


Abstract
Memory consolidation is mediated by new protein synthesis. However, the transcriptional pathways induced in neurons by behavioral training that activate gene responses have yet to be fully delineated. We have previously shown that nuclear factor κB (NF-κB) is activated in the amygdala after fear conditioning. Here we report that fear conditioning resulted in an increase in histone acetyl-transferase activity, the association between NF-κB p65 and CBP, and the increase in acetylated p65. Pretreating animals with histone deacetylase (HDAC) inhibitors prolonged the nuclear expression of acetyl-p65 and increased its DNA binding activity. Consistent with these results, HDAC inhibitors enhanced long-term but not short-term fear memory, and this effect was attenuated by κB decoy DNA, whereas scrambled DNA was without effect. This study provides evidence that HDAC-mediated deacetylation functions as an intranuclear molecular switch culminating in the termination of NF-κB transcriptional response that is involved in the formation of fear memory.
Although NF-κB is thought to play a crucial role in regulating inflammatory and immune responses (Baeuerle and Henkel, 1994; May and Ghosh, 1998; Mercurio and Manning, 1999; Perkins, 2000), it is also involved in cell survival and differentiation (O'Neill and Kaltschmidt, 1997; Yu et al., 1999; Denk et al., 2000; Castagne et al., 2001), neuronal plasticity (Albensi and Mattson, 2000; Merlo et al., 2002; Meffert et al., 2003), and peripheral myelin formation (Nickols et al., 2003). Five mammalian NF-κB subunits have been identified, p50/NF-κB1, p65/RelA, c-Rel, RelB, and p52/NF-κB2, which form homo- and heterodimers (Siebenlist et al., 1994). NF-κB is normally repressed in the cytoplasm by the endogenous inhibitor IκBα protein (Baldwin, 1996). Upon stimulation by extracellular inducers, IκB is phosphorylated, ubiquitinated, and subsequently degraded by the 26S proteasome. NF-κB then translocates into the nucleus where it can positively regulate the expression of genes (Chen et al., 1995; Verma et al., 1995; Grilli and Memo, 1999; Karin, 1999).
The action of NF-κB is regulated by reversible acetylation (Chen et al., 2001), catalyzed by a family of HATs that includes the general transcriptional coactivators CBP and p300 (Brown et al., 2000). The effect of HATs is opposed in the cell by HDACs, and p65 deacetylation promotes binding of p65 to IκB α, leading to nuclear export of p65 complex through a chromosomal region maintenance-1-dependent pathway. In addition to regulation of nuclear export, acetylation of p65 also enhanced its DNA binding affinity and transcriptional activity (Chen et al., 2002). However, it is argued that acetylation of p65 actually contributes to the mechanism of postinduction turn-off of NF-κB-mediated transcription by reducing its ability to bind κB-DNA and facilitating its IκBα-mediated export from the nucleus (Kiernan et al., 2003).
We have previously reported that NF-κB is activated in the amygdala after fear conditioning (Yeh et al., 2002). Here we provide evidence that HDAC inhibitor trichostatin A (TSA) prolongs p65 acetylation, increases NF-κB DNA binding activity, and enhances fear-potentiated startle. Importantly, these effects are attenuated by the treatment with κB decoy DNA or drug that inhibits NF-κB DNA binding activity.
Materials and Methods
Surgery
Rats (4-5 weeks old) were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and subsequently were mounted on a stereotaxic apparatus. Two cannulae made of 22 gauge stainless steel tubing (C313G; Plastic Products, Roanoke, VA) were implanted bilaterally into the lateral (LA) or basolateral (BLA) amygdala (anteroposterior, -2.8 mm; mediolateral, ±4.5 mm; dorsoventral, -7.0 mm) (Paxinos and Watson, 1986)]. A 28-gauge dummy cannula was inserted into each cannula to prevent clogging. Three jewelry screws were implanted over the skull, serving as anchors, and the whole assembly was affixed on the skull with dental cement. The rats were monitored and handled daily and were given 7 days to recover. TSA (Sigma-Aldrich, St. Louis, MO), sodium butyrate (Sigma-Aldrich), and helenalin (Calbiochem, San Diego, CA) were administered bilaterally in a volume of 0.5 to 0.8 μl at a rate of 0.5 μl/min. κB decoy and scrambled DNA were prepared and administered based on the method of Blondeau et al. (2001). In brief, double-stranded κB decoy DNA was prepared by annealing complementary single strands with the sequences of 5′-GAGGGGACTTTCCCT-3′. Control DNA with a scrambled sequence was prepared by annealing oligonucleotides of the following sequences: 5′-GATGCGTCTGTCGCA-3′. Stocks of double-stranded DNA were prepared at a concentration of 2 mM in saline. κB decoy and scrambled DNA (60 μg) were infused into the amygdala at a rate of 0.5 μl/min (two injections at 24 and 2 h before conditioning).
Fear Conditioning
Rats were trained and tested in a stabilimeter device. A piezoelectric device mounted below the stabilimeter detects and transduces the motion of the cylinder produced by the whole-body startle response of the rat (San Diego Instruments, Inc., San Diego, CA). The whole set-up was enclosed in a ventilated, sound-attenuating cabinet (length 38 cm, width 38 cm, and height 55 cm). The acoustic startle stimulus was a 50-ms white noise at the intensity of 95 dB. The visual CS was a 3.7-s light produced by an 8-W fluorescent bulb attached to the back of the stabilimeter. The unconditioned stimulus was a 0.6-mA foot shock with a duration of 0.5 s.
Acclimation. On 3 consecutive days, rats were placed in the startle test boxes for 10 min and returned to their home cages.
Matching. On two consecutive days, rats were placed in the startle box and 3 min later presented with 10 startle stimuli at a 2-min intertrial interval (ITI). Based on mean startle amplitudes in the second of these two sessions, rats were matched into groups with similar response levels.
Training. Rats were placed in the startle boxes and received 10 light/foot shock pairings with an ITI of 2 min. Unpaired controls received the same number of light and foot shock presentations, but in a pseudo-random fashion in which the unconditioned stimulus could occur at any time except at the 3.2 s after the CS.
Test. Twenty-four hours after training, rats were tested for fear-potentiated startle. This involved 10 startle-eliciting noise bursts presented alone (noise-alone trial) and 10 noise bursts presented 3.2 s after onset of the 3.7-s light (light-noise trials). The two trial types were presented in a balanced mixed order (ITI, 30 s). The percentage of fear-potentiated startles was computed as: [(startle amplitude on CS-noise - noise-alone trials)/(noise-alone trials)] × 100.
Slice Preparation and Extracellular Recordings
Male Sprague-Dawley 4- to 6-week-old rats were decapitated, and their brains were rapidly removed and placed in cold oxygenated artificial cerebrospinal fluid (ACSF) solution. Subsequently, the brain was hemisected and transverse slices of 450-μm thickness were made. ACSF solution had the following composition: 117 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 25 mM NaHCO3, 1.2 mM NaH2PO4, and 11 mM glucose. The ACSF was bubbled continuously with 95% O2/5% CO2 and had a pH of 7.4.
Extracellular field potentials were made by electrical stimulation of the external capsule, which contained fibers from the auditory cortex to the lateral amygdala, with a concentric bipolar stimulating electrode (SNE-100; David Kopf Instruments, Bern, Germany). Electrical stimuli (150 μs in duration) were delivered at a frequency of 0.05 Hz. Baseline field potentials were adjusted to ∼30 to 40% of the maximal responses.
Nuclear Extract Preparation
After the rats were killed, the brains were rapidly removed and the LA and BLA were dissected out. The tissues were ground using a Dounce grinder with a loose pestle in iced-chilled buffer (15 mM HEPES, 60 mM KCl, 1 mM NaCl, 0.25 M sucrose, 5 mM EDTA, 1 mM EGTA, 1 mM PMSF, 10 μg/ml aprotinin, 15 μg/ml leupeptin, 2 mM NaF, and 1 mM sodium orthovanadate). The homogenate was centrifuged for 10 min at 2000 rpm, and the pellet was resuspended in buffer (10 mM HEPES, pH 7.2, 15 mM MgCl2, 10 mM KCl, 1 mM PMSF, 2 mM NaF, 15 μg/ml leupeptin, and 1 mM sodium orthovanadate). After a brief vortex, they were incubated on ice for 10 min and lysed with a tight pestle. The homogenate was centrifuged at 4000 rpm for 10 min. The pelleted nuclei were resuspended in 40 to 60 μl of extraction buffer consisting of 100 mM HEPES, pH 7.2, 1.5 mM MgCl2, 1 mM EDTA, 0.8 M NaCl, 15% glycerol, 2 mM NaF, 1 mM PMSF, 15 μg/ml leupeptin, and 1 mM sodium orthovanadate and were incubated on ice for 2 to 4 h. The nuclear suspension was centrifuged at 14,000 rpm for 30 min at 4°C, and the supernatant was saved. For detection of acetyl-p65, blots were immunoprecipitated with antibody against NF-κB p65 and immunoblotted with pan-acetyl (1:2500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For detection of CBP and NF-κB p65 association, blots were immunoprecipitated with antibody against NF-κB p65 and immunoblotted with CBP (1:2500; Santa Cruz Biotechnology, Inc.).
HAT Assay
HAT activity was quantified using an immune complex kinase assay. Cells were prepared from unpaired or conditioned rats. Equal amounts of nuclear extracts were immunoprecipitated using A-22 anti-CBP antibody (Santa Cruz Biotechnology, Inc.) and protein-G agarose. Beads were washed three times with HAT buffer (50 mM Tris, pH 8.0, 1 mM EDTA, 10 mM Na-butyrate, and leupeptin at 1 μg/ml) and mixed with histone H4 substrate peptide (1 μg per assay point) in 30 μl of HAT buffer supplemented with 50 nCi of [3H]acetyl-CoA (PerkinElmer, North Point, Hong Kong). Samples were incubated at 30°C for 2 h. After centrifugation for 5 min at 14,000 rpm, supernatants (20 μl) were spotted on P81 phosphocellulose paper; supernatants were washed for 5 min in 50 ml of 50 mM NaHPO3,pH 9.0, five times, and then washed once in 50 ml of acetone for 5 min. Bound radioactivity was counted using a liquid scintillation counter.
Electrophoretic Mobility Shift Assay
EMSA was performed on the amygdala neurons isolated from unpaired control or conditioned animals at specific times after training. For binding reactions, 10 μg of protein was incubated in binding buffer [0.05% Nonidet P-40, 10% glycerol, 10 mM HEPES, pH 7.9, 50 mM KCl, 0.1 mM EDTA, 2.5 mM dithiothreitol, and 0.25 mg/ml poly(dI-dC)] for 15 min at room temperature. The double-stranded NF-κB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′) (Santa Cruz Biotechnology, Inc.) was end-labeled with T4 polynucleotide kinase and [32P]ATP. Radiolabeled oligonucleotide was added to the reaction mixture and incubated for 20 min. The reaction products were analyzed by electrophoresis in a 4% polyacrylamide gel with 0.5× Tris-borate/EDTA buffer (22.3 mM Tris, 22.2 mM borate, and 0.5 mM EDTA). The dried gels were analyzed by autoradiography after an overnight exposure.
Results
We first investigated whether fear conditioning was associated with an increase in nuclear NF-κB acetylation. Rats were exposed to a light repeatedly paired with foot shock and, at various time points after training, the tissues of LA and BLA were removed for biochemical analysis. Acetyl-p65 was quantified by immunoprecipitation with a NF-κB p65 antibody, followed by immunoblotting with an acetyl lysine antibody. Figure 1A shows that fear training led to a significant increase in acetyl-p65 in the amygdala. The increase peaked at 30 min after training and subsided within 4 h (F(6,28) = 18.78, p < 0.0001). Newman-Keuls t tests revealed the differences between control and 0.5-, 1-, and 2-h time points (p < 0.01). No significant difference was detected between control and 10 min or control and 4- or 6-h time points (p > 0.05).

Nuclear p65 acetylation is transiently increased by fear training and the increase is prolonged by histone deacetylase inhibitors. A, representative Western blots showing the transient increase in the amygdala but not the hippocampus or cortex of acetyl-p65. Rats were exposed to a light repeatedly paired with an aversive foot shock or were exposed to the light and foot shock in an unpaired pseudo-random fashion (unpaired control). Nuclear extracts from the LA and BLA at various time points after training were prepared and immunoprecipitated with anti-p65 antibody followed by immunoblotting with acetyl lysine antibody. Administration of TSA (400 nM, 0.8 μl per side) or sodium butyrate (50 μM, 0.8 μl per side) to the amygdala bilaterally 30 min before training prolonged the duration of increase. B, comparison of the immunoreactivity of nuclear acetyl-p65 from the amygdala in TSA-, butyrate-, vehicle-treated, and unpaired rats. The immunoreactivities of nuclear acetyl-p65 from the hippocampus and cortex in conditioned rats were also included. ★, p < 0.001 versus vehicle.
We determined whether an increase in nuclear p65 acetylation associated with fear conditioning occurs specifically within the amygdala or is a general phenomenon across the entire brain by quantifying p65 acetylation in the cortex and hippocampus. As illustrated in Fig. 1B, training of visual conditioned fear did not induce an increase in p65 acetylation in the cortex (F(4,15) = 1.42, p = 0.28) and hippocampus (F(4,15) = 2.34, p = 0.38), indicating that the increase is restricted to the amygdala.
We further examined whether fear conditioning led to an increase of acetylated-p65 DNA binding activity. At various periods after fear conditioning, nuclear extracts from the amygdala were prepared and incubated with biotinylated κB-oligonucleotide. Subsequently, biotinylated κB-oligonucleotide-p65 complex was isolated by using columns with immobilized monomeric avidin gel and immunoprecipitated with NF-κB p65 antibody, and then immunoblotted with pan-acetyl antibody. Figure 2 shows that fear training caused a significant increase in acetyl-p65-κB-oligonucleotide complex. The increase peaked at 2 h after training and was sustained for at least 6 h (F(4,15) = 74.9, p < 0.0001). Newman-Keuls t tests revealed the differences between control and 2-, 4-, and 6-h time points (p < 0.001).
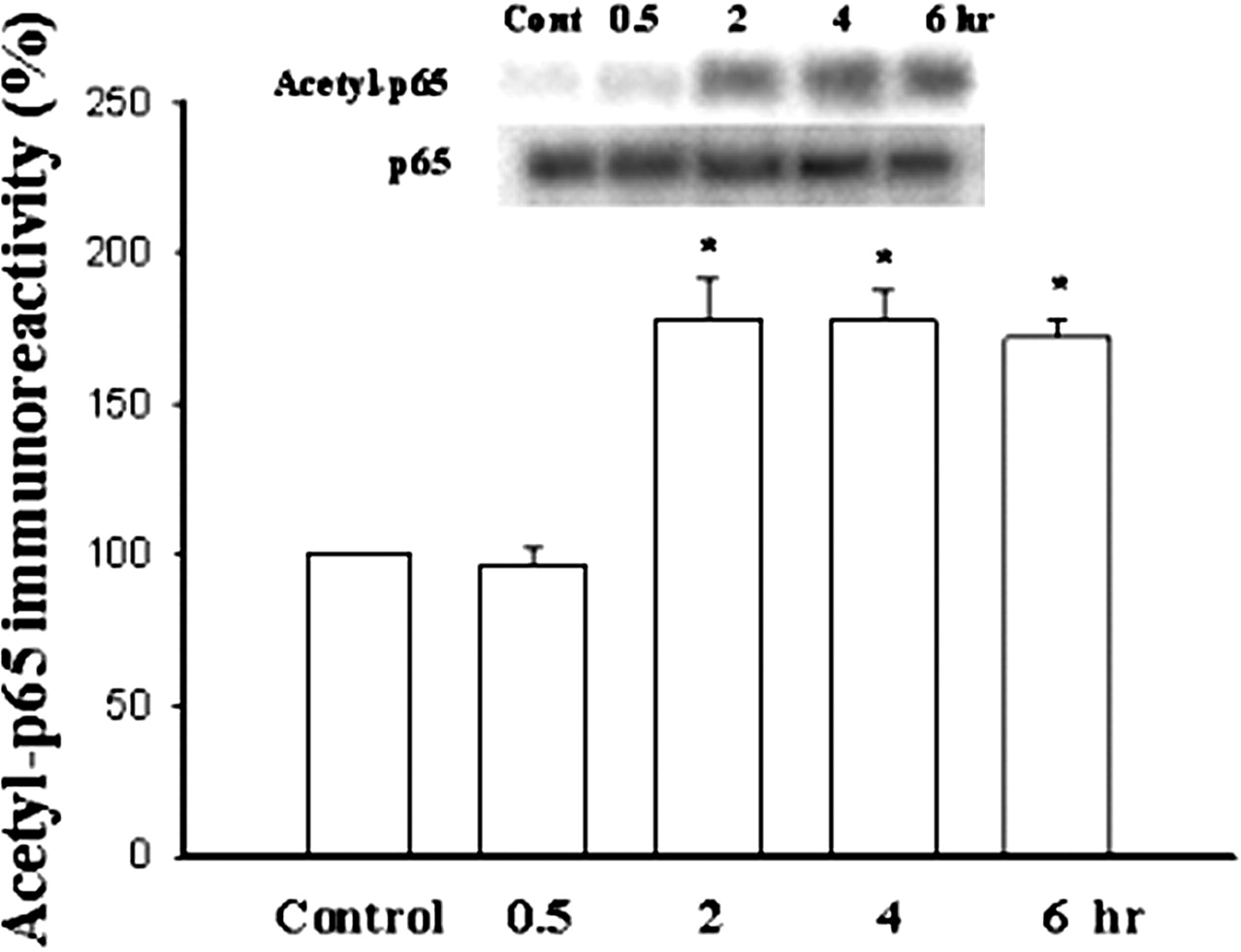
Increase in acetyl-p65 DNA binding activity after fear training. Nuclear extracts from the LA and BLA at various time points after training were prepared and treated with biotinylated-κB oligonucleotide. κB-oligonucleotide-protein complex was isolated by columns with immobilized monomeric avidin and bound proteins were either immunoblotted with NF-κB p65 antibody or eluted with 2 mM d-biotin, immunoprecipitated with anti-NF-κB p65 antibody, and immunoblotted with pan-acetyl antibody. ★, p < 0.001.
Parallel experiments were performed to examine the effect of fear conditioning on HAT activity. As illustrated in Fig. 3A, the activity of HAT was significantly increased, peaking at 0.5 to 1 h after training and sustained for at least 6 h (F(6,21) = 3.05, p < 0.05). It has been shown that p300/CBP contains HAT activity and over-expression of p300/CBP promotes p65 acetylation. We therefore investigated the interaction between CBP and p65 by performing coimmunoprecipitation experiments. At various time points after behavioral training, nuclear extracts from LA and BLA were immunoprecipitated with p65 antibody, followed by immunoblotting with CBP antibody. Figure 3B shows that the interaction between CBP and p65 was significantly increased, peaked at 0.5 to 1 h after training, and subsided within 2 h (F(7,24) = 30.9, p < 0.001).
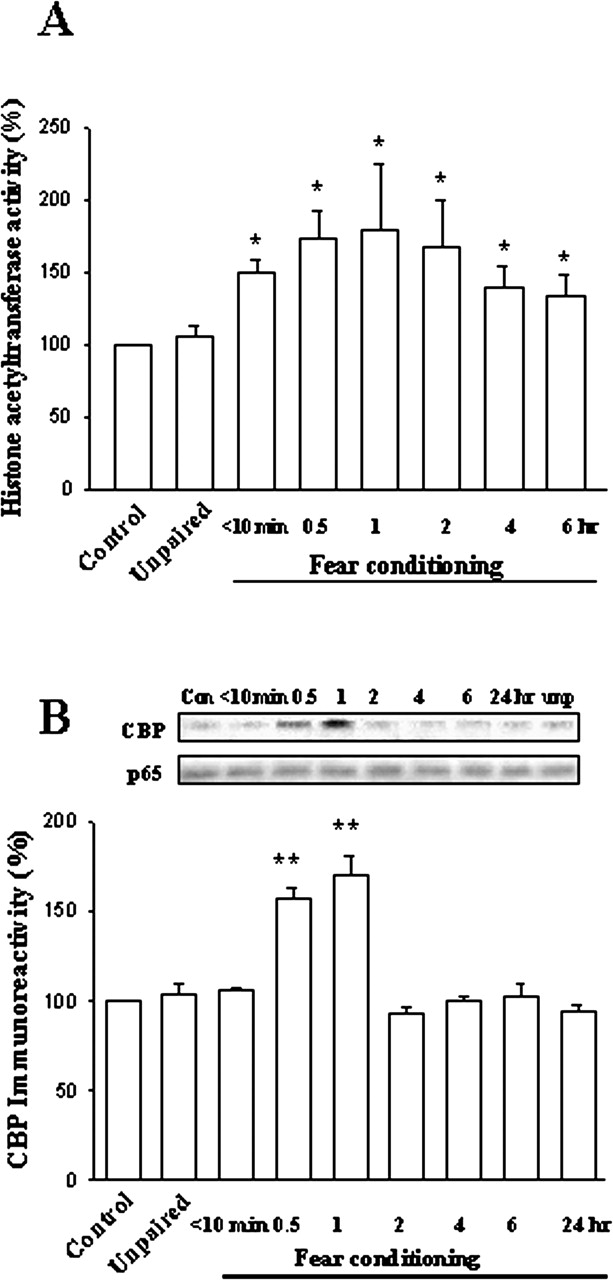
Fear conditioning stimulates HAT activity and the interaction between CBP and NF-κB. A, nuclear extracts from the LA and BLA at various time points after training were used to measure HAT activity as described under Materials and Methods. ★, p < 0.05 versus unpaired control. B, nuclear extracts were immunoprecipitated with p65 antibody and then immunoblotted with CBP. ★★, p < 0.001 versus unpaired control.
The net amount of acetylation of a protein depends on the balance between HAT and HDAC (Grunstein, 1997; Kouzarides, 1999). To determine the involvement of HDAC in deacetylation, rats were given intra-amygdalal injection of TSA (400 nM, 0.8 μl per side) 30 min before training. Densitometric analysis of five independent experiments revealed that conditioning-induced acetylation of p65 persisted for at least 6 h and was significantly different from control not receiving TSA (p < 0.001) (Fig. 1B). A similar experiment was performed with a structurally distinct HDAC inhibitor, sodium butyrate (50 μM, 0.8 μl per side). In butyrate-treated rats, conditioning-induced acetylation also persisted for at least 6 h (173.2 ± 2.7%, p < 0.001 versus control). In 293T cells, the HDAC subtype responsible for inhibition of tumor necrosis factor α-induced κB-luciferase activity is HDAC3 (Chen et al., 2001). We therefore determined the physical interaction between p65 and HDAC3 in vivo. At various time points after conditioning, nuclear extracts were immunoprecipitated using anti-HDAC3 antibody. The presence of p65 in immunoprecipitate was analyzed by Western blotting using anti-p65 antibody. As shown in Fig. 4, the interaction between HDAC3 and p65 was significantly increased at 2 h after training and sustained for at least 6 h (F(5,18) = 15.37, p < 0.001). Furthermore, the interaction between HDAC3 and p65 was abolished by treatment with TSA.

Interaction between HDAC3 and p65 is increased after fear training. At various time points after conditioning, nuclear extracts were immunoprecipitated using anti-p65 and anti-HDAC3 antibodies. The presence of p65 in immunoprecipitates was analyzed by Western blotting using anti-p65 antibody. The interaction between HDAC3 and p65 was abolished by treatment with TSA. ★, p < 0.05; ★★, p < 0.01 versus TSA.
We tested whether TSA affected NF-κB activation by using EMSA in which nuclear extracts from the amygdala were incubated with a radiolabeled NF-κB consensus DNA sequence. TSA (200 or 400 nM, 0.8 μl per side) by itself had no effect on the NF-κB DNA binding activity (Fig. 5A). However, as shown in Fig. 5B, it significantly enhanced conditioning-induced increase in DNA binding. The increase in DNA binding was markedly reduced by helenalin (50 μM, 0.8 μl per side) (Fig. 6), a sesquiterpenoid lactone that inhibits NF-κB DNA binding activity (Lyss et al., 1998). NF-κB DNA binding activity was eliminated in the presence of a 100-fold molar excess of unlabeled κB DNA (cold κB). We performed super-shift experiments to identify the NF-κB family members that were binding. The results revealed that both p65 and p50 antibodies retarded the migration of proteins interacting with NF-κB oligonucleotide.

TSA enhances conditioning-induced DNA binding of NF-κB in the amygdala. Rats were injected with TSA (200 or 400 nM, 0.8 μl per side) bilaterally, 30 min before giving 10 pairings of light and foot shock in an unpaired or paired manner. Nuclear extracts from the amygdala were analyzed with EMSA using a radiolabeled NF-κB probe. A, TSA did not affect the NF-κB DNA binding in the unpaired rats. B, by contrast, it significantly increased the binding in conditioned rats.

Supershift analysis of DNA binding activity of NF-κB. Nuclear extracts from the amygdala were analyzed with EMSA using radiolabeled NF-κB probe. Fear conditioning significantly increased NF-κB DNA binding activity, and helenalin (50 μM, 0.8 μl per side) blocked fear training as well as TSA enhancement of NF-κB DNA binding. DNA binding activity of NF-κB was eliminated in the presence of a 100-fold excess of unlabeled NF-κB probe. Supershift experiments with anti-p65 and anti-p50 demonstrated that the protein complex interacting with the NF-κB oligonucleotide contained a p65/p50 heterodimer.
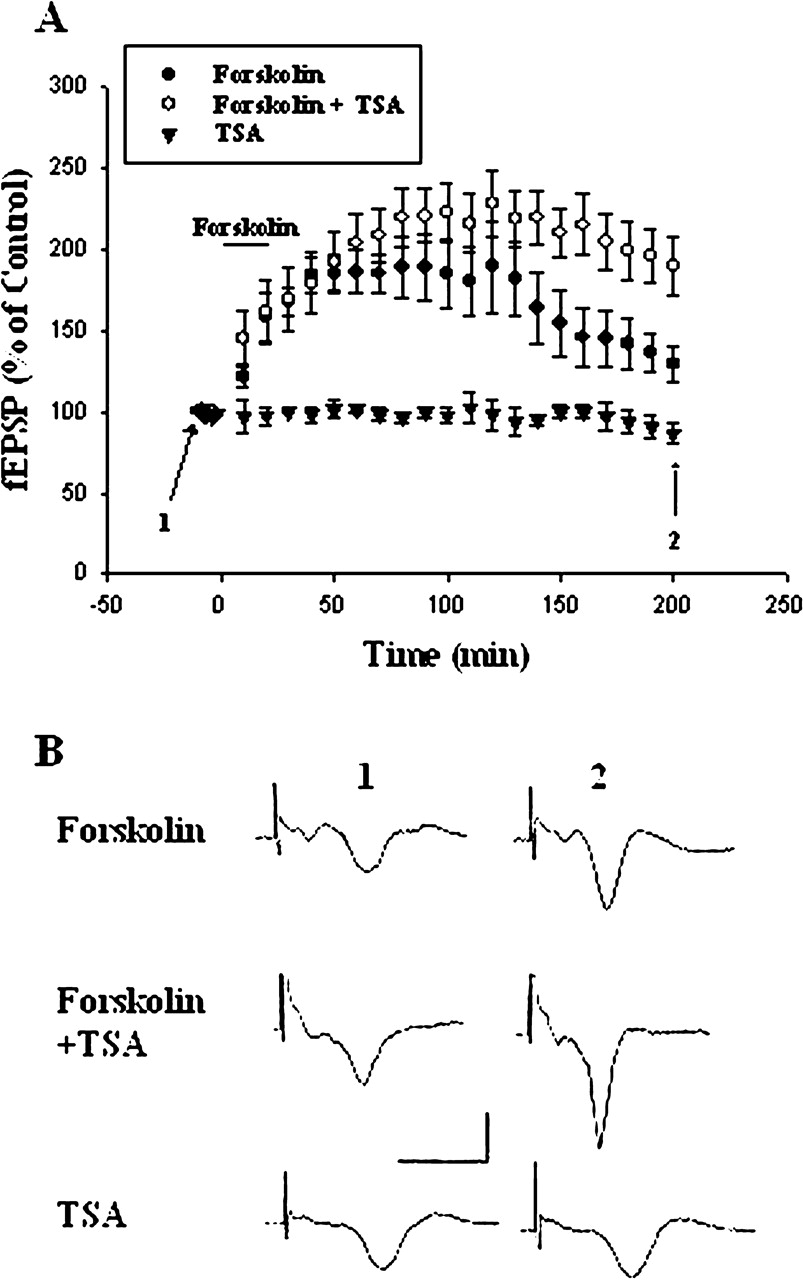
Is NF-κB acetylation functionally important for synaptic plasticity? Long-term potentiation (LTP) at sensory input synapses to the LA and BLA is a candidate mechanism for memory storage during fear conditioning (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). We determined whether block of histone deacetylase with TSA affected the LTP. In control slices, application of forskolin (50 μM) for 15 min induced a LTP that lasted for at least 3 h. The slope of fEPSP at 3 h after the washout of forskolin was 129.2 ± 10.1% (n = 6) of baseline. By contrast, in TSA (1 μM)-treated slices, the slope of fEPSP at 3 h after washout of forskolin was 192.7 ± 19.2% (n = 6) of baseline, which was significantly different from that of control (p < 0.02) (Fig. 7). Exposure to TSA alone had no long-term effect on the fEPSP (87 ± 5% of control 3 h after the washout of TSA, n = 5). Thus, TSA increased the degree of LTP.

Enhancement of LTP by TSA. A, application of forskolin (50 μM) for 15 min induced LTP that lasted for at least for 3 h. In TSA experiments, slices were incubated with TSA (1 μM) 10 min before and during the experiments. TSA significantly increased the magnitude of LTP. B, sample traces taken from experiments A at the times indicated. Calibration: 0.5 mV, 10 ms.
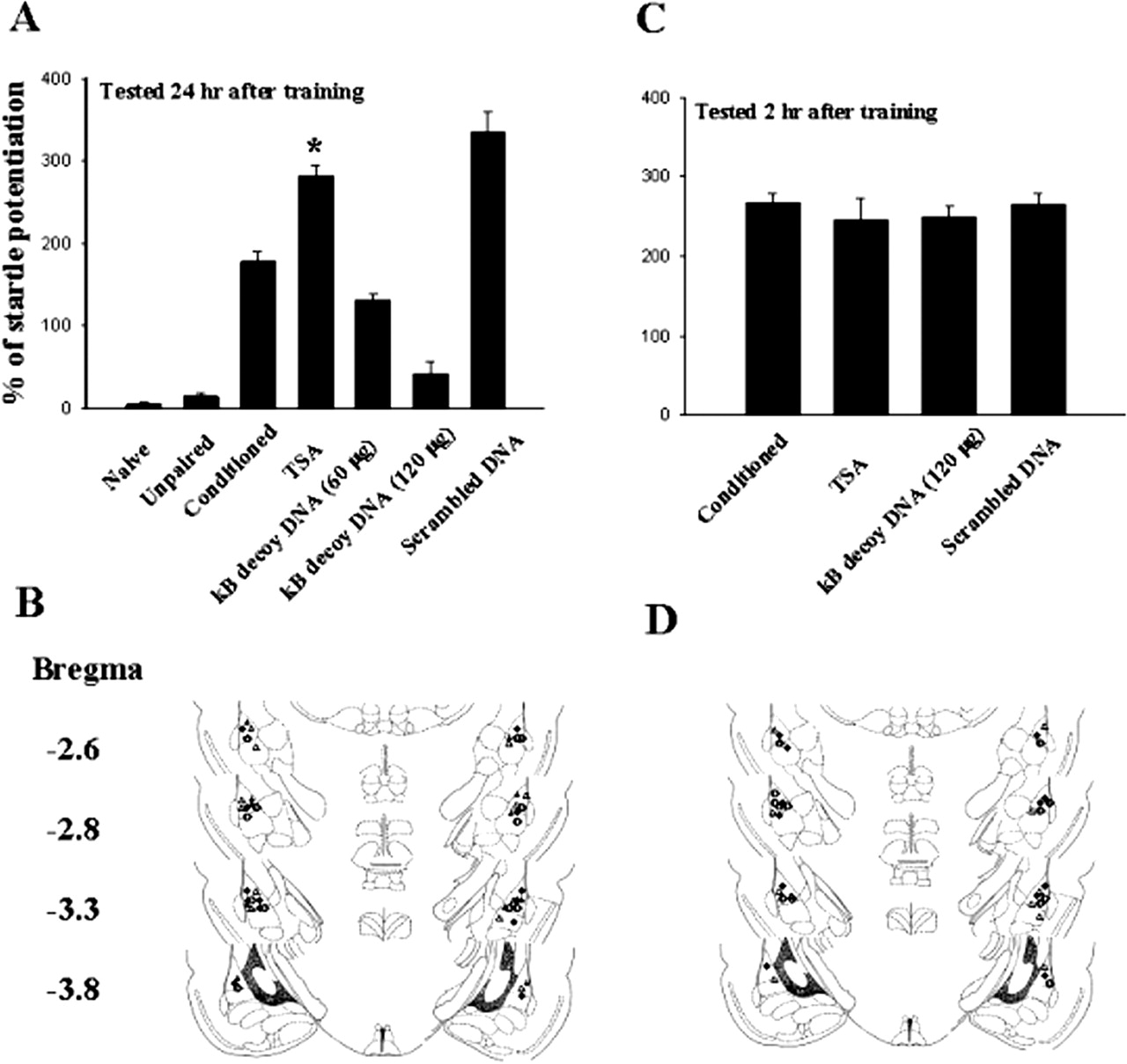
We examined whether TSA affected fear-potentiated startle. Rats were randomly assigned to three groups: fear-conditioned and two sets of controls (naive and unpaired). Figure 8A shows that, of the three groups, only rats in the conditioned group learned to associate the light with shock (178.3 ± 11.7%, n = 6). This was manifested as an increase in acoustic startle relative to naive and unpaired controls. Next, rats were pretreated with TSA (400 nM, 0.8 μl per side) and 30 min later were given 10 pairings of light and foot shock. Retention of memory was assessed 24 h later. As illustrated in Fig. 8, startle amplitude in TSA-treated rats (282.3 ± 11.8%, n = 6) was significantly higher than that of conditioned rats (p < 0.001). To confirm whether the enhancement of fear-potentiated startle by TSA was mediated by activation of NF-κB, we administered double-stranded κB decoy DNA (60 or 120 μg in 1.6 μl of saline, 0.8 μl per side) or control double-stranded DNA with a scrambled sequence bilaterally into the amygdala (120 μg in 1.6 μl of saline, 0.8 μl per side). Quantification of startle potentiation 24 h after conditioning revealed that κB decoy DNA produced a marked depression of potentiation in a dose-dependent manner (F(2,15) = 13, p < 0.0001). In contrast, rats that received scrambled DNA exhibited a normal potentiation (335.7 ± 23.7%, n = 6, p = 0.13 unpaired t test). To determine whether κB decoy DNA affected the short-term memory, additional rats were divided into four groups: conditioned, or pretreated with TSA, TSA plus κB decoy DNA (120 μg in 1.6 μl of saline, 0.8 μl per side), or TSA plus scrambled DNA (120 μg in 1.6 μl of saline, 0.8 μl per side). Behavioral tests were performed at 2 h after training. As shown in Fig. 8B, κB decoy DNA did not significantly affect short-term memory. The degree of startle potentiation in κB decoy DNA-treated rats was not different from that treated with TSA alone (t(10) = 0.14, p = 0.89) but was significantly different from that tested 24 h after training (t(10) = 7.88, p < 0.001).

Enhancement of fear-potentiated startle by TSA. A, potentiated startle reflex tested 24 h after training in rats giving TSA (400 nM, 0.8 μl per side), TSA plus κB decoy DNA (60 or 120 μg in 1.6 μl of saline, 0.8 μl per side), or TSA plus scrambled DNA (120 μg in 1.6 μl of saline, 0.8 μl per side). Fear-potentiated startle was enhanced by TSA treatment and reduced by κB decoy DNA in a dose-dependent manner, but not by scrambled DNA. ★, p < 0.001 versus conditioned. B, cannula tip placements from rats infused with TSA (○), TSA plus κB decoy DNA (60 μg, •; or 120 μg, ▵), or TSA plus scrambled DNA (•) in experiments A. C, TSA and κB decoy DNA did not affect short-term memory. Rats were given TSA (400 nM, 0.8 μl per side), TSA plus κB decoy DNA (120 μg in 1.6 μl of saline, 0.8 μl per side), or TSA plus scrambled DNA (120 μg in 1.6 μl of saline, 0.8 μl per side), and behavioral tests were performed at 2 h after training. D, cannula tip placements from rats given TSA (○), TSA plus κB decoy DNA (•), or TSA plus scrambled DNA (▵) in experiments C.
Discussion
Fear Conditioning Is Associated with an Increase in the Acetylation of p65. The nuclear function of NF-κB transcription factor is regulated by reversible acetylation. However, acetylation has been shown to exert opposite effects on the function of NF-κB depending on the type of cells studied. In HeLa cells, acetylation facilitated NF-κB removal from binding to DNA and exported to the cytoplasm to turn off transcription (Kiernan et al., 2003). Conversely, by impairing IκBα assembly, acetylation decreased nuclear export of NF-κB, resulting in the enhancement of DNA binding activity in 293T cells (Chen et al., 2002). The results presented in this study provide the first evidence that this post-transcriptional modification occurs in the brain after fear conditioning, leading to the increase in DNA binding activity.
P300/CBP has been shown to participate in the acetylation of p65 and a variety of nonhistone substrates both in vivo and in vitro (Boyes et al., 1998; Martinez-Balbas et al., 2000; Marzio et al., 2000; Chen et al., 2002). Cyclic AMP-response element-binding protein (CREB) is one of the core components in the molecular switch that converts short-term to long-term synaptic plasticity (Impey et al., 1998; Josselyn et al., 2001; Lin et al., 2001; Barco et al., 2002; Kida et al., 2002). Phosphorylation of CREB at serine 133 enhanced transcription via the recruitment of coactivators such as p300/CBP (Chrivia et al., 1993). We have shown previously that application of forskolin or tetanic stimulation, which reliably evoked long-term potentiation in the amygdala, stimulated the interaction between CREB and NF-κB (Yeh et al., 2002). Here, we demonstrated that fear training stimulates the interaction between CBP and NF-κB, suggesting that CBP may mediate the acetylation of p65. Thus, p300/CBP not only acted as CREB transcriptional activator but also cooperatively stimulated p65-mediated transcription.
Inhibition of HDAC Prolongs Nuclear Acetylation of p65 and Enhances LTP as well as Fear-Potentiated Startle. We observed that TSA, a specific inhibitor of multiple HDACs, prolonged the duration of nuclear p65 acetylation. The effect was shared by a structurally distinct HDAC inhibitor, sodium butyrate. Immunoprecipitation of the nuclear extract with antibody to p65 followed by immunoblotting with pan-acetyl antibody revealed sustained nuclear acetyl-p65 expression. In addition, TSA enhanced NF-κB DNA binding activity but did not itself stimulate NF-κB binding. In amygdala slices, TSA by itself did not affect basal synaptic transmission but enhanced LTP. In the same vein, behavioral studies showed that TSA increased, whereas κB decoy DNA reduced fear-potentiated startle tested 24 h after training. On the other hand, both TSA and κB decoy DNA had no effect on the startle reflex when tested at 2 h after training, suggesting selectivity on long-term memory formation. The lack of effect on short-term memory also ruled out the nonspecific effects, such as impaired motor activity. Taken together, these results suggest that TSA enhanced fear memory, probably via prolongation of intranuclear p65 acetylation, resulting in increased DNA binding activity.
Consistent with this hypothesis, we found that fear training stimulated the interaction between p65 and HDAC3, which was abolished in the presence of TSA. TSA broadly inhibited action of the HDACs and HDAC function, not only to deacetylate core histones but also to deacetylate various host transcription factors (Kuo and Allis, 1998; Hui Ng and Bird, 2000), altering their transcriptional activity. Therefore, an important issue is whether enhancement of fear memory by TSA could be attributed to modification of NF-κB specifically or, in a more indirect manner, alteration of chromatin structure. The fact that fear conditioning increases nuclear p65 acetylation and the increase is restricted to the amygdala indicates that it occurs in the brain region involved in the formation of fear memory. Furthermore, the control group that received light and foot shock in an unpaired fashion did not present any increase in acetyl-p65, suggesting that the increases were specific to the learning component of the task.
In summary, we have observed that fear conditioning is associated with an increase in nuclear expression of acetylp65 as well as its DNA binding activity. The enhancement and block of these effects by TSA and NF-κB inhibitor, respectively, is consistent with the notion that the duration of nuclear NF-κB action is regulated by reversible acetylation. Furthermore, behavioral tests show that HDAC inhibitors enhance fear memory. These results suggest that HDAC-mediated deacetylation functions as an intranuclear molecular switch culminating in the termination of NF-κB transcriptional response. A better understanding of these molecular events will allow the design of therapies targeted at post-traumatic stress disorders.
Acknowledgments
We thank Hsin-Yi Lu for technical assistance.
Footnotes
-
This work was supported by the National Health Research Institutes (NHRI-EX92-9202NI), Academic Excellence Program of the Ministry of Education (89-B-FA08-1-4), and China Medical University (CMU92-CI-04).
-
ABBREVIATIONS: NF-κB, nuclear factor-κB; BLA, basolateral nucleus of the amygdala; CREB, cyclic AMP response element-binding protein; CBP, CREB-binding protein; CRM-1, chromosomal region maintenance-1; CS, conditioned stimulus; EMSA, electrophoretic mobility shift assay; HAT, histone acetyl-transferase; HDAC, histone deacetylase; ITI, intertrial interval; LA, lateral nucleus of the amygdala; LTP, long-term potentiation; TNF-α, tumor-necrosis factor-α; TSA, trichostatin A; ACSF, artificial cerebrospinal fluid; PMSF, phenylmethylsulfonyl fluoride; EPSP, excitatory postsynaptic potential.
- Received August 25, 2003.
- Accepted February 9, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}