


I. Introduction
Upon stimulation of vagal nerves, acetylcholine (ACh)c is released from axonal terminals and decelerates the heart beat. This historic discovery by Otto Loewi in the 1920s established the concept of chemical synaptic transmission (Loewi, 1921; Loewi and Navaratil, 1926). Since then, many physiologists have been trying to elucidate the mechanism(s) underlying neurotransmitter (Vagusstoff)-induced bradycardia. Del Castillo and Katz (1955) first described hyperpolarization of the membrane induced by ACh in frog heart. Hutter and Trautwein (1955) measured an increase of K+ efflux across the cardiac cell membrane with vagal stimulation. Trautwein and Dudel (1958) showed an increase of K+ conductance under voltage-clamp conditions. Trautwein and colleagues analyzed the kinetics of the ACh-induced K+ current in the rabbit sinoatrial node and proposed that ACh induces activation of a specific population of K+ channels, named muscarinic K+ (KACh) channels, to decelerate pacemaker activity (Noma and Trautwein, 1978; Osterriederet al., 1981). The single channel currents of the KACh channels were recorded for the first time bySakmann et al. (1983), who showed that the channel exhibited kinetic properties that clearly differed from those of the background inwardly rectifying K+(IK1) channel in cardiac myocytes.
The next big step was the discovery that pertussis toxin (PTX)-sensitive heterotrimeric G proteins are involved in the activation of the KACh channel by M2-muscarinic and A1adenosine receptors (Pfaffinger et al., 1985; Breitwieser and Szabo, 1985; Kurachi et al., 1986a and b). Because the KACh channel could be activated by intracellular guanosine 5′-triphosphate (GTP) (in the presence of agonists) and GTPγS (even in the absence of agonists) in cell-free inside-out patches, the system seemed to be delimited to the cell membrane, which led to the proposal that the channel is directly activated by G proteins (Kurachi et al., 1986a,b,c). The G protein responsible for activation of the KACh channel was designated GK according to its function (Breitwieser and Szabo, 1985).
It was quite a surprise that the βγ subunit (Gβγ) but not the α subunit (Gα) of the GK protein, was proposed to mediate the GK-induced activation of KACh channels (Logothetis et al., 1987, 1988; Kurachi et al, 1989a), because it was strongly believed at that time that regulation of different effectors by G proteins was mediated only by Gα, although Gβγ merely served to bind to the GDP-form of Gα(Gα-GDP) to anchor the trimeric G protein to the cell membrane (Gilman, 1987). Actually, Brown, Birnbaumer, and their colleagues proposed GKα and not GKβγ as the physiological activator of KACh channels (Yatani et al., 1987, 1988; Codina et al., 1987; for review seeBrown and Birnbaumer, 1990). The dispute concerning the G protein subunit responsible for the physiological activation of KACh channels continued for nearly a decade (Itoet al., 1992; Yamada et al., 1993,1994a,b; Nanavati et al., 1990; Kurachi, 1989, 1990,1993, 1994, 1995; Kurachi et al., 1992; Clapham and Neer, 1993; Wickman and Clapham, 1995) until the functional interaction between the channel and Gβγ was shown at the molecular level with cloned G protein-gated K+ (KG) channel and/or G protein subunits (Kubo et al., 1993b; Dascal et al., 1993; Wickman et al., 1994; Reuveny et al., 1994; Krapivinsky et al., 1995a; Inanobe et al., 1995b). Now it is established that GKβγ is the physiological activator of KG channels not only in cardiac myocytes, but also in neurons and endocrine cells. Recently, it was indicated that G protein-inhibition of neuronal Ca2+ channels is also mediated by Gβγ and not by Gα (Herlitze et al., 1996; Ikeda, 1996). Efforts are now being made to elucidate the molecular mechanisms underlying Gβγ-control of KG and N-type Ca2+channels.
The importance of the G protein-activation of KGchannel system in receptor-mediated regulation of cell responses is now more widely appreciated than before because a wide variety of membrane receptors, such as M2-muscarinic, A1 adenosine, α2-adrenergic, D2dopamine, μ-, δ-, and κ-opioid, 5-HT1Aserotonin, somatostatin, galanin, m-Glu, GABAB, and sphingosine-1phosphate receptors, have been shown to use this system in inhibiting cell excitation in various organs (North et al., 1987; Lacey et al., 1988; Hille, 1992a; Grudt and Williams, 1993; Oh et al., 1995; Saugstad et al., 1996; Sharon et al., 1997; Bünemann et al., 1995; Koppen et al., 1996). In this review, we will first summarize ACh-activation of cardiac KAChchannels, the prototype of this system, and then recent progress in molecular dissection of the KG channel system.
II. Functional Analysis of G Protein-Mediated Activation of Muscarinic K+ Channels in Cardiac Atrial Myocytes
A. Time-Dependent Response of the Whole-Cell Muscarinic K+ Current to Acetylcholine
ACh added to the extracellular solution elicits a KACh channel current in cardiac atrial myocytes (fig. 1). The activation time-course is sigmoidal and takes several hundred milliseconds to reach a peak (Breitwieser and Szabo, 1988). Thereafter, the evoked current gradually decreases to a quasi-steady-state level within 1 min in the presence of high concentrations of ACh (> 0.3 μm). This reduction of cell K+ current in the continuous presence of ACh is called “short-term” desensitization (Kurachi et al., 1987b). After wash-out of the agonist, the current disappears within several seconds (deactivation). It is worth noting that in the inside-out patch configuration of the patch-clamp method, one measures KACh channel activity only in the steady-state phase. Thus, in these experiments, limited information is available regarding the desensitization of the channel.

Time-dependent response of the whole-cell muscarinic K+ channel current to acetylcholine. By using the whole-cell voltage clamp method of the patch-clamp technique, the response of the whole-cell current of a guinea-pig atrial myocyte to 11 μm acetylcholine (ACh) was measured. In the presence of normal Tyrode solution that contained 5.4 mm external K+, the cell membrane potential was clamped at −53 mV. The patch pipette contained (in mm): 150 KCl, 2 MgCl2, 5 EGTA, 5 HEPES, and 0.1 GTP (pH = 7.3). ACh was applied to the bath for the period indicated by the horizontal bar above the cell membrane current trace. An arrowhead indicates the zero current level. An upward deflection of the cell current record indicated an outwardly directed cell membrane current that would be carried by the movement of K+ ions under these circumstances.
These three phases of the response involve interactions between an agonist (i.e., ACh), an M2-muscarinic receptor, a PTX-sensitive G protein, and the KACh channel. Therefore, to understand the reaction of the KAChchannel to ACh, it is necessary to know how the receptor-generated signal is transferred to the channel through the G protein and how this signal transmission might be modulated by other factors interacting with these different reactions.
1. The G protein cyclic reaction mediating the receptor-to-channel signal transmission.
Activation of KAChchannel induced by M2-muscarinic receptor stimulation is mediated by a heterotrimeric G protein (GK) (fig. 2). The heterotrimeric G proteins are membrane-bound proteins which transduce signals from receptors to effectors such as adenylyl cyclase, phospholipase C, the KACh channel, and other ion channels (Gilman, 1987). These proteins are composed of α, β, and γ subunits (Gα, Gβ, and Gγ, respectively). Up to now, at least 16 Gα, 5 Gβ, and 11 Gγ genes have been identified (Bourne, 1997). Heterotrimeric G proteins interact with receptors through Gα. It is well known that the interaction between M2-muscarinic receptors and GKα is blocked by the toxin from Bordetella pertussis (PTX) (Ui, 1984; Kurose et al., 1986). PTX modifies covalently a cysteine residue at the carboxyl-terminal end of Gα subunits belonging to Gi, Go, and Gt families by transferring an ADP-ribose group from the nicotinamide adenine dinucleotide moiety to the cysteine residue (Gilman, 1987). Because the receptor-mediated activation of KG channels in cardiac atrial myocytes and neurons are inhibited by PTX (Pfaffinger et al., 1985; Kurachi et al., 1986a), GK seems to belong to one of these G protein families. However, its molecular identity has not been fully elucidated, although GK is proposed to be a member of the Gi class of G proteins in some systems (Kozasa et al., 1996; Takano et al., 1997)

Schematic representation of the G protein cycle involved in the activation of the muscarinic K+ channel in response to acetylcholine.
The following is the current understanding of the interaction among receptors, G proteins, and KACh channels. In the absence of agonists, most of Gα is in the GDP-bound form (Gα- GDP) (fig. 2). Gα-GDP has high affinity for Gβγ, thereby forming a heterotrimer with Gβγ (Gilman, 1987). A small fraction of Gα does release GDP even in the absence of agonists, and in turn binds GTP (GDP/GTP exchange) and becomes a GTP-bound form (Gα-GTP). Receptor stimulation substantially increases the GDP dissociation rate, which results in marked acceleration of the GDP/GTP exchange reaction. Formation of Gα-GTP leads to dissociation of Gβγ from Gα. The dissociated Gβγ, which is always a dimer under physiological conditions, interacts with the KACh channel to activate the channel. Besides the KACh channel, many effectors of G proteins have been known to be regulated by Gβγ (table1) (Clapham and Neer, 1993;Iñiguez-Lluhi et al., 1993).
G protein effectors regulated by G protein βγ subunits
Gα has a slow intrinsic GTPase activity: its Kcat value is typically 1 to 5/min (Gilman, 1987). Gα, therefore, hydrolyses the GTP on its own molecule to GDP, thereby returning to the GDP-bound form and re-associating with Gβγ. This reaction terminates the effector activation. In the continuous presence of agonists, the heterotrimeric G protein restarts the cyclic reaction by interacting with an agonist-bound receptor.
2. Activation phase.
The time to peak of the ACh-induced response of the KACh channel is dependent on ACh concentration: the higher the concentration of ACh, the faster the activation. In the presence of a maximum effective concentration of ACh, the time to peak is several hundred milliseconds. If the M2-muscarinic receptor, GK, and the KACh channel encountered by simple diffusion in the membrane, the response time requires that all these signaling molecules be within less than 1.5 μm of each other (Hille, 1992a). The molecular mechanism satisfying such a topological requirement has not been clearly identified. However, it was recently suggested that KACh channel subunits may directly interact with not only GKβγ but also GKα, trimeric GK, and the receptor and thereby might form a complex with these proteins (Huang et al., 1995; Slesingeret al., 1995) (detailed in the Sections III.F.3. and 4.).
When a recombinant KG channel corresponding to the KACh channel is expressed with M2-muscarinic receptors in Xenopusoocytes, the time course of the activation is much slower than in native atrial myocytes (Krapivinsky et al., 1995a). It was recently demonstrated that newly identified molecules known as regulators of G protein signaling proteins (RGS) serve to increase the activation rate of recombinant KG channels expressed in oocytes and a mammalian cell line (Doupnik et al., 1997; Saitoh et al., 1997). RGS proteins are the members of a multigene family that enhance the intrinsic GTPase activity of certain G proteins (mainly Gi/Go classes) probably by preferentially binding to and stabilizing G proteins in their transition state for the hydrolysis reaction (Koelle, 1997). Sixteen RGS homologues (RGS1–16) have been identified in mammals. Among them, RGS1, RGS3, RGS4, and RGS8 have been shown to shorten the time to peak of receptor-mediated activation of KG channels (Doupnik et al., 1997; Saitoh et al., 1997). Enhancement of the GTPase activity by RGS proteins leads to an increase in the off-rate of the G protein-mediated reaction (Koelle, 1997) (fig.2). This effect, at least in theory, could abbreviate the time to peak when the on-rate of the reaction is not altered by the protein (Doupniket al., 1997; Saitoh et al., 1997). In this case, the steady state KG channel activity should be decreased in the presence of a given concentration of an agonist. However, RGS proteins enhance the activation rate without changing the amplitude of the steady-state response in the KGchannel systems (Doupnik et al., 1997; Saitoh et al., 1997). One possible explanation of this phenomenon is that RGS proteins may also enhance the GDP/GTP exchange rate of GK. However, this could not be confirmed at least in an in vitro system that lacked reconstituted receptor proteins (Saitoh et al., 1997). It is still possible that RGS proteins might increase the on-rate of the GK-mediated reaction only in the presence of receptors or, alternatively, accelerate the subunit dissociation of GK. Further studies are necessary to identify the mechanism by which the RGS proteins accelerate the agonist-mediated KG channel activation without affecting the steady-state response.
3. The phase of short-term desensitization.
Short-term desensitization becomes more prominent as the concentration of ACh is increased above 0.3 μm (Kurachi et al., 1987b). This may at least partly arise from the transition of M2-muscarinic receptors from the high to low affinity-binding state due to dissociation of GKfrom receptors after agonist application (Gilman, 1987). Recent studies demonstrated that heterologous coexpression of RGS proteins with M2-receptors and recombinant KG channels reestablishes the short-term desensitization, which normally cannot be seen in the absence of RGS proteins in the reconstituted system (Doupnik et al., 1997). Therefore, this protein may also be one of the molecules responsible for the short-term desensitization of the KGchannel system. Other possible candidates for the short-term desensitization include phosphorylation of M2-muscarinic receptors by β-adrenergic receptor kinase (βARK), dephosphorylation of KACh channels and functional modulation of G proteins.
It is, however, unlikely that the phosphorylation of M2-muscarinic receptors by βARK is responsible for the short time desensitization because receptor phosphorylation occurs much slower than the desensitization (Kwatra and Hosey, 1986;Kwatra et al., 1987), and the kinase inhibitor (heparin) does not affect the desensitization time course (Mubagwa et al., 1994). However, the receptor phosphorylation by βARK may underlie the slow desensitization of KAChchannels which occurs in an order of minutes (Shui et al., 1995).
As mentioned in Section II.A., single-channel recording techniques provide only limited information about the time course of channel’s response to an extracellular ligand. This is due to the presence of agonists in the pipette solution which is going to be in contact with the cell membrane for a certain amount of time before the “giga-seal” will be formed. In most experiments therefore, short-term desensitization would have been achieved to some extent before single-channel events can be recorded. Under the conditions where a “giga-seal” could form exceptionally very rapidly, Kim (1990 and 1991) showed that the open time of KAChchannels was ∼5 msec at the beginning of the cell-attached patch recording and gradually decreased to ∼1 msec with time. Such time-dependent reduction of the channel open time might correspond to short-term desensitization. Kim (1990 and 1991) attributed this phenomenon to “dephosphorylation” of the KAChchannels in the presence of high concentrations of ACh although there is no direct evidence for phosphorylation or dephosphorylation of the channel protein. However, the open time of KAChchannels in the presence of low concentrations of ACh or even in the absence of the agonist under steady state conditions is also ∼1 msec. A possibility remains that different populations of KACh channels might be activated by low and high concentrations of ACh. A population with long open times might be less sensitive to G protein-activation due to “phosphorylation” and thus activated only by high concentrations of ACh. The “dephosphorylation” of these channels in the presence of high concentrations of ACh may then cause shortening of the open time, resulting in the decrease of the whole-cell current. The KACh channels with the short open time of ∼1 msec may be dephosphorylated and more sensitive to G protein activation. In the presence of nondesensitizing concentrations of ACh, therefore, the KACh channels with short open time would be activated preferentially. Consistent with this hypothesis, we have observed that where we had thought to have already maximally activated KACh channels with exogenously applied Gβγ subunits, the addition of mm intracellular ATP enhanced channel activity by prolonging open time (Yamada M and Kurachi Y, unpublished observation).
Huang et al. (1998) recently reported that exogenously applied phosphatidylinositol 4,5-bisphosphate (PIP2) increased the sensitivity of recombinant KG channels to Gβγin inside-out patch membranes of Xenopus oocytes. Because activation of M2-muscarinic receptors in atrial cardiac myocytes induces the phosphoinositide turnover (Quist, 1982), the resultant decrease in PIP2 content in the membrane might cause the short-term desensitization. Huang et al. (1998) also showed that intracellular ATP activated the recombinant KG channels by increasing PIP2 contents in the membrane. Therefore, the ATP-induced elongation of the open time of KAChchannels might be caused by an increase in PIP2contents in the membrane.
4. Deactivation of the response of the muscarinic K+channel.
The ACh-induced K+ current disappears quickly when the agonist is washed out from the extracellular solution (fig. 1). The rate of deactivation of the whole-cell KACh channel current was estimated as ∼30 to 200/min, which is much more rapid than either the GTP hydrolysis rate of G proteins (∼1 to 5/min) or the rate of dissociation of Gβγ from KG channel subunits (∼0.01/min) estimated in vitro (Breitwieser and Szabo, 1988; Nakajima et al., 1992;Gilman, 1987; Doupnik et al., 1997; Krapivinsky et al., 1995c). This discrepancy might in part be attributed to positive cooperativity in the interaction between the channel and GKβγ that will be described in the Section II.B.2., where even a slight decrease in free GKβγ concentration in the membrane should cause a larger reduction of the channel activity.
The deactivation of KG channels heterologously expressed in Xenopus oocytes occurs much more slowly than that of the native channel (Dascal et al., 1993; Slesingeret al., 1995). Again, RGS proteins have been found to enhance the deactivation rate of recombinant KGchannels approximately to the value of the native KACh channel (Doupnik et al., 1997;Saitoh et al., 1997). This effect of RGS proteins can be explained in terms of their increasing the GTPase activity of Gi/Go proteins (Koelle, 1997). Therefore, RGS proteins accelerate both activation and deactivation rates of KG channel systems and thus enable the systems to faithfully follow such a train of brief increases in agonist concentration as occurs in synaptic signal transmission (Doupnik et al., 1997).
In the presence of the same concentration of ACh, the apparent potency of GTP in activating the KACh channel in excised membrane patches differ depending on the intracellular anion species (Nakajima et al., 1992). The apparent potency of GTP decreases in the order: Cl− > Br− > I− > SO4− or aspartate. Because the potency of the nonhydrolyzable GTP analogue, GTPγS is not affected by intracellular anion species, the GTPase activity of GK seems to be modulated by intracellular anions. These effects of intracellular anions need to be taken into consideration because in most studies the internal side of the inside-out patch membrane is perfused with solution containing a much higher concentration of Cl− than that in the cytosol of most cells.
One related issue to be discussed here is the basal activity of the KG channel system that is observed in the absence of agonists. The native KACh channel exhibits much smaller basal activity relative to the agonistinduced maximum activity than heterologously expressed recombinant KG channels (Kurachi, 1990; Kubo et al., 1993b; Dascal et al., 1993). RGS proteins significantly reduce the basal activity of recombinant KG channels probably by activating GTPase of GK (Doupnik et al., 1997).
B. Quantitative Analysis of G Protein-Mediated Activation of the Muscarinic K+ Channel
The unique feature of the KG channel is the increase in channel activity in response to GKactivation. This response is mediated by interaction between GKβγ and a KGchannel. How they interact with each other and how the interaction leads to channel activation are intriguing questions.
The mechanism of GKβγ/KGchannel interaction has been mainly investigated in the KACh channel with inside-out patch membranes of cardiac atrial myocytes because in this system it is relatively easy to obtain many KG channels that will respond to guanine nucleotides and G protein subunits applied to the internal side of the patch membranes. One can then directly analyze the membrane-delimited activation of the KG channel by GK in detail. In the following, we discuss the results obtained from such studies. We first describe the single-channel characteristics of the KAChchannel and then go into the detail of the quantitative analysis of the GK/KACh channel interaction.
1. Single-channel characteristics of the muscarinic K+channel.
Fig. 3A shows single-channel recording of the KACh channel obtained from a cell-attached membrane of a guinea-pig atrial myocyte (Kurachi et al., 1986a). In general, K+ ions flow through K+channels depending on the electrochemical gradient for K+ ions across the plasma membrane. This gradient is the difference between the membrane potential (Vm) and the K+ equilibrium potential (EK): Vm − EK. The single-channel current flowing through a K+-selective channel can be described as follows:

Single-channel properties of the muscarinic K+ channel. A: Cell-attached recordings of muscarinic K+ channel currents from a single guinea pig atrial myocyte at different membrane potentials. The patch pipette contained 150 mm K+ and 5.5 μm acetylcholine, although K+ concentration in the bath was 5.4 mm. The resting membrane potential (Er) of the cell was −52 mV. Membrane potentials are indicated to the left of each trace as the difference from Er. Arrowheads indicate the zero current level. Downward reflection of the current record represent ion current passing inwards into the cell from the pipette. Upward deflections represent passing from the cell outwards into the pipette. B: The single-channel current-voltage relationship of the muscarinic K+ channel shown in A. The line was fitted to the data by eye, and the single channel conductance was 46 pS at potentials between Er −60 and Er +40 mV. C: Open time histogram of the muscarinic K+ channel at Er. The line is the fit of the data with a single exponential curve with a time constant of 1.35 msec. [Modified from Kurachi et al. (1986a)].
Under the conditions of the experiment shown in fig. 3A, the cell had a resting membrane potential (Er) of ∼−60 mV, although EK across the patch membrane was ∼0 mV. Therefore, the ACh-activated KAChchannel elicited inward K+ currents at potentials negative to Er + 60 mV (i.e.,Vm < EK) and outward currents at potentials positive toEr + 60 mV (fig. 3A). The outward currents were, however, very small compared with the inward currents at the corresponding potential relative to EK(compare the data at Er + 100 mV andEr + 20 mV). Thus, the KACh channel current readily flowed in the inward but not the outward direction. This occurs because intracellular Mg2+(Mg2+i) blocks the channel at the depolarized potentials (Horie et al, 1987 and 1989). Such a property is called “inward rectification,” and the K+ channels with this property are collectively termed as “inwardly rectifying” K+ (Kir) channels. All known KG channels including the KACh channel belong to this category.
The γ of the KACh channel estimated at Vm negative toEK is ∼40 pS in the presence of 145 mm extracellular K+(K+o) (fig. 3B). Based on the constant field theory, the permeability of K+through a single KACh channel has been estimated to be of the order of 10−13 cm3sec−1, a value comparable to that of the IK1 channel or the axonal delayed rectifier K+ channel (Sakmann et al., 1983;Sakmann and Trube, 1984a; Conti and Neher, 1980). Becauseγ increases approximately in proportion to the square root of the concentration of K+o([K+]o) (Sakmann et al., 1983), as is the case for the other types of Kir channels (Sakmann and Trube, 1984a), the γ is estimated as ∼8 pS at physiological [K+]o.
The mean open time of the KACh channel at potentials negative to EK is ∼1 msec (fig. 3C), which is several orders of magnitude shorter than that of the IK1 channel (Sakmann and Trube, 1984b). The open time histogram sometimes reveals less frequent opening with a longer open time. This component has been reported to appear more frequently when the internal side of inside-out patch membranes is treated with MgATP (Kim, 1990, 1991). The closed time distribution is composed of at least two distinct components with mean closed times of ∼1 and 100 msec (Sakmann et al., 1983). There are also distinct, very long closed events that cannot be reliably analyzed in single-channel recordings (Sakmann et al., 1983; Hosoyaet al., 1996). Analysis of burst behavior indicates that the KACh channel opens in bursts with a mean duration of 11 msec and consisting on average of ∼5 channel openings separated by short closed events (Sakmann et al., 1983). However, the majority of the KACh channel opening is solitary events separated by very short intervals of ∼1 msec on average (Sakmann et al., 1983). Overall, the burst behavior of KACh channel is not as evident as that of the IK1 channel.
2. Positive cooperative effect of GTP on muscarinic K+channel activity.
Activation of KAChchannels by intracellular GTP (GTPi) can be reproduced in inside-out patch membranes of atrial myocytes in the presence of ACh in the pipette (Kurachi et al., 1986a, 1990;Ito et al., 1991). Fig. 4shows the concentration-dependent effect of GTPiin the presence of different concentrations of ACh.

Concentration-dependent effect of intracellular GTP on the muscarinic K+ channel in the absence and presence of acetylcholine. A: Examples of inside-out patch experiments obtained from guinea-pig atrial myocytes. The channel currents were recorded at −80 mV with the symmetrical 145 mmK+ solutions. The concentration of acetylcholine (ACh) in the pipette was 0 or 1 μm as indicated. The bars above each trace indicates the protocol of application of different concentrations of GTP or 10 μm GTPγS to the internal side of the patch membrane. The 3- to 10-fold increase in GTP concentration resulted in a dramatic increase of N.Po of muscarinic K+ channels, indicating the existence of a highly cooperative process. B: The relation between the concentration of GTP and the N.Po of muscarinic K+ channels normalized to the maximum N.Po induced by 10 μm GTPγS in each patch. Symbols and bars are mean ± SD. The continuous lines indicate the fit of the relationship between GTP and channel activity in the presence of each concentration of ACh with the following Hill equation: where f is the relativeN.Po;Vmax, the maximumN.Po available in the presence of 10 μm GTPγS;Kd, the apparent dissociation constant of GTP; and n, the Hill coefficient. [Reproduced with permission from Ito et al. (1991)].
where f is the relativeN.Po;Vmax, the maximumN.Po available in the presence of 10 μm GTPγS;Kd, the apparent dissociation constant of GTP; and n, the Hill coefficient. [Reproduced with permission from Ito et al. (1991)].
Both in the presence and the absence of ACh, GTPiincreases the channel activity in a concentrationdependent manner (Ito et al., 1991). Channel currents in a patch membrane containing multiple KACh channels can be quantified as follows:
The Hill coefficient for the response was almost constant at ∼3 irrespective of ACh concentration (fig. 4B). Therefore, the receptor/GK/KACh channel interaction includes a certain positive cooperative process at step(s) distal to the receptor/GK interaction. Because dissociation of G protein subunits induced by GTP is a one to one reaction (Gilman, 1987), the cooperativity probably results from the GKβγ/KAChchannel interaction (Kurachi et al., 1990). Two pieces of evidence support this hypothesis. First, transducin βγ subunits applied to the internal side of inside-out patch membranes activate KACh channels reversibly (Yamada et al., 1994a). The concentration-response relationship of this reaction is also fitted by a Hill coefficient of ∼3. Second, the KACh channel partially and irreversibly preactivated by brain Gβγ exhibited apparently higher sensitivity to GTPi than the control (Yamada et al. 1993). This potentiation can be explained only by assuming that the same cooperative mechanism mediates Gβγ- and GTPi-induced channel activation. We might be able to understand how GKβγ activates the KACh channel when we can determine which kinetic parameter(s) of the KACh channel is modulated by GTPi in a positive cooperative manner.
3. Spectral analysis of the muscarinic K+ channel currents in the presence of different concentrations of intracellular GTP.
Precise and reliable analysis of the single-channel kinetics of the KACh channel is difficult because multiple KACh channels are usually included in a single membrane patch of atrial myocytes (fig. 4A). In these cases, the spectral analysis of the channel currents (an analysis based on a frequency domain) is one of the most reliable and powerful ways to assess the channel kinetics (fig. 5). The power spectrum constructed from inside-out patch recordings of the KACh channel currents is always well fitted with the sum of two Lorenzian curves irrespective of GTPi concentration (Hosoya et al., 1996). These observations indicate that the KAChchannel possesses three distinct open/closed states. Because the channel possesses a single open state (Sakmann et al., 1983), the equilibrium of the states can be described asC2↔C1↔O, where O represents the open state although C1 and C2 are closed states. It is likely that the transition among these three states is responsible for the open and closure of KAChchannel currents observed at the single-channel level (figs. 3A, 4A and5A). The corner frequencies of the two Lorenzian functions (the frequencies at which the power of the each component is the half-maximum) were constant irrespective of GTPiconcentration (fig, 5B). The ratio of the powers of the two Lorenzians at 0 Hz was also unaffected by GTPiconcentration. These results indicate that the kinetics of the fast open-close transition of the channel is not a function of GK activity. In other words, GK activates the KAChchannel without altering the channel’s fast open-close kinetics.

Spectral analysis of the muscarinic K+channel currents in an inside-out patch. A: The muscarinic K+ channel currents in the inside-out patch membrane of a guinea pig atrial myocyte. The channel currents were recorded at −60 mV with the symmetrical 150 mm K+ solutions. The patch pipette also contained 0.5 μm acetylcholine. Different concentrations of GTP were applied to the internal side of the patch membrane as indicated above each current trace. B: Power density spectra calculated from the data shown in A. Each spectrum could be fitted with the sum of two Lorentzian functions.F1 and F2indicated by arrows indicate the corner frequencies of the slow and the fast Lorentzian components, respectively. [Reproduced with permission from Hosoya et al. (1996)].
How then does GK activate the KACh channel? As GTPiconcentration was raised, the powers of both the Lorenzian components at 0 Hz became progressively larger (fig. 5B) (Hosoya et al., 1996), implying that GK increases KACh channel activity through a process too slow to be detected by spectral analysis. For reasons of simplicity, we a priori assume the presence of another transition with slow kinetics between two channel states U↔ A, whereU and A, respectively, represent “unavailable” and “available” states of the channel. In this framework, the U↔ A transition is independent of the fast transition C2↔C1↔O and the A but not theU state allows the channel to be conducting when the channel passes into the O state. Furthermore, it is hypothesized that GK causes a shift of the equilibrium towardA to increase channel activity.
Based on these assumptions, one should be able to calculate the fraction of the A state (i.e., A/(A + U)) in the presence of a given concentration of GTPi by extracting some parameters from the spectral analysis (the corner frequencies and the ration of the powers at 0 Hz) and the single-channel analysis (the single-channel open time and theN*Po value). Fig.6A shows the calculated fraction of theA state, which increased as the concentration of GTPi was raised in such a way that the concentration-response relationship could be well fitted by a Hill coefficient of ∼3. From this result we conclude that GK modulates a slow process in the KACh channel that corresponds to an increase in the number of operational ion channels in the membrane. The fast open-close kinetics of the channels seem not to be influenced by GK. Thus, N, but notPo, in equation 2 is affected by GK.

The relationship between GTP concentration and the fraction of the “available” state of the muscarinic K+channel, and the concerted allosteric model of Monod, Wyman, and Changeux. A: The fraction of the “available” state (A/(A + U)) was calculated from inside-out membrane patch experiments such as shown in fig. 5. Symbols indicate the relationship between GTP concentration and the calculated fraction of the “available” state. Lines indicate the fit of the data with the Monod, Wyman, and Changeux allosteric model with different assumed numbers of n (see Section II.B.4.). B: Schematic representation of the Monod, Wyman, and Changeux allosteric model. In this scheme, each muscarinic K+ channel is assumed to be an oligomer composed of four identical subunits (i.e.,n = 4). Each subunit is in either the tense (T) or the relaxed (R) state, which is represented by squares and circles, respectively. Each subunit in theT or R state binds with one dissociated G protein βγ subunit (solid circles) independently of each other with the microscopic dissociation constant of KTor KR, respectively. In this model, all subunits in the same oligomer must change their conformations simultaneously. Therefore, the channel can be eitherT4 or R4.T4 and R4 are in the equilibrium through an allosteric constant L. [Reproduced with permission from Hosoya et al.(1996)].
4. A possible mechanism for the G protein-mediated increase in the functional numbers of muscarinic K+ channels.
Recent studies have revealed that Kir channels, including KACh channel, have an oligomeric structure (Yanget al., 1995b; Krapivinsky et al., 1995a) that may underlie the positive cooperativity of the GKβγprotein/KACh channel interaction (Monod et al., 1965).
In the presence of a supermaximum concentration of ACh (≥1 μm), Gβγ exogenously applied to the internal side of inside-out patch membranes does not further increase the channel activity once the channel is preactivated with more than 1 μm of GTPi (Itoet al., 1992; Yamada et al., 1993). In this case, the maximum channel activity is determined by the number of KACh channels and not the GKβγ available in a patch membrane. Under these conditions, the interaction between GKβγ and KAChchannel subunits can be quantitatively assessed by analyzing the relationship between GTPi concentration and the fraction of the A state with Monod-Wyman-Changeux’s (MWC) allosteric model (fig. 6B) (Monod et al., 1965; Hosoya et al., 1996).
This model is based on the following assumptions: (a) a single KACh channel is composed of a finite number (n) of functionally identical subunits: fig. 6B illustrates the case of n = 4; (b) each subunit independently binds only one GKβγ; (c) each subunit has two distinct conformations: relaxed (R) and tense (T); (d) R and T bind GKβγ with microscopic dissociation constant KR andKT, respectively. R has higher affinity for GKβγ than T(i.e., KR <KT); (e) all subunits in an oligomer must change the conformation simultaneously. As a result, any oligomer is either Rn orTn; (f)Rn and Tn are in the equilibrium through an allosteric constant L.
According to this model, an increase in GKβγ concentration leads to an increase in the fraction of Rn [i.e.,Rn/(Rn + Tn)]. When one replacesRn and Tn of the MWC model with the A and U states of the KACh channel, the data shown in fig. 6A can be fitted with this model by changing the assumed number of n. Such analysis indicates that n must be greater than 3 to account for the data (fig. 6A) (Hosoya et al., 1996). This result is consistent with the view that Kir channels including KG possess a tetrameric structure as described in Section III.E. (Krapivinsky et al., 1995a; Yang et al., 1995b).
Therefore, we may summarize our current understanding of the interaction between GK and the KACh channel as follows. GKactivates the KACh channel by increasing the functional number of channels without modulating the fast open-close transition of the channel gate. The positive cooperativity observed in the GTPi-induced activation of the KACh channel arises from the intrinsic property of the GKβγ/KAChchannel interaction. This property can be explained in terms of the oligomeric structure of the KACh channel that is composed of more than three functionally identical subunits, each of which independently binds one GKβγ molecule. As we shall see later in Section IV., KACh channel activity is controlled not only by GK but byVm. However, ACh does not modulate the relationship between channel activity andVm (Kurachi, 1990). Therefore, the model described here is applicable to the GK-mediated activation of the KACh channel at any potential.
C. Modulation of G Protein-Mediated Activation of the Muscarinic K+ Channel
Although the GKβγ/KAChchannel interaction is the essential step of G protein-mediated activation of the KACh channel, this reaction is modulated by many factors such as intracellular ATP, Na+ ions, and arachidonic acid metabolites. Intracellular ATP has been shown to activate native and recombinant KG channels in an Mg2+i-dependent manner (Oteroet al., 1988; Heidbüchel et al., 1990;Kaibara et al., 1991; Kim, 1991; Lesage et al., 1995; Sui et al., 1996). Although the molecular mechanism underlying this phenomenon has not been unequivocally identified, PIP2 may be involved in this phenomenon (Huanget al., 1998).
The activity of KG channels pretreated with intracellular MgATP could be further enhanced by intracellular Na+ (Lesage et al., 1995; Sui et al., 1996). The site of action of Na+ is unknown. Sui et al. (1996) showed that intracellular Na+ increased the activity of the KACh channel (and also the corresponding recombinant KG channel) with an EC50 of ∼40 mm mainly by increasing the frequency of the channel’s opening. They found that priming of channels with MgATP was a prerequisite for the action of Na+. Lesage et al. (1995), however, found that 20 mm intracellular Na+ activated recombinant KG channel whether or not they had been pretreated with MgATP. This discrepancy might have occurred due to the different subunit composition of the KG channels used in these two studies. Interestingly, Sui et al. (1996)showed that a cardiac glycoside ouabain, an inhibitor of the Na+/K+ pump, induced the opening of the KACh channel. They found that theN*Po value of the channel increased although the mean open time was unchanged, indicating that the activating effect of ouabain was probably mediated by accumulation of intracellular Na+ but not a possible local increase in ATP concentration. However, they did not directly measure intracellular Na+ concentration nor reported the apparent change in the reversal potential of the KACh channel that might be expected when intracellular K+ concentration decreased due to blockade of Na+/K+ pump. Therefore, further studies may be necessary to conclude that cardiac glycosides activate the KACh channel through accumulation of intracellular Na+. This phenomenon might, at least in part, underlie the “direct” negative chrono- and dromo-tropic effects of the agent on the heart.
Arachidonic acid (AA) metabolites are known to modulate KACh channels (Kurachi et al., 1989c;Kim et al., 1989; Yamada et al., 1994b). The effect of AA is mimicked by leukotriene C4(LTC4) and specifically blocked by AA861, a 5-lipoxygenase inhibitor (Kurachi et al., 1989c). Therefore, the effect of AA may be mediated by LTC4 or its metabolites. Although the site of action of LTC4has not been clearly identified, the complete dependency of the LTC4 effect on the presence of GTPi indicates that LTC4does not directly act on the KACh channel (Kurachi et al., 1989c). In the absence of receptor agonists, GTPi usually induces only 20% of the maximum KACh channel activity in the inside-out patch membranes even when Cl− is used as an intracellular anion. However, GTPi fully activated the channel in an agonist-independent manner when the patches were pretreated with AA before patch excision (Kurachi et al., 1989c). Thus, AA metabolites may stimulate the basal turn-on reaction of GK. Stimulation of KACh channels by platelet-activating factor or α1-adrenergic receptors may be mediated by this second-messenger pathway (Nakajima et al., 1991, Kurachiet al., 1989b).
III. Molecular Analysis of G Protein-Gated K+ Channels
A. Cloning of Inwardly Rectifying K+ Channels
In 1993, the molecular structure of inwardly rectifying K+ channels (Kir) was disclosed. The cDNAs encoding an ATP-dependent Kir channel, ROMK1 (Ho et al., 1993), and a classical Kir channel, IRK1 (Kubo et al., 1993a), were isolated by expression cloning from the outer medulla of rat kidney and a mouse macrophage cell line, respectively (fig.7). The primary structure of these channels were similar with two putative membrane-spanning regions (M1 and M2) and one potential pore-forming region (H5). This structure resembles that of the S5, H5, and S6 segments of the voltage-gated K+ (Kv) channels. Because the voltage-sensor of the Kv channel subunit exists in the S4 segment that possesses repeated positively-charged amino acid residues, Kir channel subunits lack an obvious voltage-sensor region. This is consistent with electrophysiological studies that show the kinetics of Kir channels apparently depends on the difference of Vm from EK and not on Vm itself.

Evolutionary tree of Kir subunits. The tree was made using the UPGMA (Unweighted Pair Group Method with Arithmetic Mean) Tree Window in Geneworks (IntelliGenetics, Inc., Mountain View, CA).
After the cloning of ROMK1 and IRK1, the cDNAs encoding the main subunits of KG and KATPchannels (GIRK1 and BIR) were also cloned (Kubo et al., 1993b; Dascal et al., 1993; Inagaki et al., 1995a). All of these Kir channel subunits exhibit basically the same primary structure. So far, at least 11 cDNAs encoding Kir channel subunits have been isolated. The evolutionary tree of this family is depicted in fig. 7.
These cloned Kir subunit cDNAs encode proteins composed of 327 to 501 amino acids. The identity of the predicted amino acid sequences is ∼30 to 40% among the members of the different Kir subfamilies and more than 60% among those in the same subfamilies. The highest level of sequence identity (50 to 60%) is found in the H5 region and the proximal part of the C-terminal cytosolic domain. The cloned Kir channel subunits have been classified at least into four groups (Doupnik et al., 1995a): (a) IRK (Kir2.x) subfamily made of the classical constitutively active “inward rectifier” Kir channels: IRK1 (Kubo et al., 1993a;Morishige et al., 1993), IRK2 (Koyama et al., 1994; Takahashi et al., 1994) and IRK3 (Morishige et al., 1994; Makhina et al., 1994; Pärier et al., 1994); (b) GIRK (Kir3.x) subfamily, corresponding to G protein-regulated K+ channels: GIRK1 (Kuboet al., 1993b; Dascal et al., 1993), GIRK2 (Lesage et al., 1994, 1995; Isomoto et al., 1996;Tsaur et al., 1995; Stoffel et al., 1995;Bond et al, 1995; Ferrer et al., 1995), GIRK3 (Lesageet al., 1994), GIRK4 (Ashford et al., 1994;Krapivinsky et al., 1995a; Chan et al., 1996), and GIRK5 (Hedin et al., 1996); (c) KAB subfamily of ATP-dependent K+ channels (Kir1.1 and Kir4.1): ROMKs (Hoet al., 1993; Zhou et al., 1994; Yano et al., 1994; Shuck et al., 1994; Boim et al., 1995; Kondo et al., 1996) and KAB-2 (Bond et al., 1994; Takumi et al., 1995); and (d) KATP subfamily (Kir6.x), the ATP-sensitive K+ channels: uKATP-1 and BIR (Inagaki et al., 1995a,b; Sakura et al., 1995).
Recent progress in the molecular biology of Kir channels has enabled us to study the structure-function relationship of biophysics, physiological regulation, and pharmacology of these channels at the molecular level.
B. Subunits of G Protein-Gated K+ Channels
GIRK1 was first isolated from the rat atrium (Kubo et al., 1993b; Dascal et al., 1993). From a mouse brain cDNA library, two additional homologues of GIRK1 were isolated and designated GIRK2 and GIRK3 (table 2) (Lesage et al., 1994). Furthermore, it has been shown that at least three different isoforms of mouse GIRK2 are generated by alternative splicing of transcripts from a single gene, and we designated them GIRK2A, GIRK2B, and GIRK2C in the order of identification (Isomoto et al., 1997). These alternatively spliced transcripts share an N-terminal end and a central core, and differ at their C-terminal ends. GIRK2B was isolated from mouse brain cDNA library and shown to be ubiquitously expressed in various tissues (Isomoto et al., 1996). Its amino acid sequence is shorter than that of GIRK2A by 87 amino acids. The eight amino acid residues in the C-terminal end of GIRK2B are different from those of GIRK2A. GIRK2C has a C-terminus which is longer than that of GIRK2A by 11 amino acids. GIRK2C was isolated from cDNA libraries of insulinoma cells and brain (Lesage et al., 1994,1995; Tsaur et al., 1995; Stoffel et al., 1995;Bond et al., 1995; Ferrer et al., 1995).
Subunits of G protein-regulated inward-rectifying K+channels
GIRK2C was originally termed KATP-2 because it was thought to be a subunit of the KATP channel (Stoffel et al., 1995; Tsaur et al., 1995) due to its sequence similarity to cKATP-1, which was isolated by Ashford et al. (1994). However, GIRK4, which is virtually identical with rat cKATP-1, reconstitutes cardiac KACh channel with GIRK1 and does not contribute to the KATP channel as described in the Section III.D. (Krapivinsky et al., 1995a,b). Thus, it is now clear that both cKATP-1 and KATP-2 belong to the GIRK subfamily. GIRK5 was cloned from Xenopus oocytes (Hedin et al., 1996). Although its mammalian homologue has not been reported, the amino acid sequence of GIRK5 is most homologous to that of GIRK4 among mammalian GIRKs.
The GIRK clones contain various known functional motifs in their amino acid sequences that may be important for the physiological functions of the subunits in KG channels (fig.8). GIRK1 possesses an amino acid sequence homologous to the Gβγ-binding domain of βARK1 in its C-terminus, which is therefore the candidate for the site of Gβγ-binding to the KG channel (Reuveny et al., 1994). As with all the other Kir channel subunits, GIRKs possess conserved cationic residues adjacent to the C-terminal end of the M2 domain. One of these positively charged residues, arginine (R) at position 188 of ROMK1, was shown to be critically involved in PIP2-induced activation of rundown ROMK1 channels (Huang et al., 1998). Thus, it is conceivable that the corresponding residues in GIRK subunits (R190 for GIRK1; R201 for GIRK2s; R167 for GIRK3; and R196 for GIRK4) also participate in the PIP2-induced activation of KG channels. All of the GIRK clones have an arginine-glycine-aspartate (RGD) motif in their linker region between M1 and H5. This motif could be an integrin receptor-site (Hyneset al., 1992), whose role in KGchannels has not been examined yet. The characteristic feature of GIRK2C is the serine/threonine-X-valine/isoleucine (S/T-X-V/I) motif at its C-terminus end (Gomparts, 1996). This motif has been shown to be important for interactions with the PSD-95/SAP90 family of anchoring proteins, not only for Kv and NMDA receptor channels (Kim et al., 1995; Kornau et al., 1995), but also for Kir channels such as IRK3 and KAB-2 (Cohen et al., 1996; Horio et al., 1997).

Alignment of amino acid sequences of GIRK1, GIRK2A, B, C, GIRK3, and GIRK4. Positions at which all six amino acid sequences are identical are boxed. The putative transmembrane segments (M1 and M2) and pore-forming region (H5) are indicated above the sequences. Overlined sequences of GIRK1, (a), the domain in the N terminus that was shown to bind G protein βγ subunits, the GDP-form of G protein α subunits and heterotrimeric G proteins (Huanget al., 1995); (b), RGD sequence included in the integrin-binding site of fibronectin, vitronectin, and a variety of other adhesive proteins (Hynes, 1992), which is also found in other GIRK subunits; (c), the region whose amino acid sequence is similar to the G protein βγ subunit-binding domain of the β-adrenergic receptor kinase 1 (Reuveny et al., 1994); and (d) and (e), the domains in the C terminus that was shown to interact with G protein βγ subunits (Huang et al., 1995). An underlined sequence of GIRK1: the sequence similar to that of the region of adenylyl cyclase 2 which is critical for activation of the enzyme by G protein βγ subunits (N-X-X-E-R) (Chen et al., 1995; Huang et al., 1995). Numbered amino acids, (1) the phenylalanine (F137) of GIRK1 was shown to be responsible for the slow relaxation of GIRK1-containing KGchannels (Kofuji et al., 1996a), although the interaction between F137 and the corresponding serine of other GIRK subunits may be important for the larger macroscopic current amplitude yielded with coexpression of GIRK1 and other GIRK subunits than the sum of those obtained with expression of each subunit alone (Chan et al., 1996; Wischmeyer et al., 1997); (2) the amino acid residue corresponding to aspartate at position 172 of the IRK subunit which was shown to be critically involved in the Mg2+/polyamine block of IRK1 channels (Stanfield et al., 1994; Taglialatela et al., 1994; Lu and MacKinnon, 1994; Wible et al., 1994; Lopatin et al., 1994; Ficker et al., 1994; Yang et al., 1995a); (3) the residue corresponding to the arginine of ROMK1 which was shown to be involved in interaction of ROMK1 channels with phosphatidilinositol 4,5-bisphosphate (PIP2) (Huanget al., 1998); and (4) the residues corresponding to glutamate at position 225 of the IRK1 subunit that was shown to be critically involved in the Mg2+/polyamine block of IRK1 channels (Yang et al., 1995a). The double-underlined sequence of GIRK2C: the sequence including the consensus sequence (S/T-X-V/I) for interaction with PSD-95/SAP90 anchoring protein (Gomparts, 1996; and Cohen et al., 1996), although the glutamate (E) preceding the sequence was also proposed to be important for the interaction of Shaker-type K+ channels with these anchoring proteins (Kim et al., 1995).
C. Tissue Distribution of GIRK Subunits
1. Peripheral tissues.
Tissue distribution of mRNAs for GIRK subunits is summarized in table 3 (Kubo et al., 1993b; Dascal et al., 1993; Lesage et al., 1994;Stoffel et al., 1995; Bond et al., 1995; Dixonet al., 1995; Iizuka et al., 1995). In tissues other than brain, the atrium of the heart most abundantly expresses GIRK1 and GIRK4, both of which constitute the KACh channel. Both GIRK1 and GIRK4 proteins are diffusely immunostained in the atrium by antibodies specific for individual subunits (cf., fig. 15) (Iizuka et al., 1995). GIRK1 may be moderately expressed in the ventricle (Kubo et al., 1993b; Dascal et al., 1993; Karschin et al., 1994), although there seems to be a significant species-to-species difference in the level of expression of GIRK4 protein in the ventricle (Iizuka et al., 1995; Krapivinskyet al., 1995b). Iizuka et al. (1995) showed that GIRK4 immunoreactivities exist in subendocardial myocytes and also in dorsal atrial ganglia of rat. GIRK1 is also moderately expressed in other peripheral tissues except for spleen (table 3). GIRK2 and GIRK3 are rather brain-specific and barely found in peripheral tissues. However, GIRK2 (probably GIRK2C) exists in pancreatic islets (Stoffel et al., 1995), although GIRK2B mRNA is expressed ubiquitously in peripheral tissues (Isomoto et al., 1996). GIRK4 is also found in some other peripheral tissues.
Tissue expression of mRNAs for GIRK subunits

Different subcellular localization of GIRK1 proteins. A and B: Immunohistochemical analyses for the GIRK1 proteins in the rat atrium (A) and ventricle (B). Homogeneous immunoreactivity was found on the plasma membranes of the atrial, but not of the ventricular myocytes. C and D: Electron microscopic analyses for the GIRK1 immunoreactivity in the rat paraventricular nucleus of the hypothalamus. Obvious GIRK1 immunoreactivity was present at the axonal terminals (probably on the vesicles) neighboring upon a dendrite (D) (reproduced with permission from Morishige et al., 1996). All GIRK1 immunoreactivities were developed with diaminobenzidine-horseradish peroxidase method.
2. Central nervous system.
Detailed distribution of GIRK mRNAs in rat brain was analyzed with in situ hybridization and tabulated byKarschin et al. (1994, 1996) and DePaoli et al.(1994). Expression pattern of GIRK transcripts in the mouse brain is similar to that in the rat brain (Kobayashi et al., 1995). In general, GIRK1–3 mRNAs are abundantly expressed throughout the brain with overall similar distribution, although GIRK4 mRNA is expressed in the brain to a much lesser extent than other GIRK transcripts (Karschin et al., 1996; Iizuka et al., 1997).
In the rat main olfactory bulb, all GIRK1–4 mRNAs are expressed in the granular cell layer and mitral cell layer, although only GIRK3 mRNA is abundant in glomerular cells (Karschin et al., 1994, 1996;Dixon et al., 1995; Ponce et al., 1996; Iizukaet al., 1997). All GIRK1–3 mRNAs are strongly expressed in every area of neocortex and by virtually all pyramidal neurons in the cortex (Karschin et al., 1994, 1996; DePaoli et al., 1994; Dixon et al., 1995; Ponce et al., 1996). GIRK4 mRNA may be expressed in the pyramidal neurons (Iizukaet al., 1997). Basal ganglia exhibit basically very poor expression of GIRK family members (Karschin et al., 1994,1996; Ponce et al., 1996) except for amygdala nuclei where all GIRK1–3 mRNAs are abundantly expressed (DePaoli et al., 1994; Dixon et al., 1995; Karschin et al., 1996). The septum expresses all GIRK1–3 mRNAs although GIRK1 and GIRK4 mRNAs are especially abundant in lateral septal nuclei (Karschin et al., 1994, 1996; DePaoli et al., 1994). In the rat hippocampus, all GIRK mRNAs are strongly expressed by dentate gyrus granule cells and CA1-CA3 pyramidal neurons (Karschin et al., 1994 and 1996; DePaoli et al., 1994; Dixonet al., 1995; Iizuka et al., 1997). Mouse hippocampus is, however, reported not to express GIRK3 mRNA (Kobayashiet al., 1995). In the rat proximal hilus of the dentate gyrus, GIRK2 and GIRK4 mRNAs are more strongly expressed than the GIRK1 transcript (Karschin et al., 1996). Expression of GIRK4 mRNA is most prominent in CA3 pyramidal neurons (Spauschus et al., 1996). Many cells in the entorhinal cortex and the subiculum of the hippocampal formation also express GIRK4 mRNA (Spauschuset al., 1996).
In the thalamus, GIRK1 and GIRK3 mRNAs are abundantly expressed in all nuclei and especially in anterior nuclei, although GIRK2 mRNA is present at high levels in the lateral and geniculate nuclei (Karschinet al., 1994, 1996; Ponce et al., 1996). All large neurons in the rat thalamus seem to coexpress GIRKs1–4 (Karschinet al., 1996), although expression of GIRK2 mRNA was not found in mouse thalamus (Kobayashi et al. 1995). In the hypothalamus, GIRK mRNAs are not abundantly expressed (Karschinet al., 1994, 1996; Ponce et al., 1996) although GIRK1 mRNA is expressed only in the ventral medial hypothalamus (DePaoli et al., 1994). The anterior pituitary strongly expresses GIRK1 mRNA (Karschin et al., 1994).
In the midbrain, the superior colliculus contains a very high level of GIRK2 mRNA and a distinct level of expression of GIRK4 transcripts (Spauschus et al., 1996; Karschin et al., 1996;Ponce et al., 1996). The inferior colliculus contains high levels of GIRK1 and GIRK3 but no GIRK2 mRNAs (Karschin et al., 1996; Ponce et al., 1996). Red nuclei abundantly express GIRK1 and GIRK3 mRNAs and also possess GIRK4 transcripts (DePaoli et al., 1994; Karschin et al., 1996;Ponce et al., 1996; Iizuka et al., 1997). In dopaminergic neurons of the substantia nigra pars compacta and the ventral tegmental area, GIRK2 mRNA is expressed at extremely high levels (Karschin et al., 1996; Dixon et al., 1995). GIRK3 mRNA is found throughout the substantia nigra and ventral tegmental area but at significantly lower levels than GIRK2 mRNA. GIRK1 is virtually absent (Karschin et al., 1996; Ponce et al., 1996), although GIRK4 transcripts may exist in these regions (Iizuka et al., 1997).
Cerebellar granule cell layer has abundant mRNAs for all GIRK1–4 (Karschin et al., 1994 and 1996; Dixon et al., 1995; Iizuka et al., 1997). Purkinje cells express mRNAs for GIRK3 strongly and GIRK4 to a certain extent, but for GIRK1 or GIRK2 only moderately (Spauschus et al., 1996; Karschin et al., 1996; Iizuka et al., 1997). Basket cells have a moderate level of GIRK4 mRNA (Iizuka et al., 1994). The large neurons in the deep cerebellar nuclei contain high levels of GIRK1 and GIRK3 but not GIRK2 transcripts (Karschin et al., 1996; Ponce et al., 1996).
All GIRK transcripts are abundantly expressed in the brain stem (Karschin et al., 1996). Especially a high level of expression of GIRK1 mRNA is observed in pontine nucleus, trapezoid body, pontine reticular formation, superior olivary nuclei, cochlear nuclei, hypoglossal nucleus, and principal and spinal trigeminal nuclei; GIRK2, in vestibular and cochlear nuclei; and GIRK3, in lateral parabrachial nucleus (Karschin et al., 1994, 1996; DePaoliet al., 1994; Ponce et al., 1996). GIRK4 mRNA is also found in hypoglossal, trigeminal, and oculomotor nuclei (Iizuka et al., 1997). GIRK1 mRNA is absent in inferior olivary and solitary nuclei, whereas GIRK2 transcripts are not found in the trapezoid body, inferior olivary nuclei and raphänuclei (Karschin et al., 1996). Iizuka et al.(1997) found that GIRK4 mRNA is relatively strongly expressed in the choroid plexus in the lateral, third, and fourth ventricles. It is uncertain whether other GIRK transcripts exist in this tissue.
Immunohistochemical approaches revealed that the overall distribution pattern of GIRK1, 2, and 4 immunoreactivities in the rat brain is similar to that of the mRNAs for these subunits (Karschin et al., 1994 and 1996; DePaoli et al., 1994; Ponceet al., 1996; Liao et al., 1996; Iizuka et al., 1997). No studies have been done on the distribution of GIRK3 proteins in the brain to our knowledge. It is also known that there are certain discrepancies in distribution between mRNAs and proteins of GIRKs in the brain, indicating that GIRK subunits are translocated into nerve fibers, terminals, and dendrites after synthesized in the somata. The precise subcellular localization of GIRK proteins in neurons in the central nervous system shall be discussed in the section VI.B.
D. Expression of G Protein-Gated K+ Channels
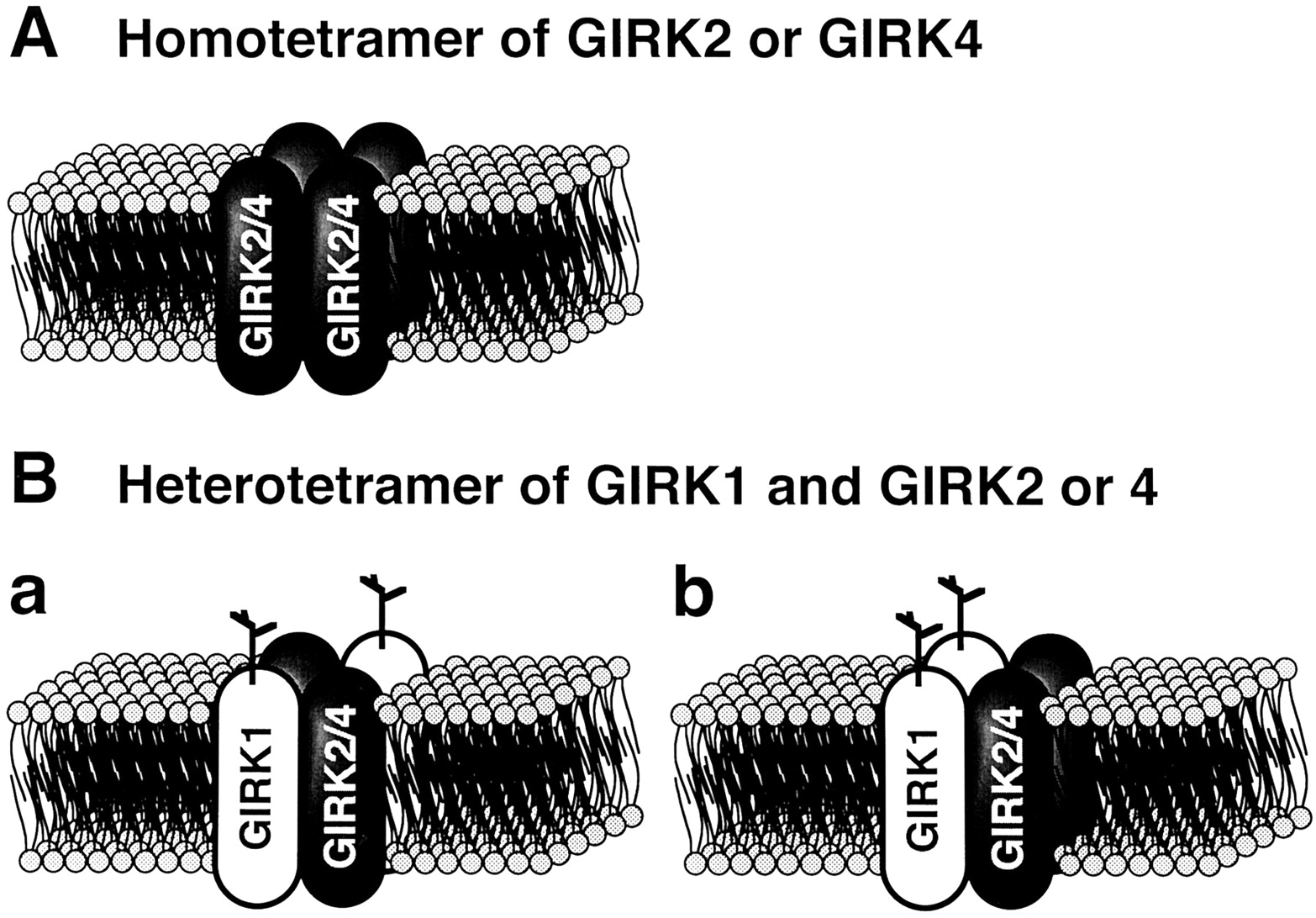
When cRNAs for GIRK1 and M2-muscarinic receptor are coinjected into Xenopus oocytes, a Kir current induced by ACh is expressed (Kubo et al., 1993b; Dascalet al., 1993). This current mimics at least some characteristics of the KACh channel current. GIRK1 expressed in Xenopus oocytes has been, therefore, successfully used to investigate the structure-function relationship of KG channels (Reuveny et al., 1994;Slesinger et al., 1995; Kofuji et al., 1996a). However, Krapivinsky et al. (1995a) proposed that the KACh channel in cardiac atria is a heteromultimer of GIRK1 and GIRK4 rather than a homomultimer of GIRK1 because GIRK1 and GIRK4 proteins are immunocoprecipitated by both specific anti-GIRK4 and anti-GIRK1 antibodies from atrial membrane preparation (fig.9A). Furthermore, coexpression of GIRK1 and GIRK4 in Xenopus oocytes yields greatly enhanced KG channel currents compared with expression of either of the subunits alone (fig. 9B). Now, it is generally believed that GIRK1 is inactive by itself because expression of GIRK1 alone fails to give rise to KG channel currents in different mammalian cell lines including CHO, COS, and HEK cells (Krapivinsky et al., 1995a; Philipson et al., 1995; Spauschus et al., 1996; Wischmeyer et al., 1997). It is likely that functional expression of GIRK1 alone is possible in Xenopus oocytes because oocytes endogenously express GIRK5 whose amino acid sequence is 78% identical with that of GIRK4 (Hedin et al., 1996). However, GIRK2 and GIRK4, but not GIRK3, may be able to form functional homomeric KG channels, although not very efficiently (Lesage et al., 1994 and 1995; Bond et al., 1995,Krapivinsky et al., 1995a; Duprat et al., 1995;Kofuji et al., 1995; Velimirovic et al., 1996;Wischmeyer et al., 1997).

Heteromultimeric structure of the muscarinic K+ channel. A: Immunological analysis for mouse atrial muscarinic K+ channel. Atrial membrane proteins were immunoblotted (IB) with anti-GIRK1C1 (lane 1) and anti-GIRK4N10 (lane 2) antibodies specific to GIRK1 and GIRK4 proteins, respectively. Some part of the GIRK1 protein, but not GIRK4 protein, seems to be glycosylated in the atrium. Lanes 3 and 4, the imunoprecipiants (IP) obtained from biotinylated mouse atrial membranes with the anti-GIRK1C1 and anti-GIRK4N10 antibodies, respectively. Both immunocomplexes of anti-GIRK1C1 and anti-GIRK4N10 seemed to be composed of three proteins that had molecular weights identical with GIRK1 and GIRK4 proteins on the gel. B: Acetylcholine- (Ach) induced K+ currents observed in Xenopus oocytes expressing M2-muscarinic receptors (m2R) plus mouse GIRK1 (m-GIRK1) and/or human GIRK4 (h-GIRK4). Acetylcholine- (1 μm) induced K+ currents at different membrane potentials were measured in the presence of 96 mm external K+and are shown under the table. The voltage-clamp protocol is depicted at the left lower corner.
Heteromultimerization of GIRK subunits occurs not only between GIRK1 and GIRK4 but within any pairs of GIRK1-4 subunits (Kofuji et al., 1995; Iizuka et al., 1995; Duprat et al., 1995; Lesage et al., 1995; Spauschus et al., 1996; Velimirovic et al., 1996; Isomoto et al., 1996; Wischmeyer et al., 1997). Coexpression of GIRK1 with either of GIRKs2-4 generally yields several- to many-fold larger macroscopic currents than the sum of those obtained with each subunit alone. However, it has not been unequivocally answered whether similar synergistic interaction also occurs between GIRKs2-4. The synergistic enhancement of current expression was reported to occur with GIRK2/GIRK4 (Duprat et al., 1995; Ferrer et al., 1995) and GIRK3/GIRK4 combinations (Spauschus et al., 1996). However, these results were not necessarily confirmed by the others (Lesage et al., 1995; Velimirovic et al., 1996; Wischmeyer et al., 1997). Coexpression of GIRK3 with GIRK2 was shown to suppress GIRK2 channel currents (Kofujiet al., 1995). It is not clear whether more than two types of GIRK subunits can be assembled into a single KG channel (Wischmeyer et al., 1997).
GIRK1/GIRK4 heteromultimeric KG channels are likely to correspond to cardiac KACh channels as mentioned in this Section. GIRK1/GIRK2 channels, however, may represent some neuronal type of KG channels for the following reasons: (a) GIRK2 mRNA is preferentially expressed in the brain (table 3); (b) GIRK1 and GIRK2 exhibit overlapping distribution in many areas of the brain at both the mRNA and protein levels (Karschin et al., 1996; Liaoet al., 1996); (c) both specific anti-GIRK1 and anti-GIRK2 antibodies coimmunoprecipitate GIRK1 and GIRK2 proteins from membrane preparations of the brain (Liao et al., 1996); (d) in the mice whose GIRK2 genes are genetically deleted (GIRK2 -/-), the substantial amount of GIRK1 proteins is concomitantly lost in the brain (Signorini et al., 1997); and (e) the hippocampal CA1 and CA3 pyramidal neurons of these mice fail to exhibit postsynaptic inhibitory KGchannel currents in response to different inhibitory neurotransmitters (Lüscher et al., 1997). Some neuronal KG channels may also be composed of GIRK1 and GIRK3 because their transcripts are also expressed together in various regions of the brain (Karschin et al., 1996). It is also possible that GIRK4 is included in neuronal KGchannels (Spauschus et al., 1996; Iizuka et al., 1997) and that some KG channels are homomultimers of GIRKs2 or 4 or heteromultimers of GIRKs2–4.
Chan et al. (1996) found that the synergistic interaction between GIRK1 and GIRK4 for KG channel current expression can be at least in part ascribed to interaction between phenylalanine at position 137 (F137) in the H5 region of GIRK1 and serine at the corresponding site (S143) in GIRK4 (fig. 8). GIRKs2 and 3 also have conserved serine at this site. They found that coexpression of the wild-type GIRK4 with the mutant GIRK4 whose S143 was replaced with phenylalanine [GIRK4(S143F)] yielded the significantly larger macroscopic current amplitude than the sum of those obtained with either of the subunits alone, as is the case for GIRK1/GIRK4 coexpression. At the single-channel level, GIRK4(S143F)/GIRK4 channels, like GIRK1/GIRK4 channels, opened in clearer bursts and exhibited a significantly longer open time than GIRK4 homomeric channels (Krapivinsky et al., 1995a; Chan et al., 1996). Thus, F137 of GIRK1 may be responsible for the larger macroscopic current amplitude of GIRK1/GIRK4 than GIRK4 channels by stabilizing the channel’s open-state conformation. Wischmeyer et al. (1997) also obtained similar results by using a mutant GIRK3 subunit bearing a mutation corresponding to GIRK4(S143F) [i.e., GIRK3(S114F)].
However, neither GIRK4(S143F) nor GIRK3(S114F) synergistically interacted with GIRK1 (Chan et al., 1996; Wischmeyeret al., 1997). GIRK1 whose F137 was substituted with serine [GIRK1(F137S)] could form a functional homomeric channel (Chanet al., 1996; Wischmeyer et al., 1997), although coexpression of GIRK1(F137S) with the wild-type GIRK1 yielded current amplitudes intermediate between those obtained with either of the subunits alone (Wischmeyer et al., 1997). These results indicate that F137 of GIRK1 on its own is inhibitory for the K+ ion flux through the channel pore possibly because of its bulky aromatic side chain (Wischmeyer et al., 1997). Thus, serine derived from other types of GIRK subunits might somehow attenuate this inhibitory effect of F137 and yield the larger current amplitudes of the heteromeic GIRK1-containing channels than those obtained with expression of GIRK1 alone.
It is, however, difficult to explain whole the aspect of the synergistic interaction between GIRK1 and other GIRK subunits only in terms of the interaction between complementary phenylalanine/serine residues in the heteromeric channel pore. For example, GIRK1(F137S)/GIRK4 channels exhibit much larger currents than the GIRK4 homomeric channels (Chan et al., 1996). GIRK1/GIRK4 channels have ∼2 times larger macroscopic currents than GIRK4(S143F)/GIRK4 channels. Thus, some region(s) other than F137 of GIRK1 must also be significantly involved in the synergistic interaction between GIRK1 and GIRK4. Similarly, GIRK1(F137S)/GIRK4 channels have substantially larger current amplitudes than homomeric GIRK1(F137S) channels (Chan et al., 1996), indicating that GIRK4 also has some effect(s) on GIRK1 which cannot be ascribed to the serine/phenylalanine interaction.
Kennedy et al. (1996) found that GIRK1 cannot translocate to the cell membrane in the absence of GIRK4. They expressed epitope-tagged GIRK1 and GIRK4 in COS cells alone or in combination and examined the localization of the subunits with immunofluorescence labeling. When expressed alone, GIRK1 was localized to the cytosol associated with intracellular intermediate filament proteins, whereas GIRK4 was primarily on the plasma membrane. GIRK1 was detected on the plasma membrane when coexpressed with GIRK4. Therefore, GIRK4 may facilitate the membrane-translocation of GIRK1 subunits. It has, however, not been shown whether GIRK2 and GIRK3 also have similar effect of translocation of GIRK1 to the membrane.
E. Tetrameric Structure
Yang et al. (1995a) found that the inward rectification of the IRK1 channel was substantially attenuated by replacement of aspartate (D) at position 172 in the M2 region with asparagine (N) and substitution of glutamate (E) at position 225 in the proximal carboxyl terminal region with lysine (K). They analyzed the subunit stoichiometry of the IRK1 channel by using the double mutation. They formed tandem tetramers or trimers consisting of different numbers of wild-type subunits and/or subunits bearing the mutation. When either the tetramer or trimer of the wild-type subunits was expressed inXenopus oocytes, the resultant channel currents exhibited the same extent of inward rectification as those obtained from expression of the wild-type monomers. This was also the case when the tetramer was coexpressed with an excess amount of the mutant monomers, indicating that the number of subunits required for a functional IRK1 channel does not exceed four. In contrast, the inward rectification of the channel currents resulting from expression of wild-type trimers was significantly attenuated by coexpression of the mutant monomers. In this case, the channel currents exhibited approximately the same extent of inward rectification as those obtained from expression of a tetramer consisting of three wild-type subunits and one mutant subunit. These data suggest that a functional IRK1 channel is formed by a tetramer of IRK1 subunits.
Biochemical measurement of the molecular weight of brain KG channel proteins suggested that these channels also have tetrameric structure (Inanobe et al., 1995a). As stated earlier, GIRK2 and GIRK4 can form functional homomeric channels (fig. 10A) (Lesage et al., 1995; Krapivinsky et al., 1995a; Duprat et al., 1995; Bond et al., 1995; Kofuji et al., 1995;Velimirovic et al., 1996). However, GIRK1 requires other GIRK subunits to form functional KG channels (Krapivinsky et al., 1995a; Duprat et al., 1995;Velimirovic et al., 1996; Hedin et al., 1996;Chan et al., 1996; Wischmeyer et al., 1997). Tucker et al. (1996) assessed the stoichiometry and relative subunit positions in the GIRK1/GIRK4 heteromeric KG channel by using tandemly linked tetramers consisting of GIRK1 and GIRK4. They found that the most efficient channel comprises two subunits of each type in an alternative array within the tetramer (fig. 10Ba). Through a similar approach,Silverman et al. (1996) found that more than one kind of subunit arrangement including (GIRK1) (GIRK1) (GIRK4) (GIRK4) may also be viable (fig. 10Bb). They also obtained similar data with GIRK1/GIRK2 heteromers.
Tetrameric structure of IRK(Kir2.x) and GIRK(Kir3.x) channels. A putative tetrameric structure of the channels composed of IRK or GIRK subunits in the cell membrane is schematically represented according to the functional and structural studies reported by Lesage et al. (1994 and 1995), Krapivinsky et al. (1995a), Velimirovic et al. (1996), Yanget al. (1995b), Silverman et al. (1996), and Tucker et al. (1996).
Tinker et al. (1996) studied the molecular mechanism of homomeric assembly of IRK1. They concluded that among IRK1, IRK2, and IRK3, the proximal C-terminus and the M2 region equally contribute to polymerization. The proximal C-terminus plays a more significant role in prevention of heteromultimerization between more distantly related channel subunits, such as IRK1 and ROMK1. Tucker et al.(1996), however, found that the core region of the GIRK subunit (M1-H5-M2) but neither the C- nor N-terminal domain was important for subunit assembly between GIRK1 and GIRK4. Thus, the mechanism of heteromultimerization of GIRK subunits may not be the same as that of IRK subunits.
F. Molecular Mechanism Underlying G Protein Activation of G Protein-Gated K+ Channels
1. Interaction between G protein βγ subunits and subunits of G protein-gated K+ channels.
a. The G protein βγ subunit-binding domains in GIRK1 subunits. GIRK1 has a significantly longer C-terminus than the constitutively active Kir channel subunits such as IRK1 (Kuboet al., 1993a,b). Reuveny et al. (1994) first suggested that the C-terminus of GIRK1 includes an amino acid sequence (between positions 318 and 455) exhibiting a certain level of similarity (∼26%) with that of the Gβγ-binding site of βARK1 (fig. 8). They also found that truncation of the C-terminus of GIRK1 at leucine (L) at position 403 but not at proline (P) at position 462 resulted in loss of functional expression of a KG channel inXenopus oocytes coexpressing Gβ1γ2. Inanobe et al. (1995b) directly demonstrated that Gβγ bound to a glutathione S-transferase (GST) fusion protein including the whole C-terminal domain of GIRK1 (between positions 180 and 501). The fusion protein also bound Gβγ when incubated with purified trimeric Gi in the presence of GTPγS but not in the presence of GDP (see also Inanobe et al., 1995a). Furthermore, the binding of Gβγ to the fusion protein was prevented by Gα-GDP but not Gα-GTPγS, indicating that the C-terminal domain of GIRK1 cannot bind with Gβγ included in the trimeric form of the G protein.
By using fusion proteins containing different deleted mutants of the C-terminal domain of GIRK1, Huang et al. (1995) narrowed down the Gβγ-binding region in the C-terminus of GIRK1 to a 190 amino acid stretch (between positions 273 and 462). Gβγ interacted with the fusion protein in ∼1: 1 stoichiometry with calculated Kd of ∼0.5 μm. They further found that the Gβγ-binding domain was composed of two separate segments between positions 318 and 374 and between positions 390 and 462 (Huang et al., 1997) (figs. 8 and 11). This latter segment did not exhibit a significant Gβγ-binding activity by itself but enhanced the Gβγ-binding activity of the other segment. The segment between residues 390 and 462 contains a short amino acid sequence similar to the asparagine-X-X-glutamate-arginine (N-X-X-E-R) motif in adenylyl cyclase 2 which is believed to be critical for regulation of the enzyme by Gβγ (fig. 8) (Chen et al., 1995; Huang et al., 1995).

Domains of GIRK1 involved in G protein-mediated regulation of GIRK1-containing KG channels. Schematic representation of approximate positions of identified domains of GIRK1 which are responsible for interaction of GIRK1 and G protein subunits or phosphatidilinositol 4,5-bisphosphate (PIP2). The approximate positions of three identified G protein βγ-binding sites (one in the N- and two in the C-terminal domain) and one trimeric G protein and/or GDP-from of G protein α subunit-binding site in the N-terminal domain are depicted (Huang et al., 1995,1997; Slesinger et al., 1995). R is the residue corresponding to the arginine of ROMK1 that was reported to play a critical role in interaction of ROMK1 channels with PIP2(Huang et al., 1998).
Interestingly, Huang et al. (1995, 1997) found that Gβγ also bound to a segment of the N-terminal domain of GIRK1 (between positions 34 and 86) (figs. 8 and11). The Gβγ-binding to the fusion protein of the N-terminus also occurred in 1:1 stoichiometry but exhibited ∼10 times lower affinity than Gβγ-binding to C-terminal fusion proteins. They also found that the fusion proteins of the N- and C-terminal domains bound together and synergistically enhanced the Gβγ-binding activity of each other (Huang et al., 1997).
The interaction between Gβγ and the cytoplasmic domains of GIRK1 may indeed underlie the Gβγ-induced activation of KG channels. Huang et al. (1995)constructed synthetic peptides possessing the partial amino acid sequence of the predicted Gβγ-binding domains of the N- and C-termini. These synthetic peptides inhibited not only the binding of Gβγ to the corresponding fusion proteins, but suppressed GIRK1-containing KG channel currents activated by Gβ1γ2. Slesinger et al. (1995) expressed chimeras of IRK1 and GIRK1 inXenopus oocytes and examined the response of the resultant channel currents to Gβ1γ2. The Gβ1γ2-induced increase in channel activity was observed only when the chimeras contained the N- (between positions 31 and 85) and/or the C-terminal (between positions 325 and 501) domain of GIRK1. A similar result was reported by using chimeras of GIRK1 and IRK2 (Kunkel and Peralta, 1995).
Yan and Gautam (1996) showed with the yeast two-hybrid system that Gβ bound with the N-terminus of GIRK1. Different types of Gβ interacted with the N-terminal domain of GIRK1 with distinct efficacies. An N-terminal fragment of 100 amino acids of Gβ interacted with the N-terminal domain of GIRK1 as effectively as the whole Gβ. This N-terminal domain of Gβ includes the region responsible for the interaction between Gβ and Gα according to the analysis of the crystal structure (Wall et al., 1995; Lambright et al., 1996). Thus, Gα-GDP might prevent Gβγ from interacting with KG channels by competing with the N-terminus of GIRK1 on the N-terminus of Gβ. Binding of Gβ to the C-terminus of GIRK1 was not clearly detected in this study. Other domains of Gβ or Gγ might, therefore, participate in the interaction between Gβγ and the C-terminus of GIRK1. At present, the whole aspect of the molecular interaction between Gβγ and KG channel subunits has not been clarified.
b. The G protein βγ subunit-binding domains in other subunits of G protein-gated K+ channels.
Homomeric GIRK2 or GIRK4 channels are also activated by Gβγ (Krapivinsky et al., 1995a; Velimirovic et al., 1996). It was also reported that GIRK4 could mediate the activation of the GIRK1/GIRK4 heteromeric channel by Gβγ (Slesinger et al., 1995; Tucker et al., 1996).
GIRKs2-4 have domains whose amino acid sequences are similar to those of the Gβγ-binding domains in the N-terminus and the proximal C-terminus of GIRK1 (between positions 34 and 86 and between residues 318 and 374, respectively) (fig. 8). However, they lack a region corresponding to that at the distal C-terminus of GIRK1 (between positions 390 and 462). Huang et al. (1997) showed that the Gβγ-binding activity was similar among the N-terminal domains of GIRKs1-4, although the C-terminal domains of GIRKs2-4 exhibited slightly lower Gβγ-binding activity than that of GIRK1. They also found that the C-terminal domain of GIRK1 interacted with the N-terminus of GIRK4 and thereby synergistically enhanced the Gβγ-binding activity (Huang et al., 1997). Therefore, the high-affinity Gβγ-binding site in the GIRK1/GIRK4 heteromeric channel might be formed through interaction of the C-terminus of GIRK1 subunit with the N-terminus of the GIRK1 and/or GIRK4 subunits. This interaction may at least in part underlie the higher channel activity yielded by coexpression of GIRK1 and GIRK4 than expression of either of the GIRK subunits alone.
2. Mechanism underlying G protein βγ subunitinduced activation of G protein-gated K+ channels.
As described in Sections II.B.3. and 4., functional analyses of the KACh channel indicate that the G protein-mediated activation of the channel results from an increase in the functional number of the channels due to the cooperative interaction of GKβγ and GIRK subunits (Hosoyaet al., 1996). However, the molecular mechanism responsible for this phenomenon has not been clearly identified.
Slesinger et al. (1995) suggested that the N-terminal domain of GIRK1 may function to suppress the Gβγ-independent basal current because chimeras of GIRK1 and IRK1 showed a lower basal activity when they included the N-terminal domain of GIRK1. Dascal et al.(1995) proposed that the C-terminal domain of GIRK1 may block the KG channel pore in a way similar to the “Shaker ball” of the Kv channels because a myristoylated cytosolic C-terminal tail of GIRK1 suppressed the GIRK1 or ROMK1 channel currents. These results suggest that the KG channel might be intrinsically inhibited by the C- and/or N-terminal domains of GIRK1, and that GKβγ might activate the channel by removing the inhibition.
Slesinger et al. (1995) found that the whole-cell channel currents of IRK1 whose distal C-terminal region was replaced with a part of the C-terminal domain of GIRK1 (between positions 325 and 501) were doubled when coexpressed with Gβ1γ2 resulted inXenopus oocytes. The doubling of the whole-cell channel current could not be explained only in terms of an increase inPo because the chimeric channel possessed aPo as high as ∼0.8 in the absence of Gβ1γ2 at the single channel level. These results support the aforementioned notion that GK activates the KAChchannel by increasing the functional number of the channels without modulating the fast open-close transitions of the channels (Hosoyaet al., 1996).
Huang et al. (1998) recently proposed that PIP2 is critically involved in Gβγ-induced activation of KG channels. They found that PIP2 by itself induced the maximum activity of heteromeric GIRK1/GIRK4 and homomeric GIRK2 channels in inside-out patch membranes of Xenopus oocytes. This PIP2-induced response seemed to result from direct interaction of PIP2 with the C-terminus of GIRK subunits because PIP2 directly bound GST-fusion protein of the C-terminus of GIRK1 in vitro and because the PIP2-induced channel activity was not inhibited by exogenously applied Giα subunits (probably in the GDP-bound form). However, the PIP2-induced enhancement of channel activity was not unique to the GIRK channels but also found in ROMK1 and IRK1 channels that had run down in the inside-out configuration. Furthermore, ROMK1 and IRK1 channels were more potently activated by PIP2 than the KG channels. However, Gβγ could not activate GIRK1/GIRK4 channels 10 min after patch excision where PIP2 was expected to be depleted from the membrane. The channels preincubated with Gβγ under these conditions, however, exhibited much higher PIP2 sensitivity than the control. Specific anti-PIP2 antibodies suppressed GIRK1/GIRK4 channel currents more slowly in the presence than the absence of Gβγ. From these results, they concluded (a) that the direct interaction of PIP2 with the C-terminus of GIRK subunits is prerequisite for KG channel activity, whereas in the absence of Gβγ, the channel has significantly lower PIP2 sensitivity than other Kir channels and (b) that Gβγactivates KG channels by increasing the PIP2 sensitivity of GIRK subunits. Therefore, from their point of view, the deactivation of KGchannels is the rundown commonly observed with different types of Kir channels, although the activation of KG channels is the reactivation of the rundown KG channels. In this context, Gβγ is a regulator of rundown/reactivation of KG channels. They suggested that the interaction between the pore-forming C-terminus of GIRK as well as other Kir subunits with PIP2 in the membrane might lead to opening of the channel pore and that the synergistic interaction between Gβγ and PIP2 might occur through the pleckstrin homology domain in the C-terminus of GIRK subunits (fig. 11). Although this hypothesis is very attractive, some precautions may be necessary to extrapolate this hypothesis into the mechanism responsible for the physiological activation of native KG channels by Gβγ. For example, in inside-out patch membranes of cardiac myocytes, IK1 channels, which may be homomeric IRK2 channels and thus are expected to be more sensitive to PIP2 than KGchannels, usually run down very promptly. However, the KACh channel, which may be less sensitive to PIP2 than the IK1 channel, can be consistently and strongly activated by Gβγ applied even tens minutes after patch excision (Kurachi, 1995). Therefore, it may be important to analyze the effect of PIP2 on the interaction between Gβγ and native KG channels.
3. Interaction between subunits of G protein-gated K+channels, Gα proteins, and membrane agonist receptors.
Huang et al. (1995) found that Gα-GDP and the entire heterotrimeric G protein can bind to fusion proteins containing the N-terminal domain of GIRK1 (fig. 11). This observation raises the possibility that membrane agonist receptors, GK, and KG channels, might form a functional complex in native KG channel systems. Namely, once GK associated with a GIRK1 subunit is activated, the dissociated GKβγ would promptly access the GKβγ-binding site on the GIRK1 subunit. The GKα-GTPdissociated from the GK, however, may be quickly converted to GKα-GDP by the aid of RGS proteins, leading to reassociation of GKα-GDP with the GIRK1 subunit. This GKα-GDP would effectively sequestrate the GKβγ from the GKβγ-binding site on the GIRK1 subunit, and the cycle would be complete. Indeed, it has been shown that KG channels coexpressed with RGS proteins inXenopus oocytes are deactivated much faster than expected from the intrinsic rate of Gβγdissociation from GIRK subunits (Doupnik et al., 1997;Krapivinsky et al., 1995c; Saitoh et al., 1997). In the classical view of receptor/G protein interaction (Levitzki, 1981), association of an agonist-bound receptor with Gα-GDP is the ratelimiting step for the receptor-mediated activation of effectors. However, the fast conversion of GKα-GTP to GKα-GDP due to the enhanced GTPi hydrolysis rate evoked by RGS proteins might not provide a sufficient time for GKα-GTP to be dissociated from receptor. In the continuous presence of an agonist bound to the receptor, therefore, the receptor would promptly restart the next round of the G protein cycle. The resultant increase in GDP/GTP exchange might balance the accelerated GTPase activity and thereby maintain the steady-state concentrations of GKα-GTP and GKβγ at a certain level, which would explain why RGS proteins do not significantly decrease the steady state response of the KG channel system (Doupniket al., 1997; Saitoh et al., 1997). A distinct but similar hypothesis of a functional receptor/G protein/effector complex was recently proposed by Ross’ group based on an extensive analysis of the reconstituted M1-muscarinic/Gq/phospholipase C-β1 system (Biddlecome et al., 1996).
The maximum response of the KACh channel is reached within several hundred milliseconds after application of ACh (fig. 1) (Breitwieser and Szabo, 1988). Neuronal KG channels are also known to respond to agonist in a comparable fast time course (Surprenant and North, 1988; Sodickson and Bean, 1996). The model of G protein/RGS protein/channel interaction described above might be applicable to the steady-state response in the continuous presence of agonists, but cannot explain this initial fast response. Slesinger et al. (1995) raised the possibility that GIRK1 may directly interact with M2-muscarinic receptors through its hydrophobic core region (M1-H5-M2). They found that a chimera of IRK1 and GIRK1 containing the C-terminus but not the hydrophobic core region of GIRK1 could be activated by coexpressed Gβ1γ2, but not through coexpressed M2-muscarinic receptors. The ability to respond to receptor stimulation was endowed by transplantation of the hydrophobic core region of GIRK1 to the chimera. When the receptor is indeed kept in the vicinity of GK through association with a GIRK1 subunit, the agonist-induced initial response is expected to be substantially accelerated. However, this hypothesis needs further verification because Kofuji et al. (1996a)showed that GIRK1 whose hydrophobic core region was substituted by that of ROMK1 could be activated through coexpressed M2-muscarinic receptors. In addition, it has not been examined whether there are additional molecules which impose a topological restriction on the receptor/GK/KG channel system which enables efficient signal transmission between the molecules. These issues remain unanswered.
4. Possible mechanisms underlying specific signal transduction in the receptor/G protein/G protein-gated K+ channel system.
In atrial myocytes, the KACh channel is activated by stimulation of M2-muscarinic and A1 adenosine receptors. However, β1-adrenergic stimulation, which would also induce dissociation of Gβγ from Gs, never activates the KACh channel in cardiac atrial myocytes.
Such specific signal transduction cannot be explained in terms of the different affinities of distinct types of Gβγ for KGchannels. Wickman et al. (1994) compared the effects of recombinant Gβγs (Gβ1γ1, Gβ1γ2, Gβ1γ5, Gβ1γ7, Gβ2γ5, and Gβ2γ7) on the KACh channel in inside-out patch membranes of atrial myocytes, and found less than 10-fold difference in potency among these Gβγs except for Gβ1γ1. Gβ1γ1 is a major component of Gβγ of the retinal G protein, transducin. Native transducin βγ subunits are more than 100-fold less potent than other Gβγs (Yamadaet al. 1994a). A lack of specificity of Gβγ in vitro was confirmed when KG channel currents in Xenopus oocytes expressing GIRK1 could be activated through coexpressed β2-adrenergic receptors (Lim et al., 1995).
An alternative explanation for receptor-specific transduction may be that a particular set of receptor, G protein, and GIRK subunits is compartmentalized into a microdomain in the native KG channel system. However, when ACh and adenosine are applied together to cardiac atrial myocytes they activate the current in a less-than-additive manner (Kurachi et al., 1986a,b; Bünemann et al., 1995). Similar phenomena have been reported for different KG channel systems (North, 1989) including stimulation of 5-HT1A and GABAB receptors in rat hippocampal CA1 pyramidal cells (Andrade et al., 1986), μ-opioid, M2-muscarinic, and GABAB receptors in rat lateral parabrachial neurons (Christie and North, 1988), and GABAB and D2 dopamine receptors in rat substantia nigra zona compacta neurons (Lacey et al., 1988). Thus, if compartmentalization existed, it would have to be formed in such a way that certain classes of receptors are excluded from a KG channel system, although the others can share the system. The molecular mechanism by which this may be achieved remains to be elucidated.
IV. Voltage-Dependent Properties of G Protein-Gated K+Channels
In previous sections, we considered the G protein-mediated activation of KG channels. In this section, we will deal with voltage-dependent properties of KGchannels. As described in the section II.B.1., all KG channels exhibit the inward rectification property where ionic currents flow through KGchannels more readily in the inward than the outward direction. The mechanism of inward rectification has been mainly studied with the constitutively-active, classical Kir channels such as IRK1 channels. Most of our understanding of the mechanism of inward rectification of KG channels is, therefore, based on these results. However, the inward rectification of KGchannels is not identical with that of the classical Kir channels especially in terms of its voltage-dependency and kinetics. In this section, we first review the present understanding of the mechanism of inward rectification of the classical Kir channels, and then deal with specific issues regarding KG channels.
A. Inwardly-Rectifying K+ Channels
1. Voltage-dependent change in inwardly rectifying K+channel activity.
K+ channels mediate a flow of K+ ions depending on an electrochemical gradient of K+ ions across the plasma membrane (i.e., Vm - EK) (Hille, 1992b). Thus, the macroscopic K+ current flowing through K+-selective channels can be expressed as:
The inward rectification of Kir channels occurs because gKdecreases as Vm increases in the positive direction. The relationship between gK andVm (G-V relationship) is usually expressed by the following Boltzmann’s equation:
Kir channels are significantly heterogenous in terms of their G-V relationship (Hille, 1992b). The constitutively-active, classical Kir channels composed of the IRK subunits have the steepest G-V relationship (strong inward rectifiers) (Kubo et al., 1993a;Takahashi et al., 1994; Morishige et al., 1994;Makhina et al., 1994; Pärier et al., 1994). The v and ΔVh values for the strong inward rectifiers are normally 5 to 10 mV and ∼−10 mV, respectively (Leech and Standfield, 1981; Hestrin, 1981; Gunning, 1983;Kurachi, 1985; Tourneur et al., 1987; Harvey and Ten Eick, 1988; Cohen et al., 1989; Silver and DeCoursey, 1990). Thus, the gK of the channels is approximately one fourth of thegKmax at EK and becomes virtually negligible around EK + 60 mV. Strong inward rectifiers can, therefore, holdVm close to EKin the absence of action potentials, but their outward currents are effectively shut off once an action potential is generated. However, ROMKs, BIR, and uKATP-1 channels are weak inward rectifiers because they only marginally rectify at highly depolarized potentials (Ho et al., 1993; Inagaki et al., 1995a; Yamada et al., 1997). In the presence of a physiological K+ gradient, they can even exhibit outwardly rectifying I-V curves as predicted by the constant-field theory. Therefore, these channels strongly impair action potential generation and fix Vm toEK. The extent of inward rectification of KG channels and KAB-2 channels is intermediate between those of the strong and weak inward rectifiers (Kubo et al., 1993b; Bond et al., 1994and 1995; Lesage et al., 1995; Krapivinsky et al., 1995a; Duprat et al. 1995; Kofuji et al., 1995; Doupnik et al., 1995b; Takumi et al., 1995; Isomoto et al., 1996; Velimirovic et al., 1996).
When Vm is suddenly shifted from one value to another, strong inward rectifiers alter gK through at least two kinetically distinct processes (Leech and Standfield, 1981;Hestrin, 1981; Gunning, 1983; Kurachi, 1985; Tourneur et al., 1987; Harvey and Ten Eick, 1988; Cohen et al., 1989; Silver and DeCoursey, 1990; Newman, 1993). Upon depolarization, gK decreases first instantaneously to a certain extent and then continues to decline slowly to the steady state level. The reverse is true for hyperpolarization of the membrane wheregK increases first instantaneously and then more slowly. The slow component of these changes is called the relaxation of the currents. This component can be fitted with a single exponential function with the time constant (τ) determined by channel-types and Vm at which the relaxation takes place. The τ of mammalian strong inward rectifiers largely falls within a range of <10 msec, having a peak around EK and smaller values at both more positive and negative potentials (Leech and Standfield, 1981; Hestrin, 1981; Gunning, 1983; Kurachi, 1985; Tourneur et al., 1987;Harvey and Ten Eick, 1988; Cohen et al., 1989; Silver and DeCoursey, 1990).
2. Mg2+ and polyamine block.
The voltage-dependent change in gK of strong inward rectifiers recently turned out to be caused by blockade of the channel pore by intracellular cations such as Mg2+i and polyamines (Matsudaet al., 1987; Vandenberg, 1987; Matsuda, 1988 and 1991;Lopatin et al., 1994; Ficker et al., 1994; Fakleret al., 1994). In other words, the apparent gating of Kir channels results from exogenous elements. Consistent with this, they lack in their primary structure the domain corresponding to the voltage-sensing S4 region of voltage-dependent K+channels (Ho et al., 1993; Kubo et al., 1993a). Polyamines are aliphatic amines such as putrescine, spermidine and spermine, which are endogenous metabolic intermediates derived from arginine. They normally exist in submillimolar concentrations in the cytosol of almost all cell-types (Watanabe et al., 1991). Putrescine, spermidine, and spermine bear 2, 3, and 4 positive charges per molecule, respectively. Thus it is likely that polyamines and Mg2+i interact with Kir channels via the electrostatic force created between their own positive charges and negatively charged amino acid residues within the channel pore (Stanfield et al., 1994; Taglialatela et al., 1994; Lu and MacKinnon, 1994; Wible et al., 1994; Lopatinet al., 1994; Ficker et al., 1994; Fakleret al., 1995; Yang et al., 1995a)
The interaction of charged substance with their receptor sites within the channel pore is affected by the transmembrane electric field (Hille, 1992b). As a result, the apparent potency of Mg2+/polyamines is greater at more depolarizedVm. The apparent potency of a blocker with a larger valance is more strongly affected byVm (Hille, 1992b). Thus, spermidine and spermine block Kir channels in a more steeply voltage-dependent manner than Mg2+i or putrescine (Lopatin et al., 1994). Therefore, it is likely that the steep voltage-dependency of gK of strong inward rectifiers mainly arises from the blockade by spermidine and spermine (Lopatinet al., 1994; Ficker et al., 1994; Fakleret al., 1994; Ishihara et al., 1989 and 1996). It has not been clarified how the effect of these intracellular cations ongK of Kir channels depends on ΔV and notVm.
Upon a sudden depolarization (or hyperpolarization), the IRK1 channel is blocked (or unblocked) by Mg2+i and putrescine quasi-instantaneously, but the effects of spermidine and spermine are slower and occur in a time-dependent manner (Ficker et al., 1994; Ishihara et al., 1996). Thus, the instantaneous and time-dependent components of the gK change may arise from the effects of Mg2+i/putrescine and spermidine/spermine blocks, respectively. When the membrane is depolarized to potentials less negative than∼EK + 20 mV, inward rectification is predominantly caused by spermine and spermidine (Ishihara et al., 1986, 1989). Upon much stronger depolarization, however, the channels may be first blocked by Mg2+i/putrescine which is followed by the block by spermidine/spermine that further suppresses gKin a time-dependent manner. The G-V relationship at these potentials is, therefore, determined by the balance between the two mechanisms with distinct voltage dependencies. As a result, the G-V relationship of strong inward rectifiers usually becomes slightly less steep atΔV between ∼+30 mV and +100 mV than expected from the data at more negative potentials (Ishihara et al., 1989;Yamashita et al., 1996). This phenomenon seems to be physiologically significant because action potentials usually cover a range of potentials between EK andEK + ∼100 mV.
3. Mg2+/polyamine block sites in the inwardly rectifying K+ channel pore.
The molecular mechanism underlying the Mg2+/polyamine block has been elucidated through comparison of the molecular structures of the strong inward rectifier IRK1 and the weak inward rectifier ROMK1 channels. At EK −20 mV, the Kd value of Mg2+ and spermine for IRK1 channels are respectively ∼1 mm and ∼10 μm (Lopatinet al., 1994), which are ∼1000 and ∼1000,000 times smaller than the corresponding values for ROMK1 channels (Nicholset al., 1994; Yang et al., 1995a). Mutagenesis studies revealed that at least two negatively-charged amino acid residues found in IRK1 (aspartate (D) at position 172 in the M2 region and glutamate (E) at position 224 in the proximal C-terminus) but not in ROMK1 are responsible for this discrepancy (fig.12) (Stanfield et al., 1994;Taglialatela et al., 1994; Lu and MacKinnon, 1994; Wibleet al., 1994; Lopatin et al., 1994; Fickeret al., 1994; Yang et al., 1995a). For convenience, we shall designate these two positions R1 and R2, respectively. The two acidic residues are likely to interact with the blocking particles independently of each other (Yang et al., 1995a). Other residues may also participate in the blocking to a lesser extent because ROMK1 exhibits weak but significant inward rectification (Ho et al., 1993). IRK2 and IRK3 also have the aspartate (D) and glutamate (E) residues at the analogous positions, although both BIR and uKATP-1 lack acidic residues at either of the sites (Takahashi et al., 1994; Morishige et al., 1994; Ho et al, 1993; Inagaki et al., 1995a andb).

Proposed Mg2+/polyamine interaction sites in Kir subunits. A schematic representation of proposed Mg2+/polyamine interaction sites in Kir subunits. R1 and R2 indicate the approximate position of the negatively charged amino acid residues in IRK1 that are responsible for the high sensitivity of the channel to intracellular Mg2+ and polyamine: aspartate at position 172 and glutamate at position 224, respectively (Stanfieldet al., 1994; Taglialatela et al., 1994;Lu and MacKinnon, 1994; Wible et al., 1994; Lopatinet al., 1994; Ficker et al., 1994; Yanget al., 1995a). The table shows the amino acid residues at the sites analogous to R1 or R2 in different types of Kir subunits. N indicates asparagine; G, glycine; D, aspartate; E, glutamate; and S, serine.
B. Inward Rectification of G Protein-Gated K+ Channels
1. Inward rectification of the muscarinic K+channel.
The KACh channel exhibits clear inward rectification irrespective of ACh concentrations and thus GK activity (figs.13Aa and b). The G-V relationship of the KACh channel can be fitted with Eq. 4 with ΔVh and v of ∼0 and ∼20 mV, respectively (fig. 13Ac). Both the values are significantly larger than those of strong inward rectifiers but much smaller than those of weak inward rectifiers. The Boltzmann’s fit deviates from the measured values at ΔV positive to ∼+40 mV. Thus, the actual G-V relationship is less steep at depolarized potentials than predicted from the data at more negative potentials as is the case for strong inward rectifiers (Ishihara et al., 1989)

Inward rectification of the muscarinic K+ channel. These data were obtained from the whole-cell recording of the muscarinic K+ channel current in a single sinoatrial node cell of rabbit. A: a, The channel current induced by 5 μm acetylcholine applied to the bath. The extracellular K+ concentration was 5.4 mm. Five hundred msec command pulses (c.p.) were applied from the holding potential of −53 mV to the potentials indicated above each trace. Note that in each trace, the command pulse induced an instantaneous jump followed by a time-dependent change (relaxation) of the channel current. This was also the case when the membrane potential was returned to the holding potential. b, The relationship between the membrane potential and the channel currents induced by different concentrations of acetylcholine. The magnitude of the currents was measured immediately (closed symbols) or 500 msec (open symbols) after application of the command pulses. The reversal potential was ∼−80 mV under each condition. c, The relationship between the membrane potential relative to the K+ equilibration potential (∼−80 mV under these conditions) (ΔV) and the macroscopic chord conductance (gK) of the muscarinic K+ channel current induced by 5 μm acetylcholine (symbols). In this graph,gK is normalized to its maximum value (gKmax) predicted by the fit of the data with Boltzmann’s equation (Equation 4 in the text) (line in the graph). The best fit was obtained with Vhand v of −0.32 and 21.0 mV, respectively. B: The voltage-dependence of the time constants of the relaxation of the muscarinic K+ channel currents induced by 0.2 μm (squares), 1 μm (triangles), and 5 μm (circles) of acetylcholine. The voltage-clamp protocol was the same as in A. The relaxation of each acetylcholine-induced current could be fitted with a biexponential function with a smaller (open symbols) and a larger (closed symbols) time constant. The inverted triangle on the voltage-axis indicates the reversal potential of the channel currents. C: The channel currents observed in the sinoatrial node cell in the presence and the absence of different concentrations of acetylcholine. Command steps were applied from −53 mV to various membrane potentials indicated to the right of each current trace, and the obtained currents are superimposed. Note that the higher concentration of acetylcholine induced not only larger currents but accelerated relaxation at every membrane potential. [Reproduced with permission from Kurachi (1990)].
Upon a sudden change in Vm, gKof KACh channel also changes first instantaneously and then time-dependently (figs. 13Aa,b). However, this relaxation of the KACh channel is much slower than that of mammalian strong inward rectifiers. This slow relaxation presumably has an important functional meaning in regulation of action potential configuration of some cell-types such as the pace-making sinoatrial nodal cells of the heart. However, this issue has not been extensively investigated to our knowledge. The relaxation of the KACh channel occurs with a time course that can be fitted with a biexponential function with τ of <10 and 50 to 150 msec (fig. 13B). The former value is similar to, but the latter is ∼5 to 10 times larger than that of mammalian strong inward rectifiers. The slow τ of the KAChchannel is less steeply voltage-dependent than the τ of the strong inward rectifiers and monotonically decreases as the membrane is depolarized (Osterrieder et al., 1981; Iijimaet al., 1985; Simmons and Hartzell, 1987; Kurachi, 1990). These results suggest that the slow relaxation of the KACh channel may arise from a molecular mechanism distinct from that underlying the relaxation of mammalian strong inward rectifiers. Furthermore, the slow τ of the KACh channel becomes smaller as the ACh concentration, therefore G protein activity is increased (figs. 13B, C).
2. Mg2+/polyamine block of G protein-gated K+ channels.
Fig. 14A shows the effect of Mg2+i and intracellular polyamines on the single KAChchannel current (Yamada and Kurachi, 1995). In these experiments an inside-out patch was obtained from a rabbit atrial myocyte with pipette solution containing 145 mm K+ and 0.5 μm ACh. The patch was excised in a solution containing 145 mm K+ and 1.4 mm free Mg2+. Application of a nonhydrolyzable GTP analogue, GTPγS induced inward KACh channel currents at −60 mV. This channel activity continued even after washout of GTPγS because the nucleotide irreversibly activated GK in the membrane. Depolarization of the membrane to +40 mV resulted in appearance of small outward currents. The unitary amplitude of these outward currents was increased after removal of Mg2+i. Under these conditions, the single-channel I-V relationship was virtually linear, indicating that γ had been reduced by Mg2+block at the depolarized potential (Horie and Irisawa, 1987, 1989). The IC50 of Mg2+i has been reported to be ∼300 μm at ΔV of +40 mV (Horie and Irisawa, 1987 and1989), whereas cytosolic Mg2+ concentrations are estimated to be in millimolar range (Hess et al., 1982;Gupta et al., 1984; Alvarez-Leefmans et al., 1986, Blatter and McGuigan, 1986). Thus, Mg2+ block is likely to underlie the physiological inward rectification of the single KACh channel conductance (γ)

Effect of spermine on the muscarinic K+ channel. All data shown were obtained from muscarinic K+ channels in inside-out patch membranes of rabbit atrial myocytes exposed to symmetrical 145 mm K+solutions. The pipette also contained 0.5 μmacetylcholine. A: Effect of 10 μm spermine applied to the internal side of the patch membrane. The protocol of perfusion of the intracellular side of the patch membrane and the change in holding membrane potential are indicated above and below the current trace, respectively. Arrowheads indicate the zero current levels. Large and small spikes observed at + 40 mV in the presence of spermine were occasional opening of ATP-sensitive K+ and muscarinic K+ channels, respectively. B: Concentration-dependent inhibitory effect of internal spermine on outward muscarinic K+ currents at + 40 mV. a, After the muscarinic K+ channels were irreversibly activated by GTPγS the indicated concentrations of spermine was applied to the intracellular side of the patch membrane in the absence of intracellular Mg2+. The N·Po of the muscarinic K+ channels was 0.096, 0.084, 0.046, and 0.014 in the presence of 0, 10, 30, and 100 nm spermine, respectively. The bars and numbers below the current trace correspond to the numbers in the graph c. b, High-speed current traces obtained from the same patch as a under the conditions indicated to the right of each current trace. c, The current amplitude histogram obtained from the same current record as a. The numbers in the graph correspond to those in a and indicate the portions of the record from which each histogram was constructed. Bin width is 0.02 pA, and the number of points in each bin is expressed as a percentage of the total number of points recorded. d, Concentration-dependent effect of spermine on the unitary current amplitude (open circles) and N·Po (closed circles). Data were normalized to the values obtained in the absence of spermine. Symbols and bars indicate the mean ± SE. Lines were drawn by eye. Unitary channel current amplitude in 1 and 10 μm spermine was measured by eye because very few openings of muscarinic K+ channels were available in this concentration range. C: Voltage-dependent change in N·Po of muscarinic K+ channels in the presence (closed circles) and absence (open circles) of 100 nm spermine. ATP (1 mm) was added to the intracellular side of patch membranes. N·Po is expressed as a percentage of the value recorded at −60 mV. Symbols and bars indicate the mean ± SE. Lines were drawn by eye. D: The time-dependent change in muscarinic K+currents in the presence or absence of 300 nm, 1 μm, and 3 μm spermine. Muscarinic K+ channels in an inside-out patch membrane were preactivated with 10 μm GTPγS, and then Mg2+ was removed from the intracellular side of the patch membrane. As shown at the top, 1 sec duration command steps to + 40 mV were applied every 5 sec from a −60 mV holding potential. Reading from left to right, progressively higher concentrations of spermine (300 nm−3 μm) were applied to the intracellular side of the patch membrane. The membrane was depolarized ∼30 to 40 times in each condition. The first and second rows show representative current traces. In each of the traces, the baseline current was subtracted with a computer. Arrowheads indicate the zero current level. Time and current scales are shown to the right beneath the second row. The third row shows the ensemble average current obtained from 20 successive pulses under each condition. The zero current level is indicated by a straight line. Each of the ensemble average current traces could be fitted by a single exponential both at + 40 and −60 mV. At + 40 mV, the time constant obtained with the best fit was 9.9 sec in the control; 1.3 sec in the presence of 300 nmspermine; 0.38 sec, 1 μm; 0.18 sec, 3 μm. [Reproduced with permission from Yamada and Kurachi (1995)].
Spermine (10 μm) applied to the internal side of the patch membrane in the absence of Mg2+i almost completely suppressed the outward currents. Fig. 14B shows the concentration-dependent effect of spermine at this potential. It is clear that spermine reduced Po in a concentration-dependent manner without altering γ (figs. 14Ba-c). The IC50 of spermine was ∼10 nm at ΔV of +40 mV (fig. 14Bd). Thus, spermine is much more potent than Mg2+i in suppressing outward KACh channel currents at this potential. Not only spermine but spermidine and putrescine also inhibit outward currents of the KACh channel although putrescine was much less potent than the other two (Yamada and Kurachi, 1995). Besides the KACh channel (a GIRK1/GIRK4 heteromer), the GIRK1/GIRK2 and GIRK2/GIRK4 heteromultimers are also shown to be blocked by Mg2+i and intracellular polyamines at Vm positive toEK (Velimirovic et al., 1996;Lesage et al., 1995).
Figure 14C shows the relationship betweenVm and Po of the KACh channel in the presence and absence of 100 nm spermine. The same concentration of spermine decreased Po more strongly asVm became more positive probably due to the voltage-dependent increase in the apparent potency of the blocker. Under these conditions, Po decreased to ∼20% at EK +40 mV. Because cytosolic concentrations of polyamines are estimated to be in a submillimolar range (Watanabe et al., 1991), these data indicate that polyamine block also underlies the physiological inward rectification of the KACh channel current by reducing Po at depolarized potentials.
When Vm was suddenly depolarized from −60 mV to +40 mV, the Po of the channel decreased in a time-dependent fashion (fig. 14Da, the upper two rows). The bottom row shows the ensemble averaged current under each condition. The decay of the outward current became faster as spermine concentration was increased, probably because the on-rate of the block increased in the presence of higher concentration of spermine (Yamashita et al., 1996). In the presence of physiological concentrations of spermine, the kinetics of polyamine block would become much faster and exceed those of the slow relaxation. Therefore, polyamine block is unlikely to be the mechanism of the slow relaxation of the KACh channel. In other words, the inward rectification and the slow relaxation of KAChchannels might be attributable to distinct mechanisms. This issue will be dealt with in Section IV.B.4.
3. The Mg2+/polyamine-binding sites in G protein-gated K+ channels.
The KG channels have an unique arrangement of negative charges at the R1 and R2 sites (figs. 8 and 12). GIRK1 has negatively-charged aspartate at R1 (D173) but uncharged serine (S) at R2 (Kubo et al., 1993b; Dascalet al., 1993). However, all GIRKs 2–5 have uncharged asparagine (N) at R1 and negativelycharged glutamate (E) at R2 (Lesage et al., 1994 and 1995; Isomoto et al., 1996; Krapivinsky et al., 1995a). Recently, Kofuji et al. (1996a) examined the functional significance of D173 of GIRK1 by replacing this residue with non-charged asparagine [GIRK1(D173N)]. The G-V relationship of KG channels inXenopus oocytes expressing GIRK1(D173N) was much less steeply voltagedependent and shifted leftward along the voltage-axis compared with that for the wild-type GIRK1: v and ΔVh were respectively ∼40 and ∼−50 mV for GIRK1(D173N), and ∼20 and ∼−30 mV for the wild-type GIRK1. These data indicate that the Mg2+i/polyamine block at D173 of GIRK1 is in fact crucial for the inward rectification of KG channels that contain GIRK1.
Although homomultimeric GIRK2 and GIRK4 channels show clear inward rectification, their gK is hardly saturated even atEK −100 mV (Lesageet al., 1994; Bond et al., 1995; Iizuka et al., 1995). However, when the G-V curve of GIRK4 channels constructed from the data shown by Chan et al. (1996) were roughly fitted with equation 4, the v andΔVh values were estimated to be ∼30 mV and ∼−40 mV, respectively. The difference in G-V relationship between GIRK1 and GIRK4 channels might be due to the distinct location of the negatively charged residues in the channel pore. However, thev and ΔVh values of GIRK1/GIRK4 heteromeric channels are ∼20 and ∼−20 mV, respectively as estimated from the data reported by Krapivinsky et al.(1995a). GIRK1/GIRK4 channels therefore have significantly smallerv and more positive ΔVh values than GIRK4 homomeric channels, and a more positiveΔVh value than GIRK1 channels. Further studies are needed to clarify how the G-V relationship of GIRK1/GIRK4 channels is determined by the negatively charged residues at R1 of GIRK1 and at R2 of GIRK4. The G-V relationship of GIRK1/GIRK4 channels is not identical with that of the native KAChchannel (Iizuka et al., 1995) and exhibits a more negativeΔVh value. These results suggest that subunit composition might not be the same between the native KACh channel and the heterologously-expressed GIRK1/GIRK4 channel, and/or that some additional factors other than GIRK1 or GIRK4 might be included in the native KACh channel.
4. Slow relaxation of G protein-gated K+ channels containing GIRK1.
No mammalian strong inward rectifiers show such slow relaxation as the KACh channel does (fig.13) (Leech and Standfield, 1981; Kurachi, 1985; Tourneur et al., 1987; Harvey and Ten Eick, 1988; Cohen et al., 1989; Silver and DeCoursey, 1990). GIRK2 and GIRK4 homomultimeric channels show fast relaxation kinetics similar to those of IRKs (Lesageet al., 1995; Krapivinsky et al., 1995a; Iizukaet al., 1995; Duprat et al., 1995; Bond et al., 1995; Kofuji et al., 1995; Velimirovic et al., 1996). The heteromeric KG channels containing GIRK1, however, exhibit the slow relaxation after a voltage step similar to that of the KACh channel (i.e., a native GIRK1/GIRK4 heteromer) (Kubo et al., 1993b;Krapivinsky et al., 1995a; Iizuka et al., 1995;Duprat et al. 1995; Kofuji et al., 1995; Doupniket al., 1995b; Velimirovic et al., 1996; Isomotoet al., 1996), although the relaxation kinetics of GIRK1/GIRK2 channels may be slightly faster than those of GIRK1/GIRK3 or GIRK1/GIRK4 channels (Wischmeyer et al., 1997). Therefore, it is likely that GIRK1 is responsible for the slow relaxation.
Kofuji et al. (1996a) recently found that a phenylalanine residue at position 137 (F137) in the H5 region of GIRK1 is responsible for the slow relaxation (fig. 8). All other GIRK subunits bear conserved serine (S) at the position analogous to F137 of GIRK1. Interestingly, this residue was identified by Chan et al.(1996) and Wisch-meyer et al. (1997) as the residue responsible for the synergistic enhancement of macroscopic current amplitudes induced by coexpression of GIRK1 and other GIRK subunits. When GIRK1 whose F137 was replaced with serine [GIRK1(F137S)] was expressed alone or in combination with GIRK3 in Xenopusoocytes, the resultant macroscopic KG channel currents no longer exhibited the slow relaxation (Kofuji et al., 1996a; Wischmeyer et al., 1997). Kofuji et al. (1996a) showed that the G-V relationships of the channel currents observed in oocytes expressing either the wild-type GIRK1 or the mutant GIRK1(F137S) alone were not significantly different. It is also noteworthy that the GIRK1(D173N), which retains F137, did not exhibit steep inward rectification (Kofuji et al., 1996a). Taken together, these results indicate that inward rectification of KG channels containing GIRK1 might be determined primarily by the Mg2+/polyamine block, although the intrinsic gating arisen from F137 might mainly serve to modulate the relaxation kinetics.
Doupnik et al. (1995b) and Kofuji et al. (1996a)conducted the envelope test for the KG channel current in Xenopus oocytes expressing GIRK1. In these experiments, the activation kinetics of the current atEK −80 mV were examined after varying durations of depolarizing prepulses. As the duration of the prepulse was elongated, a slow relaxation component of the inward tail became progressively larger until it occupied ∼50% of the total inward current with τ of ∼200 msec, whereas the faster component(s) reciprocally decreased (Doupnik et al., 1995b;Kofuji et al., 1996a). In contrast, the outward current during the prepulse was promptly suppressed by more than 90% within the initial 50 msec, and only the remaining fraction was slowly decreased (Kofuji et al., 1996a). These results suggest that at depolarized potentials, the channel currents might be first strongly and promptly suppressed by Mg2+/polyamine, and then further slightly depressed by an intrinsic gating gradually substituting for part of the Mg2+/polyamine block. Upon repolarization, the component which had been blocked by Mg2+/polyamine might be relieved first, followed by reactivation of the component affected by intrinsic gating. Such an interactive mechanism might remind one of the interaction between Mg2+i and polyamines on strong inward rectifiers (Ishihara et al., 1989). Although the latter phenomenon can be explained in terms of interaction for the common binding sites shared by Mg2+i and polyamines in IRK subunits (Yamashita et al., 1996), it is not known what molecular mechanism underlies the interaction between intrinsic gating and Mg2+/polyamine block.
There are also some other unsettled issues regarding the molecular mechanism of the slow relaxation. First, neither homomeric GIRK4(S143F) nor heteromeric GIRK1(F137S)/GIRK3(S114F) channels exhibit the slow relaxation (Chan et al., 1996; Wischmeyer et al., 1997), indicating that interaction of F137 with some other part(s) of the same GIRK1 subunit is prerequisite for the slow relaxation. Furthermore, Wischmeyer et al. (1997) found that GIRK1/GIRK1(F137S) “homomeric” KG channels showed the fast relaxation kinetics. Thus, it may be that a GIRK1-containing KG channel can exhibit the slow relaxation only when such intramolecular interaction occurs in all the GIRK1 subunits included in the channel. These results indicate that we are still not aware of all the molecular mechanisms underlying the slow relaxation of GIRK1-containing KG channels. Second, the kinetics of the slow relaxation may vary depending on the subunit composition of KG channels containing GIRK1. The slow relaxation of GIRK1/GIRK4 channels is much faster than that of the KG channel yielded with expression of GIRK1 alone in Xenopus oocytes (Iizuka et al., 1995). GIRK1/GIRK2 channels may have even faster relaxation kinetics than GIRK1/GIRK3 or GIRK1/GIRK4 channels (Wischmeyer et al., 1997). It is not yet known how GIRK subunits other than GIRK1 regulate the slow relaxation. Finally, the relaxation kinetics of the KACh channel become faster in the presence of higher ACh concentrations (figs. 13B and C). Such a receptor- (or GK-) dependent change in the slow relaxation kinetics cannot be reproduced in oocytes expressing either GIRK1 alone (Doupnik et al., 1995b) or GIRK1 plus GIRK4 (Iizuka et al., 1995). The agonist- (or GK-) dependent regulation of the kinetics might, therefore, require some unidentified factor(s) existing in cardiac myocytes but not in oocytes. Further studies are necessary to fully elucidate the molecular mechanism underlying the slow relaxation of native KG channels containing GIRK1.
V. Pharmacological Properties of G Protein-Gated K+Channels
The pharmacological properties of KGchannels including the KACh channel have not been extensively investigated. However, it is well known that like other Kir channels (Hagiwara and Takahashi, 1974; Hagiwara et al., 1976, 1978; Gay and Stanfield, 1977; Standen and Stanfield, 1978;Constanti and Galvan, 1983; Harvey and Ten Eick, 1989), cesium and barium ions effectively block various native KGchannels (Carmeliet and Mubagwa, 1986; Gähwiler and Brown, 1985;Inoue et al., 1988; Mihara et al., 1987;Surprenant and North, 1988; Pennefather et al., 1988;Penington et al., 1993; Lacey et al., 1987 and1988; Gerber et al., 1991; Dousmanis and Pennefather, 1992;Sodickson and Bean, 1996). These cations, when applied extracellularly, block KG channels in a steeply voltage-dependent manner with higher potency at more negative membrane potentials (but see Sodickson and Bean, 1996). Thus, the cations seem to go through the transmembrane electric field before binding to their receptor sites in the channel pore. Depending on experimental protocols and cell type, the reported Kd values of these cations for KG channels vary but largely fall into a concentration range of 10-3-10-2m for cesium ions and 10−5to 10−4m for barium ions at negative membrane potentials. These values are ∼10 times more than those required to block the classical Kir channels (Hagiwara and Takahashi, 1974;Hagiwara et al., 1976, 1978; Gay and Stanfield, 1977;Standen and Stanfield, 1978; Constanti and Galvan, 1983; Harvey and Ten Eick, 1989). Similar values were reported for the cesium or barium blockade of the recombinant KG channels (Velimirovic et al., 1996; Lesage et al., 1995;Bond et al., 1995). Although barium ions are known to block the classical Kir channel in a clear time-dependent manner (Hagiwaraet al., 1978; Constanti and Galvan, 1983), some KG channels were reported to be instantaneously inhibited by the ions (Carmeliet and Mubagwa, 1986; Dousmanis and Pennefather, 1992, but see Velimirovic et al., 1996). KG channels are also known to be blocked by millimolar concentrations of rubidium ions (Katayama et al., 1997; Mihara et al., 1987; Surprenant and North, 1988).
Recent studies indicated that cesium, rubidium, and strontium ions block a recombinant strong inward rectifier IRK1 channel at least in part through binding to a negatively charged aspartate at the R1 position in the putative channel pore (D172) (Reuveny et al., 1996; Abrams et al., 1996). As we have already seen in Section IV.A.3., this acidic residue is conserved among various constitutively active Kir channels and may serve as a receptor site for the intracellular Mg2+ and polyamines as well (fig. 12) (Stanfield et al., 1994; Taglialatela et al., 1994; Lu and MacKinnon, 1994; Wible et al., 1994;Lopatin et al., 1994; Ficker et al., 1994; Yanget al., 1995a). GIRK1 but not GIRKs2–4 bear this residue. This may at least in part explain the lower sensitivity of KG channels to these cation blockers than classical Kir channels.
KG channels are also fully blocked by quinidine and quinine with the IC50 of 10 μm(Kurachi et al., 1987a; Nakajima et al., 1989;Katayama et al., 1997). Verapamil also inhibits the ACh-activated cardiac KACh channel with the IC50 of 1 μm although its effect is partially mediated by suppression of the M2-muscarinic receptor/GKsystem (Ito et al., 1989). To our knowledge, KG channels are rather insensitive to other well-known channel blockers such as tetraethylammonium, 4-aminopyridine, apamine, charybdotoxin, disopyramide, and procainamide (Inoue et al., 1988; Lacey et al., 1987 and 1988;Nakajima et al., 1989; Katayama et al., 1997).
VI. Localization of the G Protein-Gated K+ Channel in Different Organs
GKβγ seems to be the physiologically functional arm of GK activating KG channels not only in the heart but also in the brain and endocrine organs. However, the molecular mechanisms of G protein-regulation of ion channels have been found to be more complicated than we had thought. In AtT20 cells which had been transfected with the α2A-adrenergic receptor, adrenergic agonists can inhibit the Ca2+ current and adenylyl cyclase and activate a K+ current (Surprenant et al., 1992). A point mutation of the receptor removes activation of the K+ current, but not inhibition of Ca2+ current and adenylyl cyclase. This indicates that the G protein coupling to the K+ channel is different from that to the Ca2+ channel and adenylyl cyclase although the receptor is the same. G proteins may thus be more specific to each receptor and to each signaling system than we are currently assuming or than we can determine in vitro. In Xenopus oocytes, however, when β2-adrenergic receptors, Gs protein, and GIRK1 are coexpressed, β-adrenergic agonists could induce activation of KG channel current (Lim et al., 1995). Accordingly, the affinity of particular G protein subunits for the KG channel may not be sufficient to explain specific activation of KACh channel by GK. Actually, various combinations of recombinant Gβγ (except for Gβ1γ1) have similar efficacy and potency in activating KACh channels (Wickmanet al., 1994). However, the receptor specificity in cardiac atrial myocytes is well documented by extensive studies (Kurachi, 1995). It is often argued that receptor specificity could arise from compartmentalization of the appropriate receptors and channels, although little evidence has been shown for such compartmentalization. We do not know whether different mechanisms underlie receptor specificity in different organs. In other words, we have not yet fully answered the question how information specifically passes from a membrane receptor to the effector, the KGchannel, via G proteins.
Because the signal transduction mechanisms are not necessarily the same among heart, neurons, and endocrine cells, it is worthwhile at present to summarize the observations on the localization of KG channels in these organs. Apparently, GIRK1 and GIRK4 immunoreactivities diffusely distribute in the cell membrane of cardiac myocytes, whereas those of GIRK1 and/or GIRK2 are localized to specialized segments of neuronal membrane, such as presynaptic axonal termini and postsynaptic dendritic regions (fig.15). Thus, we may tentatively classify the system based on the apparent distribution into two categories: (a) homogeneously distributed system and (b) localized system. In these systems, different mechanisms may underlie signal transduction.
A. Cardiac Atrial Myocytes
The cardiac KACh channel is the prototype of KG channels and is a heteromultimer of GIRK1 and GIRK4 (Krapivinsky et al., 1995a). Immunohistochemistry using a specific antibody showed that GIRK1 is homogeneously localized on the cell membrane of atrial but not ventricular myocytes (fig. 15A). This is consistent with the electrophysiological studies of cardiac myocytes. The electrophysiological experiments also suggested that some topological restriction may exist in cardiac atrial myocytes, because KACh channels in the cell-attached membrane patch are activated by ACh or adenosine when they are applied to the pipette solution, but not when they are added to the bathing solution (Soejima and Noma, 1984).
It was found that either Gi- or Gs-coupled receptors, when expressed together with GIRK channels in Xenopus oocytes, can activate the channels (Lim et al., 1995). This may indicate that under the conditions where compartmentalization does not exist, as inXenopus oocytes, the βγ subunits released from Gs may be able to activate KG channels. Because β-adrenergic agonists never activate these channels in cardiac myocytes, there must be some mechanism to guarantee the specificity of the native GK/KACh channel system in cardiac atrial myocytes. Further studies are needed to elucidate the mechanism.
B. Neurons
1. Differential cellular and subcellular distribution of GIRK subunits.
Cellular and subcellular localization of GIRK1, 2, and 4 subunits in the rat and mouse brains has been studied with specific antibodies against individual GIRK proteins (Ponce et al., 1996; Liao et al., 1996; Iizuka et al., 1997). No corresponding studies have been done on GIRK3 proteins to our knowledge. The followings are the summary of currently available information.
Liao et al. (1996) showed that antibodies against GIRK1 or GIRK2 proteins immunocoprecipitated both GIRK1 and GIRK2 proteins from membranes of the cerebral cortex, indicating the presence of GIRK1/GIRK2 heteromultimers in this region. In the mice whose GIRK2 genes were genetically deleted (GIRK2 -/-), virtually all GIRK1 and GIRK2 proteins disappeared from the cerebral cortex, indicating that most of GIRK1 proteins are assembled with GIRK2 subunits (Signoriniet al., 1997). GIRK1 and GIRK2 immunoreactivities are found in the somata and apical dendrites of pyramidal cells in the somatosensory cortex (Ponce et al., 1996; Liao et al., 1996). The GIRK1 immunostaining is stronger in the dendrites than in the somata (Ponce et al., 1996), whereas strong GIRK2 immunoreactivity is found both in the cell body and apical dendrite of layer V pyramidal neurons (Liao et al., 1996). GIRK4 immunoreactivity is also exist in the cerebral cortex (Iizukaet al., 1997).
In the layer IV of the neocortex, fiber-like immunostainings of GIRK1, but not GIRK2, protein can be seen in addition to the staining of pyramidal neurons (Ponce et al., 1996; Liao et al., 1996). Ponce et al. (1996) argued that these stainings correspond to those of the terminals of thalamocortical projections, the axons of thalamic relay neurons, because the stainings markedly reduced on the side ipsilateral but not contralateral to thalamus lesioned with kainic acid. Consistent with this, neurons in most thalamic nuclei express GIRK1 mRNA strongly (Karschin et al., 1994 and 1996), and the somata of relay neurons and neuropiles in thalamus are stained with anti-GIRK1 antibodies (Ponceet al., 1996). The presence of GIRK subunits in presynaptic compartments is also suggested in other regions. Morishige et al. (1996) found that in the paraventricular nucleus of the hypothalamus, GIRK1 immunoreactivity seems as “beads on a string,” a typical staining pattern of nerve terminals. Electron microscopy immunohistochemistry revealed that the immunoreactivity resides in terminal buttons but not in the dendrites (figs. 15B and C). Liaoet al. (1996) also found that GIRK1 and GIRK2 immunoreactivities exist in axon-like fibers in the lateral septal region.
In the hippocampus, virtually all GIRK1 subunits seem to form heteromultimers with GIRK2 proteins (Liao et al. 1996;Signorini et al., 1997). GIRK1 immunoreactivity is strongest in the stratum lacunosummoleculare and the adjacent stratum radiatum, followed in intensity by the deep portion of the stratum radiatum and the stratum pyramidalis, whereas the stratum oriens is only weakly stained (Ponce et al., 1996; Liao et al., 1996; Drake et al., 1997). Drake et al.(1997) examined the detailed subcellular localization of GIRK1 immunoreactivity in CA1 pyramidal neurons with an electron microscope. They found that the immunoreactivity almost exclusively exists in perisynaptic areas of the fine dendrites and dendritic spines of pyramidal neurons locating in the stratum lacunosum-moleculare and the stratum radiatum (Drake et al., 1997). In the dentate gyrus, immunostaining of GIRK1 is strong in the molecular layer, moderate in the granular layer, and sparse in the polymorphic layer (Ponce et al., 1996; Liao et al., 1996; Drake et al., 1997). An overall immunostaining pattern of GIRK2 in the hippocampus is similar to that of GIRK1 (Liao et al. 1996).
Strong immunoreactivities of GIRK1 and GIRK2 but not GIRK4 are found in the cerebellar granule cell layer (Ponce et al., 1996; Liaoet al. 1996; Iizuka et al., 1997). The immunoreactivities are largely confined to the glomeruli where granule cell dendrites form synapses with mossy fiber terminals (Ponce et al., 1996; Liao et al. 1996). Electron microscopy immunohistochemistry revealed strong immunostaining of GIRK1 protein on distal dendrites of granule cells surrounding unstained mossy fiber terminals (Ponce et al., 1996). Purkinje cells have weak or no GIRK1 and GIRK2 immunoreactivities but a certain level of GIRK4 immunoreactivity throughout the soma (Ponce et al., 1996;Liao et al. 1996; Iizuka et al., 1997). Deep cerebellar nuclei exhibit distinct immunostaining of GIRK1 and GIRK2 proteins in a clear somatodendritic pattern (Ponce et al., 1996; Liao et al. 1996). Axons of basket cells surrounding the proximal segment of Purkinje cell axons are likely to be relatively rich in GIRK4 immunoreactivity (Iizuka et al., 1997). Although GIRK1 and GIRK2 proteins are coimmunoprecipitated from cerebellar membranes (Liao et al., 1996), a significant amount of GIRK1 proteins remains in the cerebellum of the GIRK2-/- mice (Signorini et al., 1997), indicating that GIRK2 is not the sole partner of GIRK1 in the cerebellum. This observation is consistent with the fact that GIRK2 mRNA is expressed only by granule cells in the cerebellum, whereas significant amounts of GIRK1 transcripts are found in the other regions of the cerebellum as well (DePaoli et al., 1994; Karschin et al., 1994 and 1996).
Substantia nigra and the ventral tegmental area are unique in that abundant GIRK2 subunits exist although little or no GIRK1 protein is expressed (DePaoli et al., 1994; Ponce et al., 1996; Liao et al., 1996). GIRK4 is expressed in these regions at the mRNA level but not at the protein level (Iizuka et al., 1997). GIRK2-specific immunoreactivities are found in both the cell body and dendrites of dopaminergic neurons in the pars compacta and in the dendrites in the pars reticulata (Liao et al., 1996; Yoshimoto et al., 1997).
Immunostaining of GIRK1 proteins in the brain stem exhibits a clear somatodendritic pattern in several nuclei including the superior olive, the nucleus of the trapezoid body, the reticular formation and the central nuclei of the inferior colliculus (Ponce et al., 1996). GIRK2 immunoreactivities are also found in these neurons basically in a somatodendritic pattern (Liao et al., 1996). Immunoreactivity of GIRK4 is also found in the neuronal fiber plexus in the inferior olivary, pontine, and facial nuclei (Iizuka et al., 1997).
These immunohistochemical analyses delineate a great deal of complexity of neuronal KG channel systems in the brain. To summarize, GIRK1, 2, and 4 subunits exist mostly in the somatodendritic subcellular compartment and sometimes in the axon-like fibers, although some neurons may have these proteins in both the compartments.
2. Functional significance of differential subcellular distribution of GIRK subunits.
Differential subcellular distribution of GIRK subunits in neurons would be intimately correlated with the functional task of KG channels in each neuron. For example, in the CA3 hippocampal pyramidal neurons, a selective GABAB agonist baclofen causes both presynaptic and postsynaptic inhibition in the wild-type mice but only presynaptic inhibition in GIRK2 -/-mice (Lüscher et al., 1997). These results indicate that GIRK1/GIRK2 channels in these neurons are selectively activated by the postsynaptic GABABreceptor because of the postsynaptic localization of the GIRK subunits in these neurons. In hippocampal CA1 pyramidal neurons, Drake et al. (1997) found that GIRK1 immunoreactivity is found mainly associated with the dendritic membranes in the vicinity of the asymmetric (stimulatory) but not symmetric (inhibitory) type of synapses. These observations raise the possibility that the GIRK1-containing KG channels in these neurons might serve to modulate the propagation of neuronal inputs originated at the excitatory synapses to the soma, as well as the back propagation of the action potential from the soma to the synapses on a synapse-to-synapse basis. To more concretely identify the functional roles of KG channels in these neurons, it is important to determine the type(s) of neurotransmitter receptors, G proteins and nerve terminals associated with these channel subunits.
It is surprising that GIRK subunits also exist in the axon of some neurons (Ponce et al., 1996; Morishige et al., 1996; Liao et al., 1996). In fact, many G protein-coupled receptors inhibit release of neurotransmitter presynaptically (Nicoll, 1988; North, 1989; Thompson et al., 1993; Wu and Saggau, 1997). In most cases, however, the presynaptic inhibition has been ascribed to inhibition of presynaptic Ca2+channels and/or direct inhibition of exocytotic machineries (Scanzianiet al., 1992; Thompson et al., 1993; Dittman and Regehr, 1996; Takahashi et al., 1996; Wu and Saggau, 1997). The functional role of the presynaptic KGchannels needs to be clarified in future studies.
C. Endocrine Cells
Electrophysiological studies indicate that KG channels activated by somatostatin and/or dopamine exist in endocrine cells of anterior pituitary lobe (Pennefather et al., 1988; Einhorn and Oxford, 1993). This system would be essential for the inhibitory regulation of hormone secretion. However, there is no information available on subcellular distribution of GIRK subunits in these and other endocrine cells so far.
VII. Weaver Mutant Mice and the GIRK2 Gene
Weaver mice have been studied intensively over the past 25 years for insights into the normal processes of neuronal development and differentiation (Hess, 1996). Homozygous animals (wv/wv mice) suffer from severe ataxia due to death of cerebellar granular cells. The animals also represent a model of Parkinsonism because dopaminergic input to the striatum is lost during the first few weeks after birth due to the death of dopaminergic neurons in the substantia nigra. Male wv/wv mice are sterile: spermatogenesis fails to proceed normally past the third postnatal week leading to a complete failure of sperm production.
Recently, it was shown that wv/wv mice have their neurological abnormalities because of a point mutation of guanine 953 to adenine in the GIRK2 gene (Patil et al., 1995). This mutation causes a change of the corresponding amino acid from glycine (G) at position 156 to serine (S), which is in the ion selectivity filter of the potassium channel in the H5 region. In this way, the “finger print” K+ channel sequence of glycine-tyrosine-glycine (G-Y-G) is altered to serine-tyrosine-glycine (S-Y-G) in GIRK2 with the weaver mutation (GIRK2wv). This results in a striking change in the selectivity of homomultimeric GIRK2 channels. Wild-type GIRK2 channels are highly selective for K+ ions with the permeability ratio PNa/PK of <0.05, and are virtually impermeable to Cs+ (Lesage et al., 1994; Slesinger et al., 1996; Kofuji et al., 1996b). However, homomultimeric GIRK2 wv channels allow K+, Na+, Rb+, and Cs+ to permeate with PNa/PK of 0.5 to 0.95, PRb/PK of ∼0.8 and PCs/PK of 0.9 to 1.0, but are still impermeable to Ca2+, N-methyl-d-gulcamine, and anions (Slesingeret al., 1996; Kofuji et al., 1996b; Navarroet al., 1996). Thus, the weaver mutation renders GIRK2 channels nearly nonselective among monovalent cations.
The weaver mutation also has another unexpected effect on GIRK2 channels. Compared with wild-type GIRK2 channels, GIRK2wv-channels have a large G protein-independent basal current and a small G protein-induced increase in a current amplitude (Slesinger et al., 1996; Kofuji et al., 1996b;Navarro et al., 1996). This may be because the pore mutation impairs the gating which is crucial for the regulation of channel activity in response to G protein activity. However, Slesinger et al. (1996) found that as a larger amount of GIRK2wvcRNA was injected into Xenopus oocytes, expressed GIRK2wv-channels exhibited a larger basal current and a smaller response to G protein stimulation. Thus, it is also possible that the Na+ influx through GIRK2wv-channels increases the intracellular Na+ concentration, which in turn directly activates GIRK2wv-channels and occludes the channels’ response to G protein stimulation (Lesage et al., 1995; Suiet al., 1996).
As with the wild-type GIRK2 subunits, GIRK2wv subunits form a heteromultimeric channels with GIRK1 (Slesinger et al., 1996; Liao et al., 1996). However, coexpression of GIRK1 and GIRK2wv in Xenopus oocytes yields current amplitudes not larger than the sum of those obtained with either of the subunits alone. Liao et al. (1996) found that in thewv/wv brain, the amounts of both GIRK1 and GIRK2 proteins were reduced although the amount of the unglycosylated form of GIRK1 proteins increased compared with the control. Thus, some fraction of the GIRK1/GIRK2wv complex might be retained in the endoplasmic reticulum and not be expressed to the membrane. Furthermore, GIRK1/GIRK2wv channels also lack K+ selectivity and have an impaired response to G protein stimulation (Slesinger et al., 1996; Kofuji et al., 1996b; Navarro et al., 1996; Liao et al., 1996). All these in vitro studies indicate that the effect of the weaver mutation can be pleiotropic depending on the expression level of GIRK2wv subunits and the presence of other types of GIRK subunits coexisting with GIRK2wvsubunits.
During the early postnatal development of the mouse cerebellum, the external granule cell layer (EGL) consists of a mitotically active outer layer and a later developing inner postmitoic layer (Goldowitz and Smeyne, 1995). As development proceeds, the postmitotic granule cells migrate out of the EGL to the internal granule cell layer (IGL). In the wv/wv cerebellum, the granule cells undergo an apoptotic process before the migration. GIRK2 mRNA can be already detected in granule cell precursors in the prenatal mouse cerebellum (Kofuji et al., 1996b). Both GIRK1 and GIRK2 immunoreactivities are found in the mouse cerebellum by the postnatal day (PND) 3 and in the EGL and the newly forming IGL on PND4 (Slesingeret al., 1996). On PND19 and PND27, an anti-GIRK2 and anti-GIRK1 antibodies give a nearly uniform staining in the cerebellum of wv/wv mice compared with the discrete staining pattern in the wild-type littermates (Liao et al., 1996). This abnormal staining pattern of the wv/wvcerebellum corresponds to a loss of granule cells in the mutant mice by these ages. Therefore, the temporal expression pattern of GIRK2 and GIRK1 in the mouse cerebellum consists with the time course of the development of the neuronal deficits in the wv/wvcerebellum.
However, how the malfunction of GIRK2-containing KG channels leads to the death of the cerebellar granule cells has not been unequivocally identified. The simplest explanation is that the basal or neurotransmitter-induced Na+ influx caused by the mutation imposes a heavy metabolic burden on granule cells and thereby causes the premature death or prevention of differentiation of the neurons. Kofuji et al. (1996b) found that charged channel blockers MK-801 and QX-314 and a Ca2+ channel blocker verapamil more potently inhibited GIRK1/GIRK2wv channels than wild-type GIRK1/GIRK2 channels. They also found that these blockers potently inhibited the aberrant, constitutively-active Na+conductance in cultured wv/wv granule cells and promoted the survival and differentiation of the neurons in vitro. These results strongly suggest the causal relationship between the Na+ current caused by the weavermutation and the deterioration of wv/wv granule cells. Slesinger et al. (1996) reported thatXenopus oocytes injected with GIRK2 wv cRNA also died much faster than those injected with the wild-type GIRK2 cRNA and that the survival period of oocytes was shorter when the larger amount of GIRK2 wv cRNA was injected. However, Surmeier et al. (1996) reported that they could not detect such aberrant Na+ currents in their culturedwv/wv granule cells. Instead, they found that somatostatin and a metabotropic glutamate receptor agonist trans-ACPD induced significantly smaller KG channel currents in these cells than in the wild-type granule cells. Accordingly, they speculated as follows. In the postnatal cerebellum, the postmitotic, premigratory granule cells are known to be exposed to elevated levels of extracellular glutamate, which may trigger the migration of granule cells from the EGL. On this occasion, the GIRK2-containing KG channels in the wild-type granule cells may be activated by glutamate and serve to counteract the depolarization caused by stimulation ofN-methyl-d-aspartate glutamate receptors. However, wv/wv granule cells may be continuously depolarized by glutamate and die due to excessive Ca2+ entry because they lack the KG channels. It is difficult to reconcile these different two observations. However, Signorini et al. (1997)found that the morphology of the cerebellum and midbrain dopaminergic neurons and the fertility of GIRK2 -/- mice are different from those of the wv/wv mice and indistinguishable from those of the wild-type (GIRK2 +/+) mice. Thus, the loss of GIRK2-containing KG channels in the wv/wvmice may not be the primary cause of the weaver phenotype. They further argued that the cerebellum of the heterozygouswv/- mice is histologically more similar to wv/+ mice than that of +/+ or wv/wv mice. This observation favors the hypothesis that the gain-of-function and gene dosage mechanisms are responsible for the developmental defects inweaver mutants.
GIRK2 proteins are widely expressed in different regions in the brain as described in the section VI.B. However, only limited regions such as the cerebellar cortex, substantial nigra, and hippocampus are severely affected by the weaver mutation (Hess, 1996). Even within the same regions, damages are not homogeneous. For example, granule cells in the lateral cerebellar hemispheres are more resistant to theweaver mutation than midline neurons. Such inhomogeneity could be accounted for by several factors including the pleiotropic effects of the mutation depending on the expression levels of GIRK2 and the other GIRK subunits (Slesinger et al., 1996) as well as the environmental factors that determine the inherent vulnerability of each neuron (Hess, 1996). Further studies are necessary to answer why some neurons are more susceptible to the weaver mutation.
VIII. Conclusions
Until 1993, the G protein-activation of inwardly rectifying K+ channel systems was mainly studied in cardiac myocytes with electrophysiological techniques. The recent rapid progress in the molecular biology of KG channels has disclosed an unimagined complexity of this channel system. Although many aspects of regulation of the KG channels have been elucidated by the efforts of many laboratories listed in this review, there also have emerged many unclarified but possibly important mechanisms that may underlie the physiological regulation of KG channels in organs that include heart, brain, and endocrine tissues. We cannot yet explain the molecular mechanisms responsible for receptor-specific control of KGchannels. Because the expression of the different GIRK genes and coupling to different receptor subtypes occurs throughout the central nervous system, the role of the KG channel and its regulation by G proteins in neural systems requires more attention than it has received to date. Phenomena described for KACh channels in cardiac atrial myocytes, such as desensitization, deactivation, and cross-talk with other signaling systems have not yet been examined at all in other tissues.
Acknowledgments
We are grateful to Dr. Ian Findlay (University of Tours, Tours, France) for critical reading of this manuscript. This study is supported by grants from the Ministry of Education, Science, Sports and Culture of Japan, “Research for the Future” Program of The Japan Society for the Promotion of Science (96 L0302), and the Human Frontier Science Program (RG0158/1997-B).
Footnotes
-
↵FNa Present address: Mitsuhiko Yamada, Department of Cardiac Physiology, National Cardiovascular Center, Research Institute, 5-7-1 Fujishiro-dai, Suita, Osaka 565-8565 Japan.
-
↵FNb Address for correspondence: Yoshihisa Kurachi, Department of Pharmacology II, Faculty of Medicine, Osaka University, 2-2 Yamada-oka, Suita, Osaka 565-0871, Japan.
- Abbreviations:
- ACh
- acetylcholine
- KACh channel
- muscarinic K+ channel
- IK1
- the background inwardly rectifying K+channel in cardiac myocytes
- PTX
- pertussis toxin
- GTP
- guanosine 5′-triphosphate
- GTPγS
- guanosine 5′-O-(3-thiotriphosphate)
- GK
- the heterotrimeric G protein responsible for the physiological activation of the KACh channel
- Gβγ
- βγ subunits of G protein
- Gα
- α subunits of G protein
- Gα-GDP
- the GDP-bound form of Gα
- GKβγ
- βγ subunits of GK
- GKα
- α subunits of GK
- KG channel
- G protein-gated K+ channel
- Gβ
- β subunits of G protein
- Gγ
- γ subunits of G protein
- Gα-GTP
- the GTP-bound form of Gα
- RGS
- G protein signaling protein
- βARK
- β-adrenergic receptor kinase
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- Vm
- the membrane potential
- EK
- the potassium equilibration potential
- i
- the single-channel current amplitude
- γ
- the single-channel conductance
- Er
- the resting membrane potential of cells
- Mg2+i
- intracellular Mg2+
- Kir channel
- the inwardly rectifying K+ channel
- K+o
- extracellular K+
- [K+o]
- extracellular K+ concentration
- GTPi
- intracellular GTP
- I
- the macroscopic current amplitude
- N
- the number of functional channels
- Po
- the single-channel open probability
- MWC allosteric model
- Monod-Wyman-Changeux’s allosteric model
- AA
- arachidonic acid
- LTC4
- leukotriene C4, Kv channel, the voltage-gated K+ channel
- KATP channel
- the ATP-sensitive K+ channel
- GST
- glutathione S-transferase
- gK
- the macroscopic chord conductance
- G-V relationship
- the relationship between gK and Vm
- gKmax
- the maximum gK
- ΔV
- Vm relative toEK
- ΔVh
- ΔV at which gK is the half maximum
- v
- the slope factor in the Boltzman’s equation
- τ
- a time constant
- IC50
- the half-maximum inhibitory concentration
- Px
- the permeability for ion species x
- EGL
- the external granule cell layer
- IGL
- the internal granule cell layer
- and PND
- the postnatal day
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
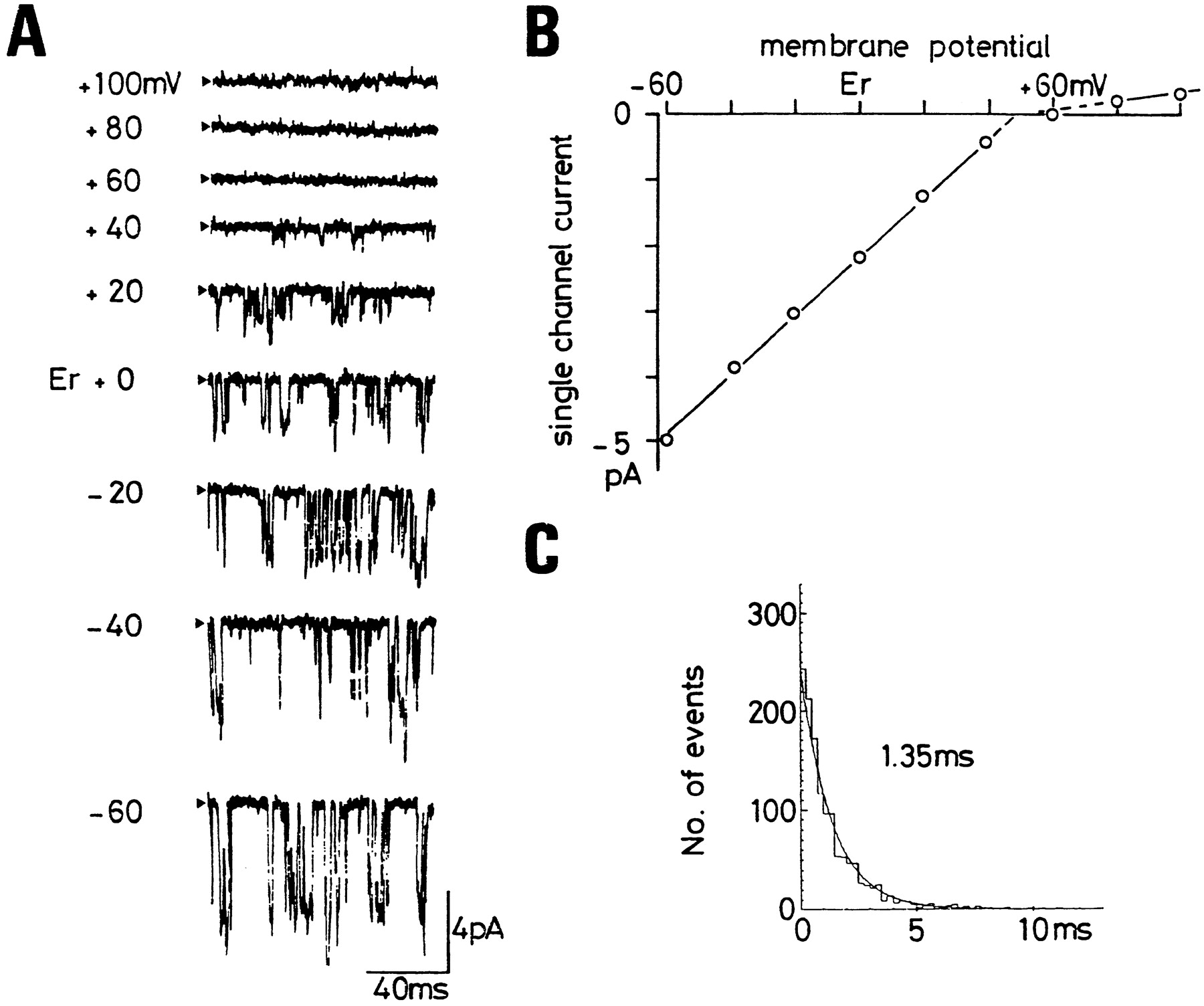{kind=link}
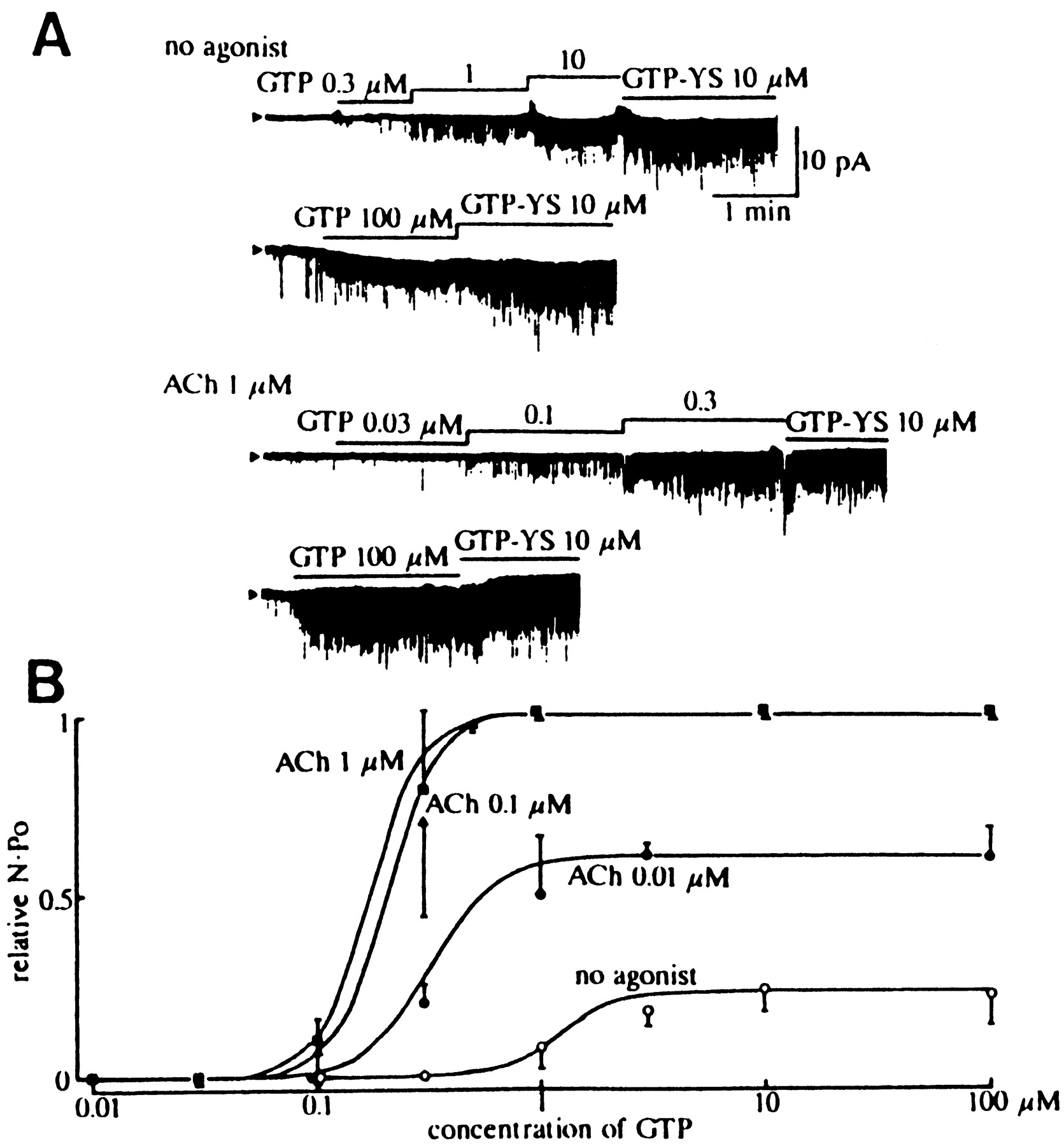{kind=link}
{kind=link}
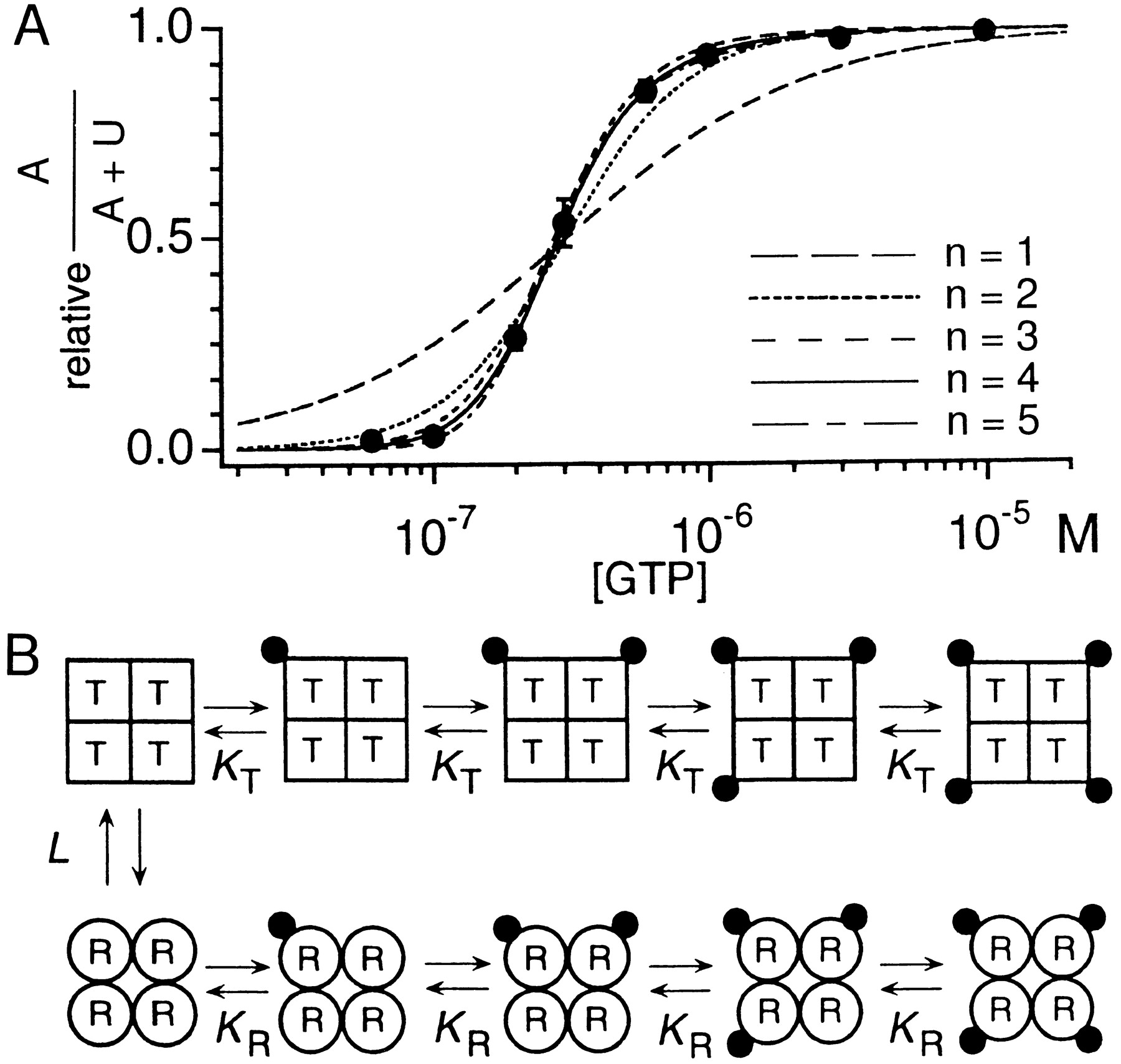{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
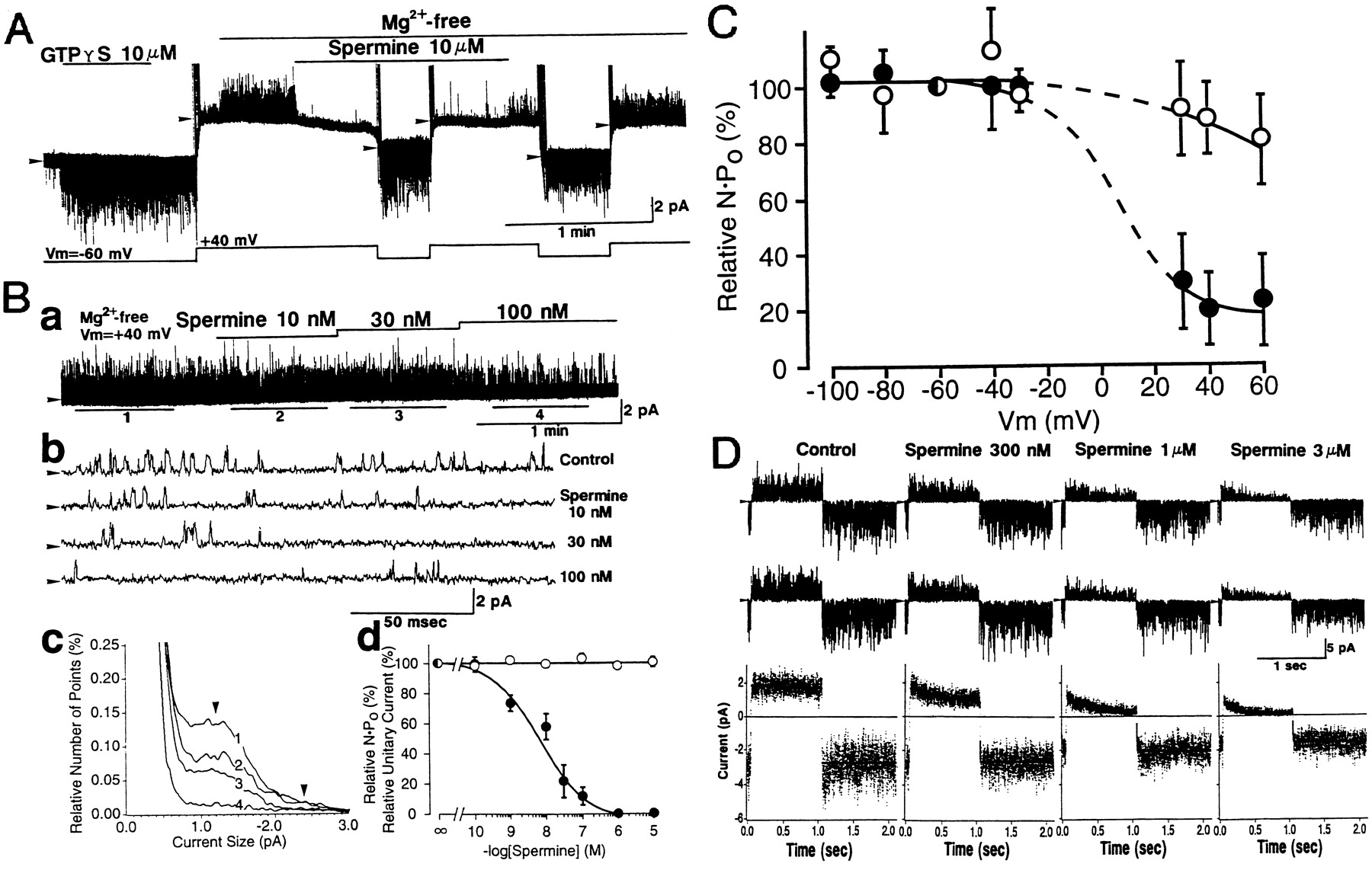{kind=link}
Jump to section
- Article
- I. Introduction
- II. Functional Analysis of G Protein-Mediated Activation of Muscarinic K+ Channels in Cardiac Atrial Myocytes
- III. Molecular Analysis of G Protein-Gated K+ Channels
- IV. Voltage-Dependent Properties of G Protein-Gated K+Channels
- V. Pharmacological Properties of G Protein-Gated K+Channels
- VI. Localization of the G Protein-Gated K+ Channel in Different Organs
- VII. Weaver Mutant Mice and the GIRK2 Gene
- VIII. Conclusions
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters