-mediated currents in our system was ∼12 msec, which is consistent with previous reports (31). Control solution was also continuously gravity fed directly into the chamber.
Recording solutions.
In the recording chamber, cells were bathed in standard external solution containing 145 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 11 mmglucose, and 10 mm HEPES, titrated to pH 7.35 with NaOH. To determine the Ca2+-versus-K+ reversal potential, standard external solution was replaced with solution containing 110 mm CaCl2, 5.4 mmKCl, 25 mm HEPES, and 11 mm glucose, titrated to pH 7.3 with Ca(OH)2. Glutamate and glycine were diluted from 100-mm stock solutions (kept at −20°) into the extracellular solution just before recording. ZnCl2 was prepared from a 1-mm stock solution. In all recordings, 50 μm glycine was added to both the control and glutamate-containing extracellular solutions. The solution in the recording pipette contained 145 mm KCl, 5.5 mm1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, 0.5 mm CaCl2, 2 mmMgCl2, 2 mm tetraethylammonium chloride, 4 mm MgATP, and 10 mm HEPES, titrated to pH 7.2 with KOH.
Data analysis.
Currents were sampled at 333 Hz and acquired and analyzed using pCLAMP software and the Axopatch 200A amplifier (Axon Instruments, Foster City, CA). Results were calculated as mean ± standard error. Sets of different results were compared using the Student’s t test or analysis of variance, and significant differences were determined at 95% confidence intervals. Curve fitting was accomplished by a least-squares regression routine using commercial software (Axum, Trimetrix, WA). Glutamate dose-response measurements were fitted to the logistical function 1/{(1 + (EC50/[glutamate])n}, where [glutamate] is the concentration of glutamate, EC50is the concentration at 50% of the maximal response, and nis the slope factor. To determine dose response for zinc inhibition, we used the function 1/{1 + ([zinc]/IC50)n}, where [zinc] is the zinc concentration, IC50 is the 50% blocking concentration, and n is the slope factor.
Materials.
NR1a and NR2B (both gifts from S. Nakanishi, Kyoto University, Kyoto, Japan) and NR2A [from mouse brain (also called ε1); a gift from M. Mishina, University of Tokyo, Tokyo, Japan] were subcloned into pRK5, a mammalian expression vector containing the cytomegalovirus promoter. (±)-2-Amino-5-phosphonopentanoic acid and memantine were purchased from Research Biochemicals (Natick, MA). Tissue culture material was obtained from Canadian Life Technologies (Burlington, Ontario, Canada) and all other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Results
Electrophysiological properties of recombinant NMDA receptors.
Transfected cells were continuously superfused with nominally Mg2+-free extracellular recording solution containing a saturating concentration (50 μm) of glycine. Current responses to rapid application of 100 μm glutamate (for 2–3 sec) were recorded in the whole-cell mode under voltage clamp at −60 mV. The characteristics of NR1/NR2A- and NR1/NR2B-mediated currents recorded under these conditions are shown in Table1. As previously reported by other investigators (32), these two receptor subtypes exhibited similar calcium permeability but different macroscopic kinetics of the glutamate-evoked current response. To characterize more fully the macroscopic kinetics of agonist-induced desensitization, we applied agonist pulses of relatively long duration. NR1/NR2B currents desensitized with an apparent single exponential time constant (τD) of 715 ± 71 msec (20 cells), but the extent of desensitization over a 3-sec glutamate application was relatively low (30 ± 3%) (Fig.1A and Table 1). On the other hand, 20 ± 3% of the NR1/NR2A current decayed with a fast time constant (τDF) of 148 ± 18 msec, 48 ± 3% desensitized with a time constant similar to that of NR1/NR2B (τDF = 651 ± 41 msec), and the remainder seemed to be nondesensitizing on the 3-sec time scale (32 cells; Fig. 2, A, C, and D; Table 1). Interestingly, in nominally Ca2+-free extracellular recording solution, macroscopic desensitization of NR1/NR2A currents followed a single exponential time course, with a time constant (Table 1) similar to the single τD value measured for NR1/NR2B and the slower component seen for NR1/NR2A in 1.8 mm CaCl2. Moreover, the extent of desensitization at the end of a 3-sec agonist application was only 29 ± 2%, similar to that seen for NR1/NR2B. Our results of a quantitative comparison of the macroscopic kinetics of NR1/NR2A and NR1/NR2B desensitization are consistent with the qualitative data of Monyer et al. (32).
Characterization of glutamate-evoked current responses mediated by NR1/NR2A and NR1/NR2B
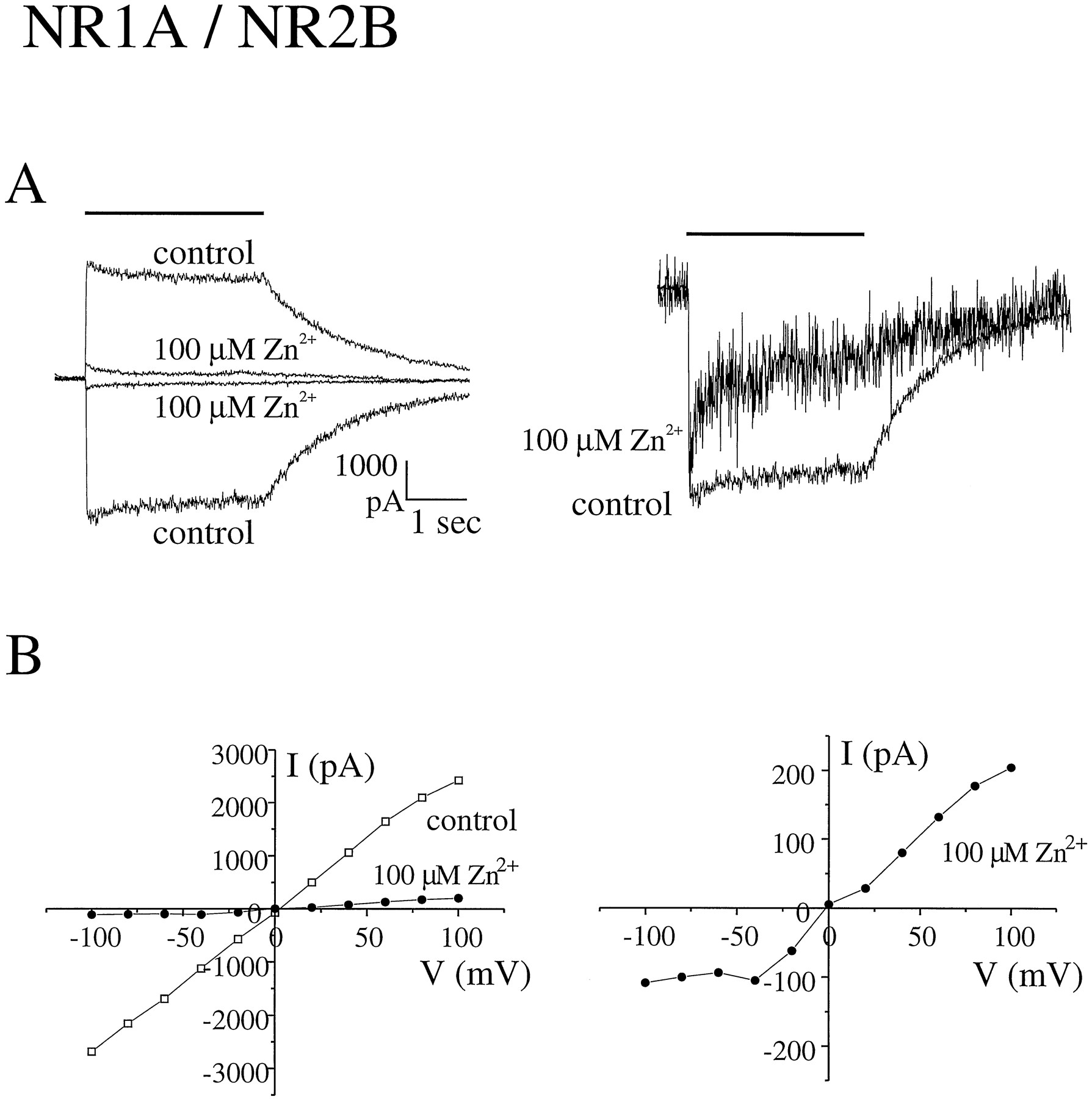
Extracellular zinc inhibits peak glutamate-evoked current responses mediated by NR1/NR2B. A, Current responses to rapid application of 100 μm glutamate (+50 μmglycine) were recorded at holding potentials of −60 mV and +60 mV in the absence of zinc (control) and presence of 100 μm zinc. Right, zinc-inhibited current scaled to same amplitude as control to illustrate effect on macroscopic desensitization kinetics. Bars, duration of glutamate application. Current traces represent the average of two consecutive responses. B, Current-voltage relationships recorded from the same cell before (control) and during application of zinc. Right, current-voltage curve in the presence of 100 μm zinc at higher gain. Current responses to 500-msec pulses of 100 μm glutamate (+50 μm glycine) were recorded in standard extracellular solution at holding potentials ranging from −100 to +100 mV in 20-mV increments. The leak current was subtracted, and the peak current at each holding potential was determined. Note that extracellular zinc did not alter the Na+-versus-K+ reversal potential but did result in a slope conductance of nearly zero at hyperpolarized holding potentials.

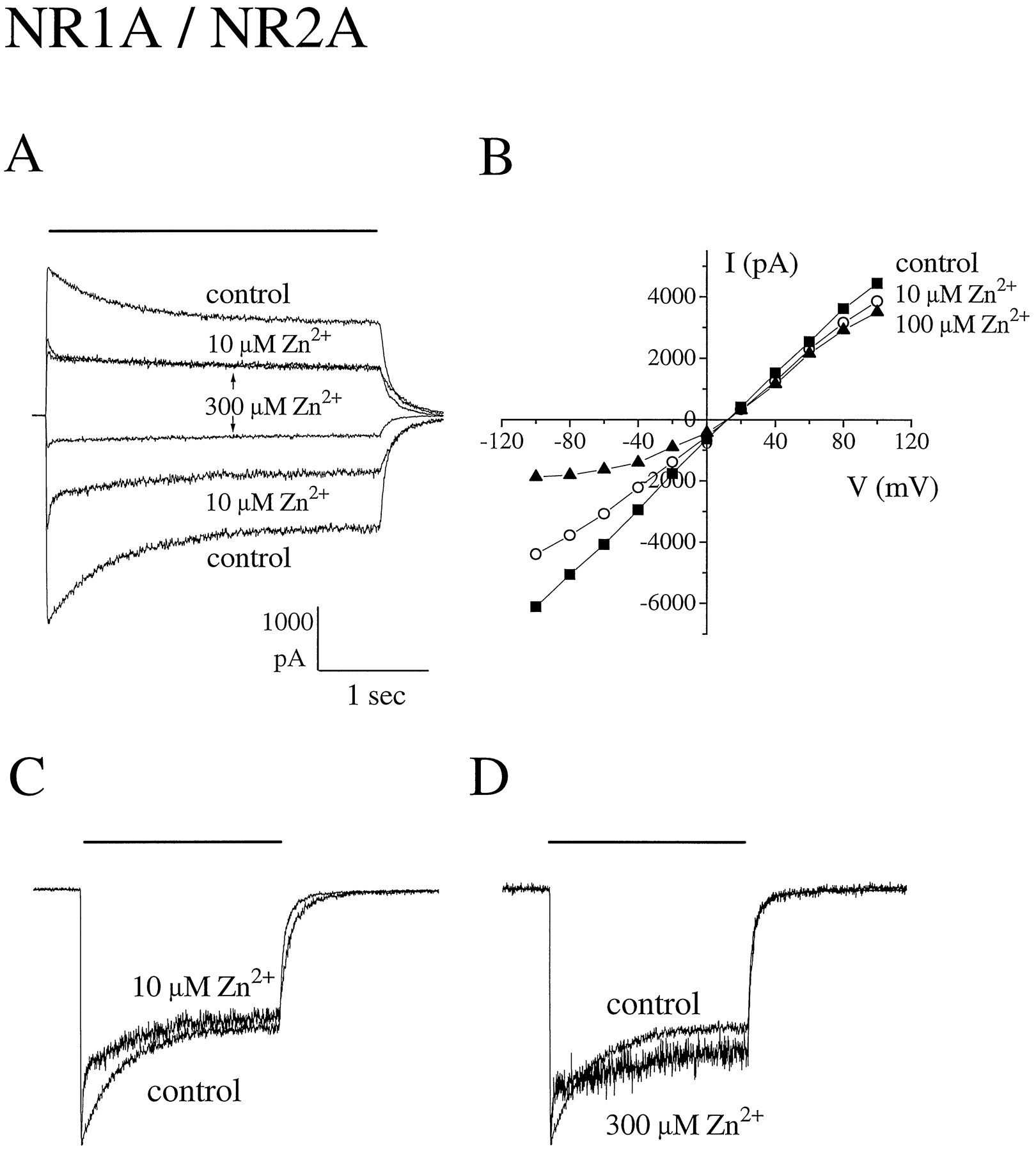
Extracellular zinc inhibits peak glutamate-evoked current responses mediated by NR1/NR2A. A, Current responses to rapid application of 100 μm glutamate (in the presence of 50 μm glycine) were recorded at holding potentials of −60 and +60 mV in the absence of zinc (control) and presence of the indicated concentrations of zinc. B, Current-voltage relationships recorded from a different cell before and during application of zinc (▪, control; ○, 10 μmZn2+; ▴, 100 μm Zn2+). Current-voltage curves were generated as for Fig. 1B. Note that zinc did not alter the Na+-versus-K+ reversal potential but that the current-voltage curve in the presence of 100 μm Zn2+ approaches zero slope conductance at hyperpolarized holding potentials, resulting in apparent outward rectification. C and D, Zinc-inhibited currents scaled to same amplitude as control to illustrate effect on macroscopic desensitization kinetics. Bars, duration of glutamate application. A, C, and D, values represent the average of two consecutive responses.
Currents mediated by NR1/NR2A are more sensitive to zinc than are those mediated by NR1/NR2B.
We determined the effect of extracellular zinc on glutamate-evoked current responses recorded from NR1/NR2A and NR1/NR2B transfected cells. Zinc concentrations were identical in control and agonist solutions for these experiments, so zinc was pre-equilibrated with receptors before agonist application. As previously reported (21), we observed a decrease in holding current (increase in membrane resistance) with the addition of >1 μm zinc but did not further investigate this effect. Although zinc inhibited currents mediated by both subtypes of NMDA receptors in a dose-dependent manner (Figs. 1- 3), NR1/NR2A and NR1/NR2B exhibited important qualitative and quantitative differences in sensitivity to zinc inhibition.

Dose response for zinc inhibition of peak glutamate-evoked current mediated by NR1/NR2A and NR1/NR2B. Current responses to rapid application of 100 μm glutamate (+50 μm glycine) were recorded as described in legends to Figs. 1 and 2 in the presence of different concentrations of zinc. Peak current amplitude recorded during zinc application (I peak) was normalized to that recorded from the same cell before zinc application [I peak (control)]. The curves were fitted by a logistical equation (see text). For NR1/NR2A, the higher potency binding site absorption curve showed an IC50value of 5.0 ± 1.6 nm and the slope factorn was 1.1; the second, lower potency binding site absorption curve indicated an IC50 value of 79 ± 18 μm and a slope factor of 1.3. Of the control peak current, 38% was inhibited by zinc binding at the higher potency site, and the remainder was inhibited at the lower potency site. For NR1/NR2B, IC50 = 9.5 ± 3.3 μm andn = 0.72. Points, mean ± standard deviation for 4–11 different cells.
Inhibition of NR1/NR2B currents by zinc was rapid, achieving a steady state level at each different zinc concentration in <30 sec (the minimum interval between agonist pulses). Zinc inhibition of the peak glutamate-evoked current could be fitted with a single binding site absorption curve with an IC50 value of 9.5 ± 3.3 μm (mean ± standard deviation) (Figs. 1 and 3) and was fully reversible with a time constant for recovery of peak current of 30–60 sec (not shown).
In contrast, two distinct binding sites for zinc inhibition were apparent for NR1/NR2A. The first site exhibited high zinc sensitivity, with an IC50 value of 5.0 ± 1.6 nm (mean ± standard deviation), but maximal inhibition was only ∼40% of the peak current (Figs. 2 and 3). Furthermore, with each new addition of zinc in the concentration range covered by this first site (0.001–10 μm), a stable level of peak current inhibition was achieved only after 60–90 sec, and the initial (within 30 sec) level of peak current inhibition seemed to be more extensive than the equilibrium inhibition level (at 60–90 sec). At >10 μmzinc and at a holding potential of −60 mV, a second site became apparent, so that the remaining ∼60% of the peak current was blocked with an IC50 value of 79 ± 18 μm (mean ± standard deviation) (Figs. 2 and 3). At all zinc concentrations tested (0.0001–1000 μm), recovery of NR1/NR2A peak currents on zinc wash-out showed a two-phase time course: ∼50% of the current recovered with a time constant similar to that for NR1/NR2B (30–60 sec), but the remainder recovered on a time scale of tens of minutes (data not shown).
As shown in Fig. 4, the glutamate dose-response curve generated in the absence of zinc for recordings from NR1/NR2A-transfected cells revealed EC50 values of 9.9 ± 1.1 and 1.3 ± 0.1 μm, with slope factors (n) of 1.1 and 1.5 for peak and steady state currents, respectively. Our EC50 value for the glutamate-evoked steady state current is in agreement with those of previous studies (33, 34). However, our observation that the EC50 value for the NR1/NR2A peak current is ∼8-fold higher than that for the steady state current has not been reported previously. Fig. 4 also demonstrates that inhibition of NR1/NR2A peak current by zinc was noncompetitive at both the high and low potency zinc binding sites.
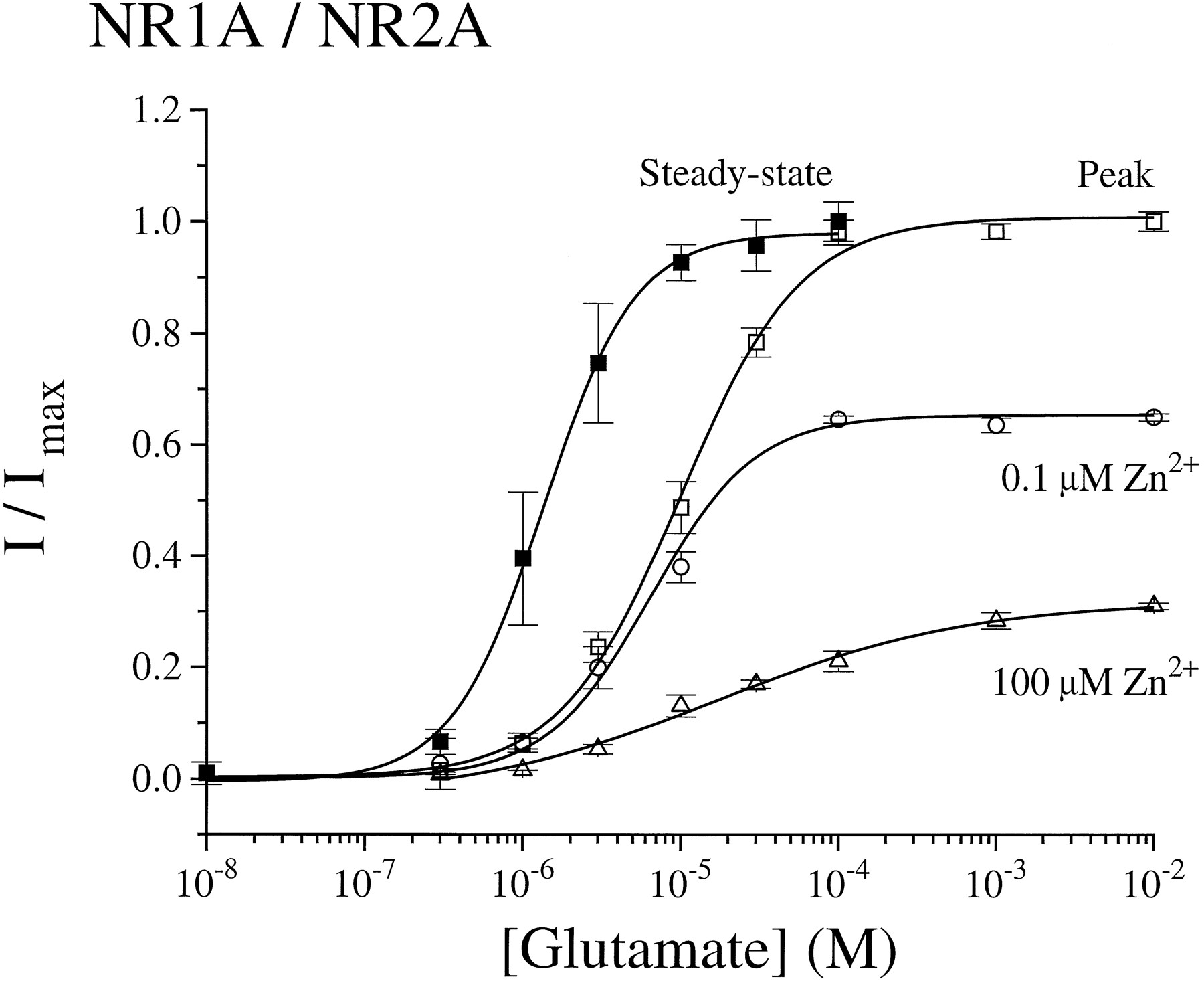
Zinc inhibition of glutamate-evoked currents is noncompetitive. Current responses to rapid application of different concentrations of glutamate (+50 μm glycine) in the presence or absence of 0.1 and 100 μm zinc were recorded from NR1/NR2A-transfected cells. Peak, peak current (I) normalized to the peak current obtained in response to 1 mm glutamate (Imax). Steady state,steady state current (I) at different glutamate concentrations normalized to the steady state current response to 100 μm glutamate (Imax). The glutamate dose-response curves in the absence of zinc were fitted by a logistical equation (see text) in which EC50 = 1.3 ± 0.1 μm ( n = 1.5 for steady state current) and 9.9 ± 1.1 μm ( n = 1.1 for peak current).Curves in the presence of zinc represent peak currents (I) normalized to peak currents obtained in response to 1 mm glutamate (in the absence of zinc) (Imax). Points, mean ± standard error for three to seven different cells.
A comparison of the glutamate-evoked current-voltage relationships in the presence and absence of zinc showed that at concentrations of ≤10 μm, zinc inhibition was voltage independent for both NR1/NR2A and NR1/NR2B currents (Fig. 5). Furthermore, there was no change in the Ca2+-versus-K+reversal potential with application of 1 or 10 μm zinc for NR1/NR2A and NR1/NR2B, respectively (Table 1). However, at concentrations of ≥30 μm, zinc inhibition demonstrated voltage dependence (Figs. 1, 2, and 5). Moreover, at concentrations of ≥100 μm zinc, current-voltage relations approached zero slope conductance in the voltage range of −100 to −40 mV, which is similar to that observed in the presence of extracellular Mg2+ and suggestive of voltage-dependent channel block (3). For NR1/NR2B, this second, voltage-dependent binding site contributed little to overall inhibition by zinc, since >80% of the peak glutamate-evoked current was already blocked at 30 μmzinc (Fig. 3). In contrast, more than half of the initial peak current mediated by NR1/NR2A at −60 mV was inhibited by zinc binding at the second, lower potency, voltage-dependent site, but there was little further inhibition of glutamate-evoked current observed at >10 μm zinc at holding potentials of +60 mV (Figs. 2, 3, and5). Taken together, these data suggest that the major binding site for zinc inhibition of NR1/NR2B and the high potency binding site for zinc inhibition of NR1/NR2A are outside of the pore; however, at high micromolar concentrations, zinc binds to a site within the channel of both receptor subtypes to produce voltage-dependent block.
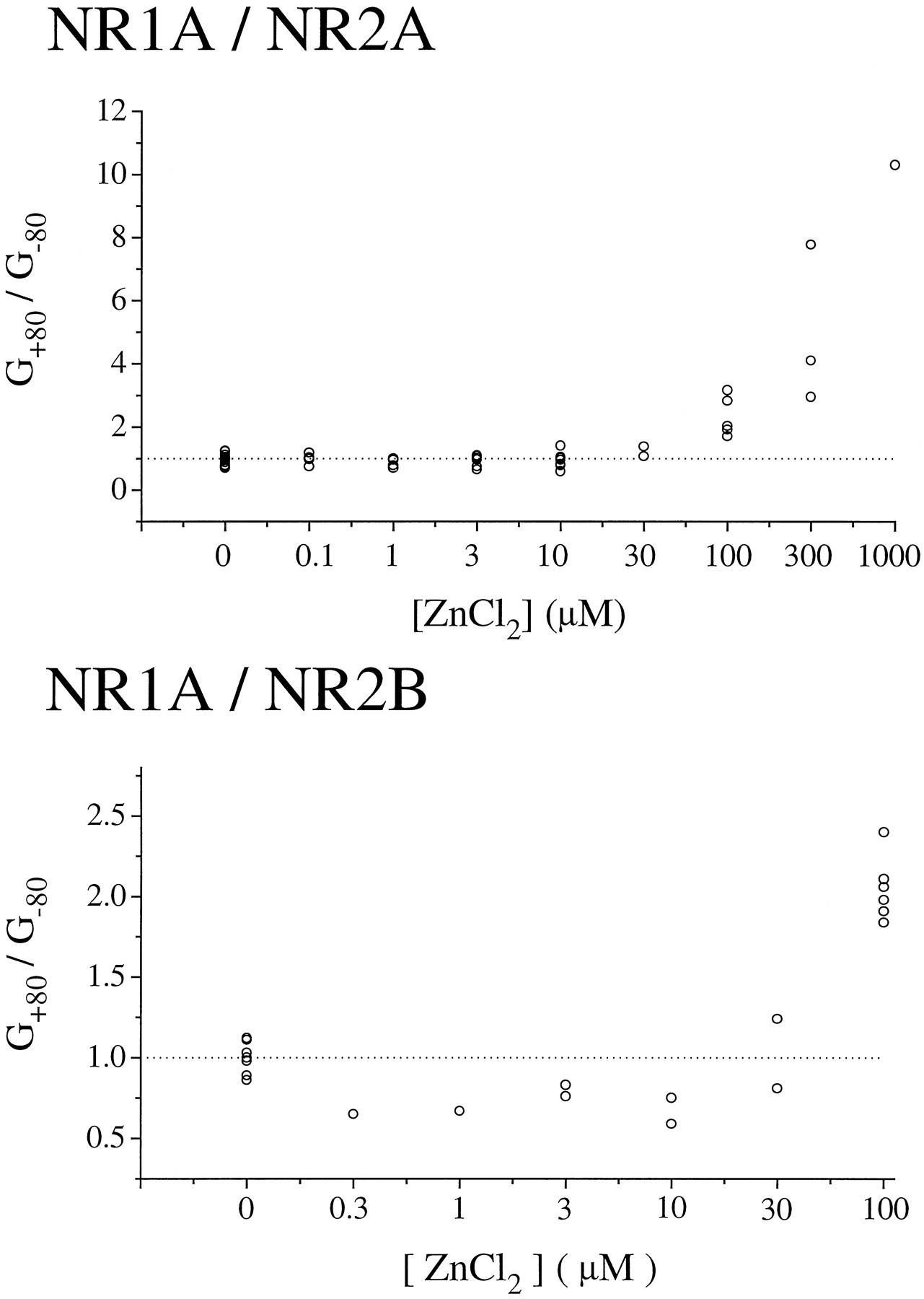
Voltage-dependent block by zinc at high micromolar concentrations. Peak current-voltage curves in the presence of zinc were generated as described in legend to Fig. 1. The ratio of conductances at +80 to −80 mV (G +80/G −80) were calculated and plotted versus zinc concentration. Note that zinc concentrations at >30 μm cause apparent outward rectification of the current-voltage curve for both NR1/NR2A and NR1/NR2B. Points, individual cells.
Extracellular zinc also accelerated agonist-induced macroscopic desensitization for both receptor subtypes in a dose-dependent manner (Figs. 1, 2, and 6). For NR1/NR2A, only the fast component of macroscopic desensitization (τDF) was affected. At zinc concentrations of 0.03–10 μm, τDF progressively decreased to a minimum value of just 59 ± 8 msec at 10 μm zinc (seven cells) (Fig. 6). Moreover, the percentage of current decaying with the faster time constant increased from ∼20% in the absence of zinc to a maximum of ∼40% at zinc concentrations of 0.1–10 μm. At >10 μm zinc, no further reduction in τDF was observed. For NR1/NR2B in the absence of zinc, glutamate-evoked current decay was well fit by a single exponential with a macroscopic time constant of 715 ± 71 msec (20 cells) (Table 1). With the addition of 1 μm zinc, this single exponential decay was accelerated to a time constant of 376 ± 98 msec (three cells), whereas at zinc concentrations of >10 μm, a second, faster component of decay became prominent. As exemplified in Fig. 1A, 34 ± 6% of the peak current decayed with a macroscopic τDF of 73 ± 19 msec (five cells) at 100 μm zinc.

Zinc accelerates desensitization of glutamate-evoked current responses mediated by NR1/NR2A. Current responses to 100 μm glutamate (+50 μm glycine) were recorded as before (Figs. 1 and 2) in the presence of the indicated concentrations of zinc. The time constant of the fast component of desensitization (τDF; see Table 1), determined in the presence of zinc (τZn), was normalized to that measured from the same cell before zinc application (τcontrol). Bars, mean ± standard error for four to eight different cells. ∗, Significant difference from control by the paired t test (two-tailed;p < 0.05). ∗∗, Significant difference from control (p < 0.005).
It is unlikely that the effects of zinc on desensitization of NR1/NR2A receptors are due to preferential binding to the desensitized state of the receptor because the IC50 value for zinc inhibition of steady state current at the higher potency site was calculated to be 14 ± 1 nm, which is ∼3-fold higher than that found for peak current inhibition (5.0 ± 1.6 nm). To test the possibility that zinc interacts with the site responsible for Ca2+-dependent desensitization (35, 36), we analyzed the effects of zinc on the macroscopic kinetics of NR1/NR2A desensitization in extracellular recording solution that was nominally Ca2+-free. In response to 1 mm glutamate (with 50 μm glycine), macroscopic desensitization followed a single exponential time course with τD of 770 ± 87 msec (six cells). With the addition of zinc at concentrations of 0.1–10 μm, a faster component of desensitization became apparent, characterized by a progressively decreasing value for τDF with increasing zinc concentration [τDF = 176 ± 19 msec at 0.1 μm zinc (six cells) and 52 ± 2 msec at 10 μm zinc (three cells)]. Thus, the dose-dependent acceleration of NR1/NR2A desensitization by zinc was qualitatively similar in the absence and presence of extracellular calcium.
Recent work suggests that at least a portion of neuronal NMDA receptors in the forebrain may be composed of heteromers containing NR1 with both NR2A and NR2B (37, 38). Therefore, we investigated the effects of zinc on glutamate-evoked currents recorded from HEK 293 cells cotransfected with NR1, NR2A, and NR2B cDNAs. As before, whole-cell currents were recorded in response to 3-sec applications of 100 μmglutamate in the presence of 50 μm glycine, at a holding potential of −60 mV. Currents showed macroscopic desensitization (I2s/Ipeak = 0.40 ± 0.06) that was not significantly different (p = 0.211) from that exhibited by the NR1/NR2A complex (I2s/Ipeak = 0.33 ± 0.03) but quite distinct (p < 0.01) from that of NR1/NR2B-mediated currents (I2s/Ipeak = 0.73 ± 0.03) (Fig.7A). Despite the similarity to NR1/NR2A with respect to desensitization, currents recorded from 13 of 16 NR1/NR2A/NR2B-transfected cells showed two distinct time constants for deactivation after removal of glutamate. The faster component (114 ± 20 msec) was not significantly different (p= 0.11) from the single exponential decay constant observed for NR1/NR2A currents (97 ± 5 msec), whereas the slower component (882 ± 138 msec) was not significantly different (p = 0.58) from that of NR1/NR2B currents (954 ± 44 msec) (Fig. 7B). Moreover, for the majority of NR1/NR2A/NR2B cells, these two components contributed approximately equally to current deactivation. Unlike the majority of NR1/NR2A/NR2B-transfected cells, currents recorded from 3 of 16 such cells closely resembled those from NR1/NR2B-expressing cells. Currents from each of these cells exhibited a single exponential time course for deactivation with time constants of 502, 513, and 751 msec. In addition, glutamate-induced macroscopic desensitization was similar to that of NR1/NR2B (I2s/Ipeak ∼ 0.75). Taken together, these results suggest that at least 13 of 16 cells transfected with NR1, NR2A, and NR2B cDNAs expressed all three subunits, whereas 3 of 16 cells may have expressed predominantly NR1 and NR2B.

Glutamate-evoked currents recorded from NR1/NR2A/NR2B-transfected cells exhibit properties of both NR1/NR2A and NR1/NR2B. A, The current amplitude after 2 sec of glutamate application was measured and normalized to the peak current amplitude in recordings from cells transfected with NR1 and NR2A (2A; 24 cells); NR1, NR2A, and NR2B (2A-2B; 14 cells); and NR1 and NR2B (2B; 21 cells). B, Time course of current decay after removal of glutamate was fit to either a single exponential curve (2A, 34 cells, and 2B, 28 cells) or a biexponential curve (2A-2B; 13 cells for the fast and 16 cells for the slower component) using pCLAMP software, and the time constant(s) are shown. In the majority of 2A-2B cells, the two time constants made approximately equal contributions to current deactivation. C, Peak glutamate-evoked current amplitude recorded in the presence of 0.1 μm and 100 μmzinc (IZn) was normalized to peak current amplitude recorded in the absence of zinc (Icontrol) in 7–12 NR1/NR2A-transfected cells (2A), 4–8 NR1/NR2B-transfected cells (2B), and 13 NR1-NR2A-NR2B-transfected cells (2A-2B).Bars, mean ± standard error. ∗∗, Significant difference from the value for NR1/NR2A (p < 0.01). # and ##, Significant difference from the value for NR1/NR2B (#,p < 0.05; ##, p < 0.01) by one-way analysis of variance.
To compare the zinc sensitivity of these NR1/NR2A/NR2B-transfected cells with that of cells expressing either NR1/NR2A or NR1/NR2B, we analyzed the magnitude of peak glutamate-evoked current inhibition in the presence of 0.1 and 100 μm zinc. At these two zinc concentrations, peak current inhibition of NR1/NR2A differs most sharply from that observed for NR1/NR2B (see Fig. 3). Specifically, at a concentration of 0.1 μm, zinc inhibits 0.33 ± 0.05 of the NR1/NR2A peak current but only 0.06 ± 0.01 of the NR1/NR2B current. On the other hand, 100 μm zinc inhibits 0.72 ± 0.04 of the NR1/NR2A peak current, whereas 0.93 ± 0.03 of the NR1/NR2B peak current is inhibited (Fig. 7C). The pooled data from 13 of 16 NR1/NR2A/NR2B-transfected cells (those that exhibited biophysical properties consistent with expression of all three subunits; see above) showed that peak current inhibition by 0.1 and 100 μm zinc was intermediate between that observed for NR1/NR2A and NR1/NR2B (Fig. 7C). Although the mean zinc inhibition seemed to more closely resemble that of NR1/NR2A at 0.1 μm and that of NR1/NR2B at 100 μm, an analysis of variance indicated that inhibition of NR1/NR2A/NR2B current by 100 μm zinc significantly differed from that of both NR1/NR2B and NR1/NR2A (p = 0.038 andp = 0.008, respectively) and that inhibition by 0.1 μm zinc significantly differed from that of NR1/NR2B (p = 0.007) but not that of NR1/NR2A (p = 0.072).
Discussion
Differential sensitivity of heteromeric NMDA receptor subtypes to zinc inhibition.
In this study, we have shown that whole-cell current responses to rapid agonist application recorded from either NR1/NR2A- or NR1/NR2B-transfected HEK 293 cells are sensitive to zinc inhibition. Our results indicate that zinc accelerates macroscopic desensitization and inhibits peak current at a binding site that is independent of membrane voltage but that zinc sensitivity is ∼2000-fold higher for NR1/NR2A (IC50 = 5 nm) than for NR1/NR2B (IC50 = 9.5 μm). However, zinc inhibition at this voltage-independent binding site is only partially effective for NR1/NR2A, blocking ∼40% of the current, whereas a second, voltage-dependent binding site with an IC50 value of 79 μm blocks the remaining current at normal resting membrane potentials.
These results represent the first demonstration that zinc differentially inhibits whole-cell currents of the NR1/NR2A versus NR/NR2B receptor subtype expressed in a mammalian cell line. A previous report described differential effects of zinc on currents mediated by splice variants of homomeric NR1 expressed in Xenopus laevisoocytes (39), including potentiation of NR1 subunits lacking the 21-amino acid amino-terminal insert (e.g., NR1a, the splice variant we used in our study) by zinc concentrations <1 μm. However, the authors mentioned (without showing data) that 1 μm zinc inhibited heteromeric NMDA receptors composed of NR1 with NR2A, NR2B, or NR2C, which is consistent with our results. In addition, a very recent study has shown dose-dependent zinc inhibition of 45Ca2+ influx into L(tk−) cells stably transfected with NR1/NR2B (IC50 = 4 μm, ∼2-fold lower than our result) (40). This difference may be explained by the competitive effect of Ca2+ on zinc inhibition of NMDA receptor currents (19); Grimwood et al. (40) measured the IC50 for zinc inhibition in the absence of Ca2+, whereas our experiments were carried out in 1.8 mm external Ca2+. In cells stably expressing NR1/NR2A, Grimwoodet al. (40) observed dose-dependent inhibition of45Ca2+ influx for zinc concentrations of 0.3–10 μm with maximal inhibition of ∼60%; however, they noted partial relief of inhibition at 100 μm zinc (back to just 45% inhibition). The lack of further inhibition of45Ca2+ influx at zinc concentrations at >10 μm (40) likely reflects the fact that in their system activation of NMDA receptors resulted in membrane depolarization and thus loss of voltage-dependent inhibition by zinc at the second binding site on NR1/NR2A.
Anatomical studies have shown that NR2A and NR2B, along with NR1, are the predominant NMDA receptor subunits expressed in the cerebral cortex and hippocampus (27). Previous electrophysiological studies in neurons from these regions have indicated that NMDA receptor-mediated currents are inhibited by zinc (5, 17). In three different patch-clamp recording studies from cultured neurons, the IC50 value for zinc inhibition was determined to be 12 and 13 μm (hippocampal neurons; Refs. 19 and 20) and ∼3 μm (cortical neurons; Ref. 21). Although zinc inhibition in the hippocampal neurons seemed to fit a single binding site absorption curve, the variability seen for the 1 μm range in the first study (19) was perhaps greater than expected, and inhibition by zinc concentrations of <5 μm was not shown for the second study (20). Moreover, zinc inhibition in the cortical neurons could not be fit to a single binding site absorption curve (21).
It is possible that the results of previous studies in neurons reflect, in part, a mixed population of pure NR1/NR2A and NR1/NR2B receptor subtypes. However, recent evidence suggests that NMDA receptors composed of all three subunits may be the predominant complex in the forebrain (37, 38). In recordings from HEK 293 cells cotransfected with NR1, NR2A, and NR2B DNAs, we found that the majority of cells exhibited currents with two deactivation time constants, suggestive of approximately equal contributions from NR2A and NR2B, but that macroscopic desensitization was dominated by NR2A. Inhibition by zinc in the nanomolar range seemed to reflect a stronger contribution from the NR2A subunit, whereas zinc inhibition at high micromolar concentrations suggested a more dominant role for NR2B. Because previous studies suggest that complexes combining all three subunits (“heterotrimeric”) are preferred in cells expressing significant levels of NR1, NR2A and NR2B (or NR2C) (38, 41), we predict that heterotrimeric receptors may be characterized by zinc sensitivity higher than that of NR1/NR2B and by more pronounced zinc inhibition at 100 μm than that of NR1/NR2A. However, further experiments, including single-channel recordings from membrane patches, are required to determine with certainty the zinc dose response for inhibition of heterotrimeric receptors.
Mechanism of zinc inhibition of NMDA receptor current.
Our data indicate that zinc inhibits NMDA receptor channel function in a noncompetitive manner at two different binding sites. Previous studies have also described noncompetitive antagonism by zinc of NMDA receptors (5, 22). Moreover, our finding that the low potency site for NR1/NR2A (and NR1/NR2B) is sensitive to membrane potential is consistent with two previous single-channel recording studies that noted voltage-dependent channel block by zinc at concentrations in the range of 30–100 μm (20, 21). These authors also concluded that zinc binds within the pore of NMDA receptor channels at concentrations of >10–30 μm. In contrast, the high potency site and major binding site for zinc inhibition of NR1/NR2A and NR1/NR2B, respectively, seem to be outside the pore and are also separate from the glutamate agonist binding site. It is interesting to note that only ∼40% of NR1/NR2A-mediated current is inhibited by zinc at this site, suggesting that zinc may modulate the single-channel properties of NR1/NR2A without completely eliminating channel openings. In support of this hypothesis, single-channel recordings from hippocampal neurons had shown that zinc selectively decreased NMDA-evoked large conductance openings (both opening frequency and open time) without affecting the smaller conductance states (20). Further studies are required to determine the site and mechanism of voltage-independent zinc inhibition.
Apart from inhibition of peak current, we have shown that zinc accelerates the onset of agonist-induced macroscopic desensitization of both NR1/NR2A and NR1/NR2B receptor channels. A variety of molecular mechanisms may underlie the effect of zinc on the macroscopic kinetics of NMDA receptor desensitization. Zinc may promote entry into a new closed state (e.g., due to open channel block) or alter the microscopic rate constants that determine the equilibrium among open, closed, and desensitized states. Specifically, zinc might decrease channel open time and/or open frequency by increasing the rate of entry into the desensitized or closed states or, alternatively, by decreasing the rate of entry to the open state or recovery from the desensitized state. We have no evidence for preferential zinc binding and stabilization of the desensitized state, as the IC50 for zinc is similar for both peak and steady state current. Furthermore, open channel block by zinc at concentrations of <10 μm cannot explain acceleration of macroscopic desensitization because the effect is equally profound at depolarized and hyperpolarized holding potentials. Although glycine binding has been shown to regulate the extent of NMDA receptor desensitization (so that steady state to peak current ratio varies with glycine concentration; Ref. 6), the rate of onset of macroscopic desensitization has not been shown to be regulated by glycine concentration. Moreover, our experiments were performed at saturating glycine concentrations. Therefore, as Westbrook and Mayer (5) had already suggested for zinc inhibition of peak current, it is unlikely that competition at the glycine binding site underlies the effect of zinc on macroscopic desensitization. Finally, our data show that the dose-dependent zinc-induced acceleration of fast macroscopic desensitization is qualitatively similar (and quantitatively the same at 10 μm zinc) in the presence and absence of 1.8 mm calcium, suggesting that zinc acts via a mechanism independent of the actions of calcium. Additional experiments, especially single-channel recordings to determine the effect of zinc on microscopic rate constants, would help to further define the role of zinc in modulating macroscopic desensitization of NMDA receptors.
Significance of NR1/NR2A versus NR1/NR2B sensitivity to zinc inhibition.
The differential sensitivity of heteromeric NMDA receptor subtypes to zinc inhibition may have important implications for synaptogenesis, synaptic plasticity, and excitotoxic neuronal death. Although NR2B expression is abundant during embryonic stages, NR2A expression is insignificant until the early postnatal period (32). The lower sensitivity of NR1/NR2B to zinc inhibition would contribute to maximization of NMDA receptor currents during this critical period of synaptogenesis. Further, our results indicate that even at concentrations of <0.1 μm, zinc may play an important role in modulating NMDA receptor responses in neurons expressing predominantly NR1/NR2A. However, at zinc concentrations in the high micromolar range and under conditions of significant membrane depolarization, such as may occur during ischemia, only NR1/NR2B-mediated currents would be completely eliminated. On the other hand, if the predominant NMDA receptor complex in adult forebrain is composed of NR1/NR2A/NR2B, then these receptors may exhibit 80–90% inhibition by 100 μm zinc in combination with sensitivity to nanomolar concentrations of zinc. Thus, relative expression levels of NR2A versus NR2B in regions of the cortex or hippocampus in which zinc is abundant may play a role in determining the importance of NMDA receptor currents in triggering long term potentiation or excitotoxic neuronal death.
Acknowledgments
We thank Dr. G. Westbrook for many useful comments and suggestions; Drs. T. Murphy and S. Duffy for critical reading of the manuscript; Dr. C. Price for helpful discussions; P. Lee, G. Kenner, and T. Luo for technical assistance; and S. Sturgeon and M. Thejomayen for assistance with manuscript preparation. We are grateful to Profs. S. Nakanishi and M. Mishina for the cDNA clones.
Footnotes
- Received August 15, 1996.
- Accepted February 21, 1997.
-
Send reprint requests to: Dr. Lynn Raymond, Division of Neurological Sciences, Department of Psychiatry, University of British Columbia, 2255 Wesbrook Mall, Vancouver, BC V6T 1Z3, Canada. E-mail:lynnr{at}unixg.ubc.ca
-
This work was supported by a Medical Research Council of Canada Scholarship (L.A.R.), Medical Research Council Operating Grant MT-12699 (L.A.R.), and a Huntington’s Disease Society of America Postdoctoral Fellowship (N.C.).
-
N.C. and A.M. contributed equally to this work.
Abbreviations
- NMDA
- N-methyl-d-aspartate
- HEK
- human embryonic kidney
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- τD
- exponential time constant
- τDF
- fast time constant
- I2s
- current amplitude after 2-sec exposure to agonist
- Ipeak
- peak agonist-evoked current amplitude
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}