


Abstract
Sphingosine 1-phosphate (S1P) is a bioactive sphingolipid metabolite involved in many critical cellular processes including proliferation, survival, and migration, as well as angiogenesis and allergic responses. S1P levels inside cells are tightly regulated by the balance between its synthesis by sphingosine kinases and degradation. S1P is interconvertible with ceramide, which is a critical mediator of apoptosis. It has been postulated that the ratio between S1P and ceramide determines cell fate. Activation of sphingosine kinase by a variety of agonists increases intracellular S1P, which in turn can function intracellularly as a second messenger or be secreted out of the cell and act extracellularly by binding to and signaling through S1P receptors in autocrine and/or paracrine manners. Recent studies suggest that this “inside-out” signaling by S1P may play a role in many human diseases, including cancer, atherosclerosis, inflammation, and autoimmune disorders such as multiple sclerosis. In this review we summarize metabolism of S1P, mechanisms of sphingosine kinase activation, and S1P receptors and their downstream signaling pathways and examine relationships to multiple disease processes. In particular, we describe recent preclinical and clinical trials of therapies targeting S1P signaling, including 2-amino-2-propane-1,3-diol hydrochloride (FTY720, fingolimod), S1P receptor agonists, sphingosine kinase inhibitors, and anti-S1P monoclonal antibody.
I. Introduction
Sphingosine 1-phosphate (S1P)1, originally considered to be merely the end metabolite of all sphingolipids, is now under the spotlight with important new roles as a signaling molecule (Spiegel and Milstien, 2003) (Fig. 1). Sphingolipids are structural components of all eukaryotic cell membranes. In the plasma membrane, they are commonly believed to protect the cell surface by forming the mechanically stable and chemically resistant outer leaflet of the lipid bilayer. All sphingolipids contain a sphingoid longchain base (sphingosine) backbone, linked to a fatty acid molecule through an amide bond. S1P is produced from sphingosine (2-amino-4-octadecene-1,3-diol; an aliphatic 18-carbon amino alcohol with an unsaturated hydrocarbon chain), by sphingosine kinases (Fig. 2). The discoveries that S1P regulates cell growth (Zhang et al., 1991; Olivera and Spiegel, 1993) and suppresses apoptosis (Cuvillier et al., 1996) triggered the interests of many researchers to investigate S1P as a bioactive lipid mediator. This interest has led to literally thousands of articles linking S1P to a myriad of essential cellular process besides the aforementioned affects on cell growth and survival, including to name just a few, cytoskeletal rearrangements and cell motility (Wang et al., 1999; Lee et al., 2001; Rosenfeldt et al., 2001; Graeler et al., 2002; Sugimoto et al., 2003), invasion, angiogenesis, and vascular maturation (Lee et al., 1999; Wang et al., 1999; English et al., 2000; Liu et al., 2000b; Garcia et al., 2001), and trafficking of immune cells (Spiegel and Milstien, 2003; Cyster, 2005). One of the reasons that such a simple molecule can play such diverse roles is that it functions not only inside cells (Olivera and Spiegel, 2001; Kohno et al., 2006) but also as a ligand of cell surface receptors after it is secreted into the extracellular millieu (Spiegel and Milstien, 2003) (Fig. 1). Gene deletion studies and reverse pharmacology have provided evidence that many of the biological effects of S1P are mediated via five specific G protein-coupled receptors (GPCRs), now designated S1P1–5 (Fig. 3).
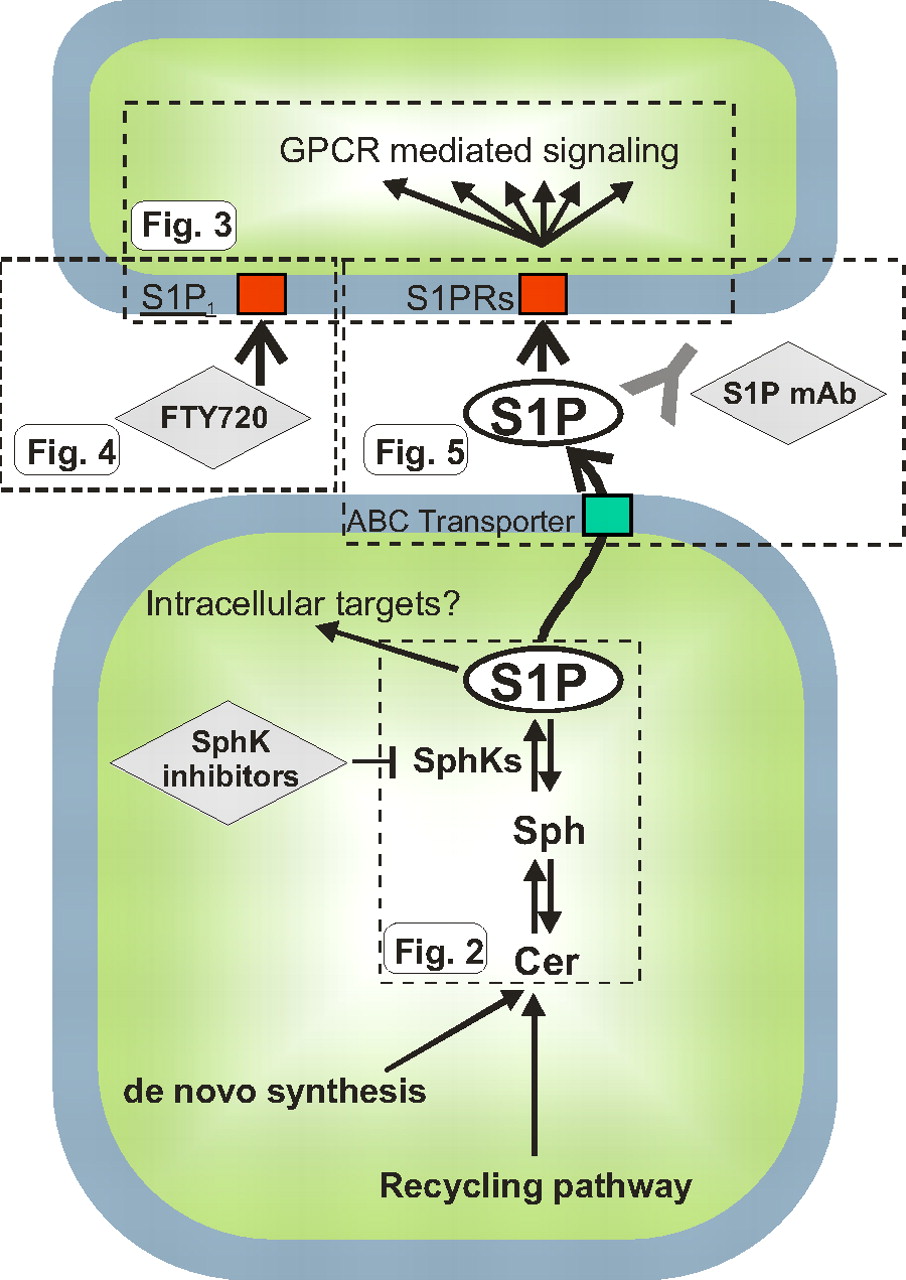
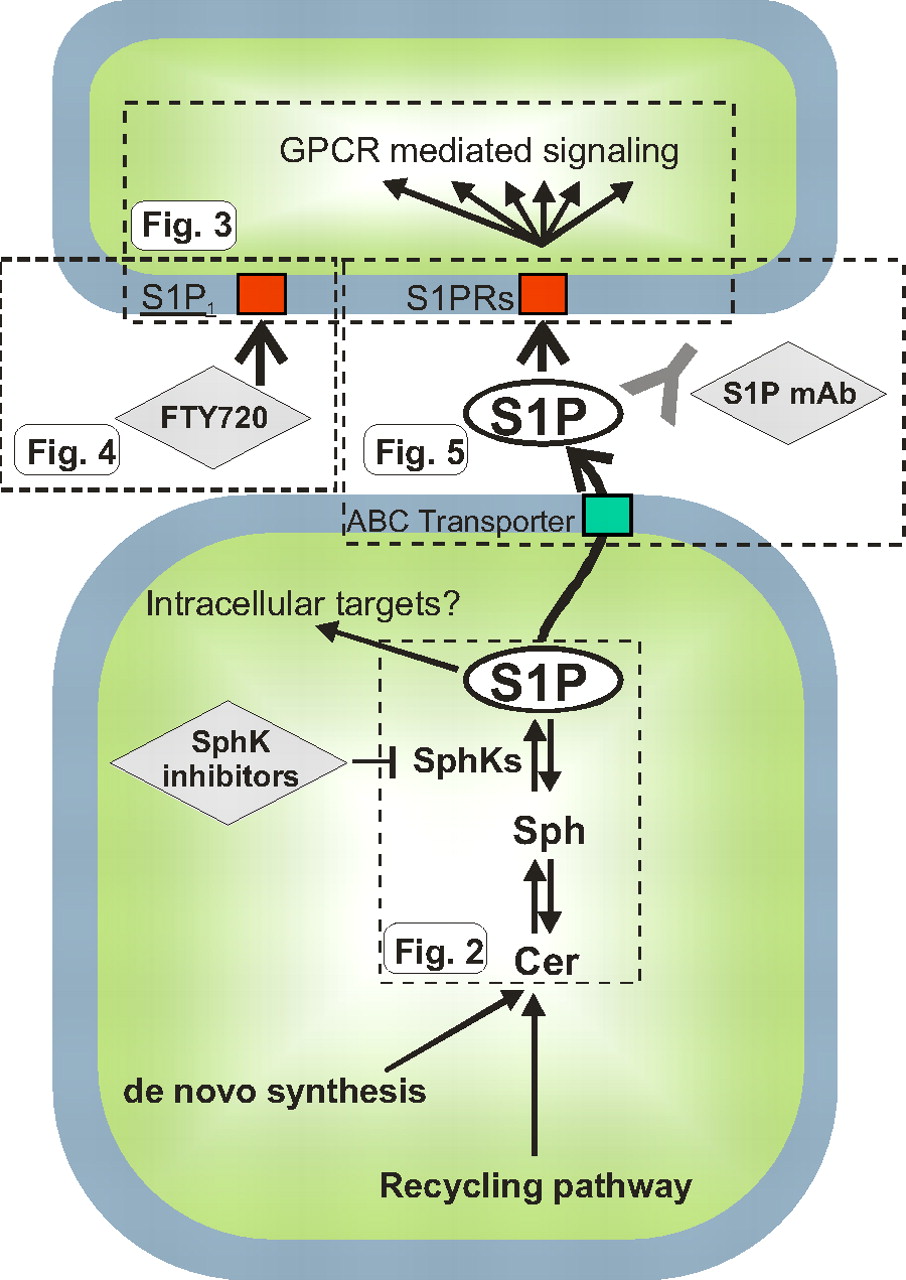
“Inside-out” signaling of S1P. The scheme depicts the metabolism and actions of S1P in broad strokes. S1P is produced by phosphorylation of sphingosine by sphingosine kinases, emerging chemotherapeutic targets. Several lines of evidence suggest that S1P can act intracellularly on as yet unknown targets. S1P can also be exported from cells via ABC transporters and act on cell surface S1P receptors in autocrine or paracrine manners. This extracellular S1P has been targeted by a monoclonal antibody (sphingomab) to block its proliferative and angiogenic effects. In addition, a therapeutic agent directed toward S1P1, FTY720 (fingolimod), is currently being developed for treatment of MS. The flags, labeled Fig. 2, Fig. 3, Fig. 4, and Fig. 5, indicate the portion that is shown in more detail in the respective figures.

Structures and formation of interconvertible bioactive sphingolipid metabolites. The relative concentrations of the bioactive sphingolipid metabolites, S1P, sphingosine, and ceramide represent a rheostat that determines cell fate. S1P is antiapoptotic and progrowth, whereas its precursors, sphingosine and ceramide are proapoptotic and antiproliferative.
Relatively high concentrations of S1P are constitutively present in body fluids and at lower levels in tissues. Increased production of S1P has been linked to various pathological conditions suggesting that it may be a target for therapy for disorders such as cancer, atherosclerosis, and autoimmune diseases such as multiple sclerosis.

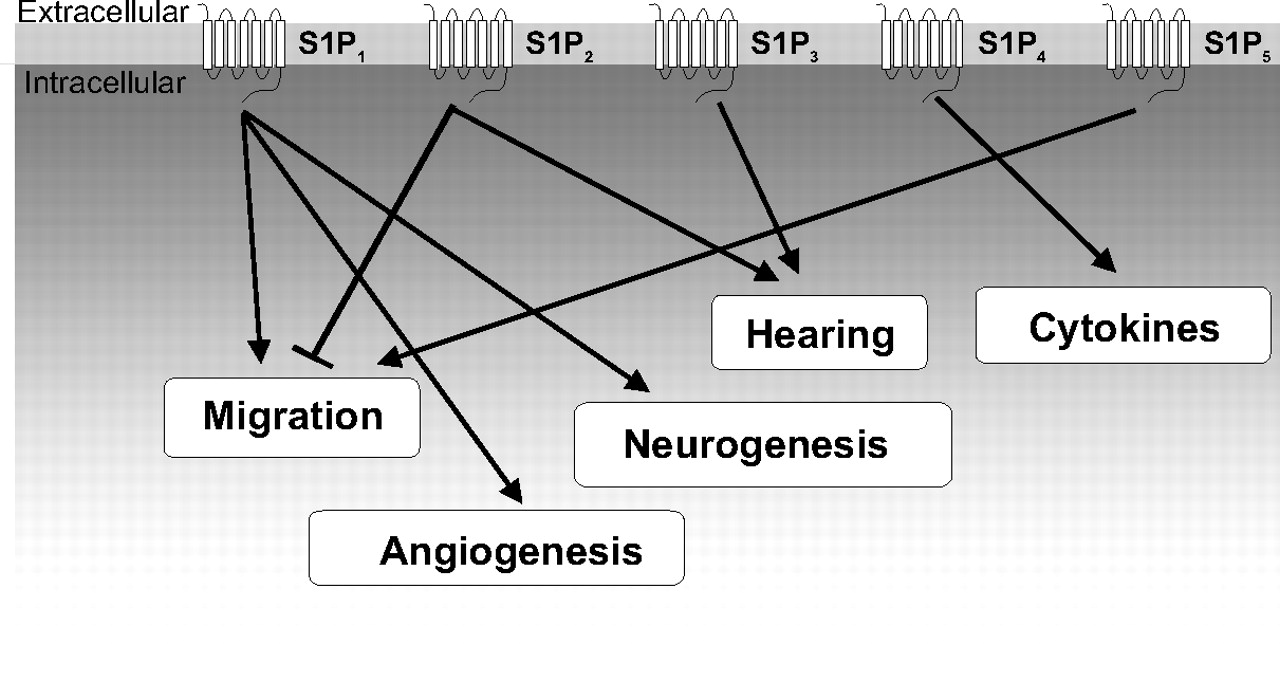
S1P receptors and the major downstream biological processes that they regulate. S1P receptors have been implicated in the regulation of a wide variety of cellular and biological processes including lymphocyte trafficking, cell migration, angiogenesis, neurogenesis, and others.
In this review we summarize current knowledge of S1P synthesis and metabolism, mechanisms of sphingosine kinase activation, and the role of S1P receptors and their downstream signaling. Furthermore, we present updates of recent experimental results and clinical trials with regard to therapeutic agents that target S1P signaling, including FTY720 (fingolimod), S1P receptor agonists, sphingosine kinase inhibitors, and anti-S1P monoclonal antibody.
II. Biosynthesis and Metabolism of Sphingosine 1-Phosphate
Sphingosine, the direct precursor of S1P, is not produced by de novo biosynthesis but only as a consequence of metabolism of sphingolipids and subsequent deacylation of ceramide. Its conversion to S1P requires the enzymatic activity of sphingosine kinases (Fig. 2). There are two pathways that lead to ceramide production: the endocytic sphingolipid recycling pathway and de novo sphingolipid biosynthesis. Sphingomyelinases are responsible for turnover and recycling of sphingomyelin, hydrolyzing it to ceramide and phosphorylcholine. There are three major types of sphingomyelinases characterized by their pH optima as acidic, neutral, and alkaline forms, which accordingly have differential cellular localizations. Acidic sphingomyelinase is localized to the lysosomal compartment (Quintern et al., 1989) and can be secreted to the extracellular space. Neutral sphingomyelinases have various subcellular locations including the plasma membrane, endoplasmic reticulum (ER), Golgi, and nucleus. The localization of alkaline sphingomyelinase is more restricted, and it is mainly expressed in the intestinal tract and the bile where it participates in sphingomyelin digestion in a bile salt-dependent manner (Duan, 2006). The seminal discovery that stress factors and proinflammatory cytokines rapidly increase sphingomyelinase activity and ceramide levels, leading to cell death (reviewed in Obeid et al., 1993) has been confirmed and extended by numerous reports, implicating these events in apoptotic responses to chemotherapeutic agents, ultraviolet and γ-irradiation, and many other cellular stresses (Kolesnick and Hannun, 1999; Hannun and Obeid, 2002). As is the case with sphingomyelinases, there are also acidic, neutral, and alkaline forms of ceramidases, the hydrolases that deacylate ceramide to sphingosine (el Bawab et al., 2002).
The de novo biosynthesis pathway of sphingolipids is initiated at the cytosolic leaflet of the ER, where the enzymes required for ceramide synthesis are located. The initial and rate-limiting reaction is the condensation of l-serine with palmitoyl-CoA to form 3-ketosphinganine, catalyzed by serine palmitoyltransferease (Futerman et al., 1990; Merrill, 2002). 3-Keto-sphinganine is then reduced to dihydrosphingosine by 3-ketodihydrosphingosine reductase and subsequently N-acylated to dihydroceramide by a family of ceramide synthases with different fatty acyl-CoA substrate preferences. Dihydroceramides are then converted to various chain-length ceramides by a desaturase. The de novo formation of ceramide can be induced by several factors including tumor necrosis factor-α, hypoxia, and some chemotherapeutic agents (Huwiler et al., 2000; Ogretmen and Hannun, 2004). Which of these pathways is the dominant one supplying ceramide depends on the cell type, and specific conditions cannot be generalized.
The ceramide transport protein CERT, a cytoplasmic protein with a phosphatidylinositol-4-phosphate-binding domain, transports dihydroceramide and/or ceramide from the ER to the Golgi apparatus by nonvesicular transport (Hanada et al., 2003). Ceramides are converted to sphingomyelins by sphingomyelin synthase on the luminal side of the Golgi or to glucosylceramides (GlcCer) on the cytosolic surface of the Golgi (van Meer and Holthuis, 2000). Two recent studies (D'Angelo et al., 2007; Halter et al., 2007) have implicated FAPP2, a protein known to play a key role in intracellular protein transport, in transport of GlcCer between Golgi compartments (D'Angelo et al., 2007). Consequently, GlcCer is released and translocated into the lumen of the distal Golgi. Protein transport from this compartment to the cell surface is spatially and temporally linked to ongoing GlcCer synthesis and transport. However, it was demonstrated that FAPP2 can also mediate backward transport of GlcCer from the Golgi complex to the ER, where it is processed to complex glycosphingolipids (Halter et al., 2007). Although many details remain unresolved, together these two studies imply an unexpected function for FAPP2 in glycosphingolipid synthesis, and, as a consequence, in protein trafficking.
When sphingosine undergoes ATP-dependent phosphorylation by sphingosine kinases [sphingosine kinase 1 (SphK1) or sphingosine kinase 2 (SphK2)], S1P is produced (Fig. 2). S1P levels in cells are tightly regulated by the balance between its synthesis and degradation, which is the case for many other signaling molecules. S1P can be dephosphorylated back to sphingosine by two specific S1P phosphatases (SPP1 and SPP2), which belong to the family of magnesium-dependent, N-ethylmaleimide-insensitive type 2 lipid phosphate phosphohydrolases that reside in the ER (Le Stunff et al., 2002; Ogawa et al., 2003). S1P can also be irreversibly degraded by a pyridoxal phosphate-dependent S1P lyase to hexadecenal and phosphoethanolamine, with the latter subsequently being reused for the biosynthesis of phosphatidylethanolamine.
Among the sphingolipid metabolites, S1P, sphingosine, and ceramide have drawn considerable attention over the last decade as critical mediators of cell survival and death (Spiegel and Milstien, 2003). Not only do S1P and ceramide exert opposing effects on cell survival but also the fact that these sphingolipid metabolites are interconvertible with each other gave birth to the concept of the sphingolipid biostat (Fig. 2), which postulates that the dynamic ratio between S1P and ceramide determines the fate of the cell (Cuvillier et al., 1996). SphK1 converts sphingosine to S1P, thereby enhancing cell growth and survival, and plays a crucial role in regulation of this balance. Conversely, S1P phosphatases tilt the balance from S1P to sphingosine and ceramide. Whereas ceramide levels increase in response to many stress stimuli including a wide array of chemotherapeutic agents, suppression of apoptosis is associated with increased S1P levels and decreased ceramide (Spiegel and Milstien, 2002). Importantly, inhibition of SphK1 not only decreases S1P levels, it also markedly increases ceramide levels (Spiegel and Milstien, 2002).
III. Mechanisms of Sphingosine Kinase Activation
The two mammalian isoforms of sphingosine kinase, SphK1 and SphK2, have five conserved domains (C1–C5) with a unique catalytic domain contained within C1 to C3 (Alemany et al., 2007). The ATP-binding site [SG-DGX(17–21)K(R)] is present within C2. SphK1 does not possess any hydrophobic transmembrane domains, whereas SphK2 has four predicted transmembrane domains. Although both SphK1 and SphK2 share overall homology and produce the same product, S1P, they display different catalytic properties, subcellular locations, tissue distribution, and temporal expression patterns during development and possibly have unique and specific functions. SphK1 prefers d-erythro-sphingosine as a substrate, whereas SphK2 phosphorylates a wider range of sphingoid base substrates, including phytosphingosine and dihydrosphingosine. In contrast to pro-survival SphK1, both SphK2 overexpression (Liu et al., 2000a; Igarashi et al., 2003) and down-regulation (Van Brocklyn et al., 2005; Sankala et al., 2007) have been reported to result in inhibition of cell growth and induction of apoptosis. Moreover, SphK1 and SphK2 have opposing roles in regulation of ceramide biosynthesis (Maceyka et al., 2005). Despite the fact that either SphK1 or SphK2 single knockout mice develop and reproduce normally, the double knock-out completely eliminates S1P and is embryonically lethal because of severely disturbed neurogenesis, including neural tube closure, and angiogenesis (Mizugishi et al., 2005). This observation suggests that each isoform of SphK may at least partially compensate for the lack of the other, but S1P is critical for neurogenesis and angiogenesis during development.
SphK1 can be stimulated by a wide variety of growth factors including platelet-derived growth factor (PDGF) (Hobson et al., 2001), vascular endothelial growth factor (VEGF) (Shu et al., 2002), epidermal growth factor (EGF) (Sarkar et al., 2005), hepatocyte growth factor (Duan et al., 2004), cytokines (tumor necrosis factor-α) (Xia et al., 2002; Pettus et al., 2003; Pitson et al., 2003), steroid hormone (estradiol) (Sukocheva et al., 2003, 2006), and GPCR ligands (acetylcholine) (van Koppen et al., 2001), lysophosphatidic acid (Delon et al., 2004), S1P itself (Meyer zu Heringdorf et al., 2001), and many other factors (reviewed in Spiegel and Milstien, 2003; Taha et al., 2006a; Alvarez et al., 2007).
There are several mechanisms by which SphK1 can be activated: 1) phosphorylation and translocation to the plasma membrane, 2) interaction with acidic phospholipids, and 3) possible association with other proteins. All or parts of these mechanisms may be required for full activation of SphK1. Translocation to the plasma membrane places SphK1 in the vicinity of its substrate, sphingosine, which mainly resides there. Furthermore, this translocation enables localized production of S1P in the vicinity of its cell surface receptors, which might account for the specificity of its actions through different S1P receptors (Johnson et al., 2002; Pitson et al., 2005). Phosphorylation on Ser225 by ERK2 is essential for enhancing the membrane affinity and plasma membrane selectivity of SphK1 (Stahelin et al., 2005). Recent studies suggest that membrane recruitment of SphK1 also involves protein-lipid interactions. Thr54 and Asn89 in the putative membrane-binding domain of SphK1 appear to be necessary for selective binding of phosphatidylserine and membrane targeting (Stahelin et al., 2005). However, it is important to note that in many cases, the involvement of SphK1 activation and subsequent formation of S1P in biological processes has only been demonstrated indirectly using blunt pharmacological approaches that block both SphK isoforms, such as the pan SphK inhibitors, N,N-dimethylsphingosine (DMS) and dl-threo-dihydrosphingosine (DHS).
Much less is known of the functions and regulation of SphK2. Recent reports indicate that EGF and the protein kinase C activator, phorbol ester, also stimulate SphK2 (Hait et al., 2005, 2007). Both of these activate ERK1, which in turn binds to SphK2 and phosphorylates it on Ser351 and Thr578, increasing its enzymatic activity (Hait et al., 2007). Moreover, others have suggested that phosphorylation of SphK2 is catalyzed by protein kinase D, which leads to its nuclear export for subsequent cellular signaling (Ding et al., 2007). In contrast, cross-linking of the IgE receptor on mast cells was reported to activate both SphK1 and SphK2, leading to their translocation to the plasma membrane (Olivera et al., 2006), and stimulation of both is required for mast cell functions (Olivera et al., 2007).
IV. Sphingosine 1-Phosphate Receptors
Since the first report identifying S1P as the ligand of the orphan GPCR, endothelial differentiation gene 1 (EDG1) (Lee et al., 1998), the number of receptors that bind S1P and dihydro-S1P with high affinity has expanded to five: S1P1/EDG1, S1P2/EDG5, S1P3/EDG3, S1P4/EDG6, and S1P5/EDG8 (Spiegel and Milstien, 2000; Chun et al., 2002). Through these receptors that S1P can regulate many cellular processes, such as cytoskeletal rearrangements and cell motility (Wang et al., 1999; Lee et al., 2001; Rosenfeldt et al., 2001; Graeler et al., 2002; Sugimoto et al., 2003), invasion, angiogenesis, and vascular maturation (Lee et al., 1999; Wang et al., 1999; English et al., 2000; Liu et al., 2000b; Garcia et al., 2001), and lymphocyte trafficking and actions (Schwab and Cyster, 2007) (Fig. 3; Table 1).
S1P receptors and their G protein coupling, signaling pathways, tissue distribution, physiological actions, and agonists/antagonists S1P receptors were originally designated as belonging to the EDG family. The accepted nomenclature is now S1P(subscript 1–5), with the numbers indicating order of discovery and characterization.
S1P receptors are ubiquitously but differentially expressed on all cells. The specific repertoire of S1P receptors that are expressed, together with their differential coupling to various heterotrimeric G-proteins (guanine nucleotide binding proteins) that regulate numerous downstream signaling pathways, is responsible for the ability of S1P to regulate diverse physiological processes in a highly specific manner. “Inside-out” signaling of S1P is crucial for many of these functions, such as migratory responses of fibroblasts toward PDGF (Hobson et al., 2001) and chemotaxis of mast cells toward antigen (Jolly et al., 2004). For example, PDGF induces translocation of SphK1 to the leading edge of cells, promoting localized formation of S1P, which in turn, stimulates S1P1 spatially and temporally in a manner important for the directional movement of cells toward this chemoattractant (Hobson et al., 2001; Rosenfeldt et al., 2001). Interestingly, cross-talk between PDGF and S1P2 can function in the opposite way, counteracting cell movement (Goparaju et al., 2005). Therefore, a delicate balance between transactivation of S1P1 and S1P2 by PDGF is a crucial factor that determines net cell movement toward PDGF.
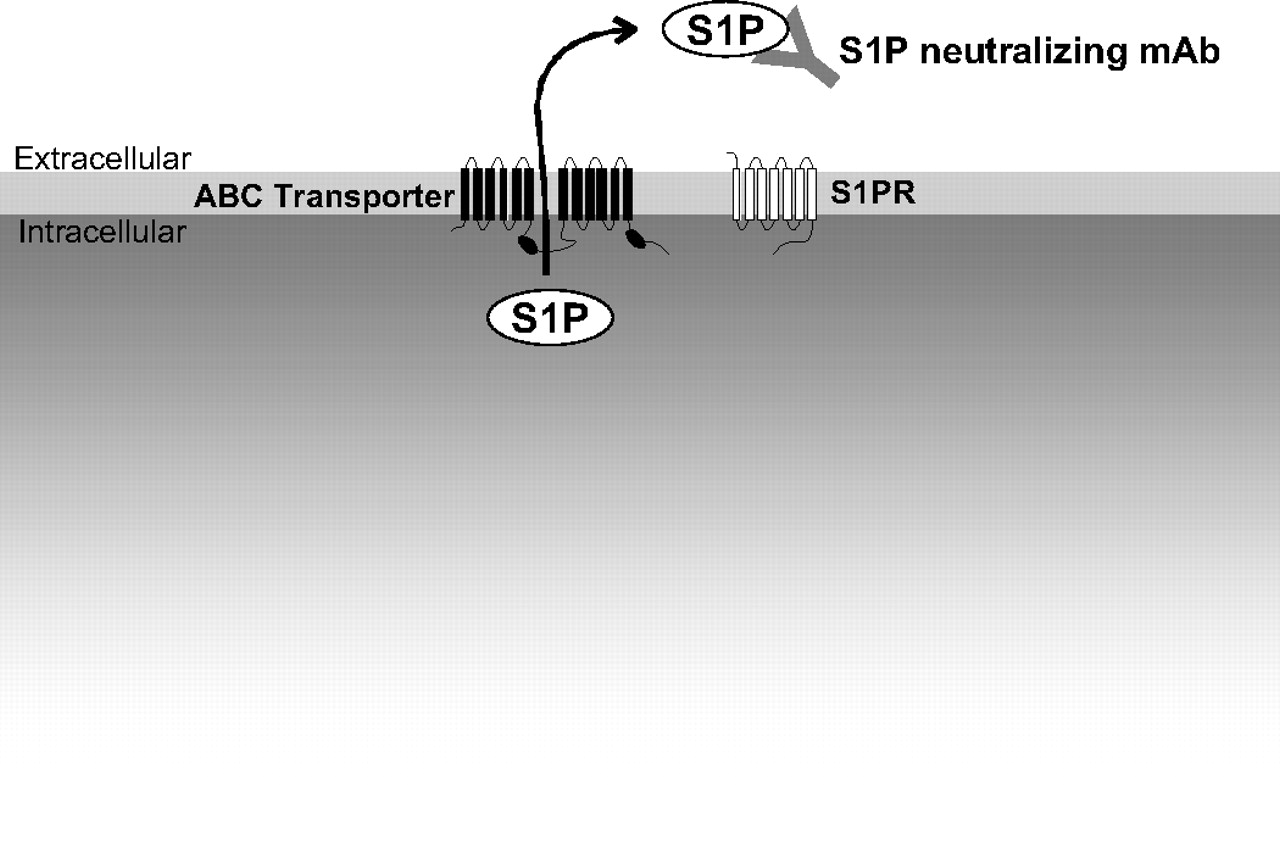
One mechanism for export of S1P from the inside to the outside of mast cells has recently been elucidated (Mitra et al., 2006) (Fig. 4). It was shown that export of S1P from activated mast cells was mediated by a member of the ATP-binding cassette family of transporters, ABCC1, independently of degranulation (Mitra et al., 2006). In agreement, it has recently been shown that thrombin stimulation of S1P secretion from platelets is also mediated by an ABC transporter (Kobayashi et al., 2006; Anada et al., 2007). Moreover, the ABCA1 transporter is critical for release of S1P from astrocytes (Sato et al., 2007). Collectively, these studies suggest that members of the large family of ABC transporters might be important for export of S1P out of cells.

Export of S1P and neutralization with a monoclonal antibody. S1P is produced inside cells by sphingosine kinases and can be exported by ABC transporters. After externalization, a newly developing monoclonal antibody directed toward S1P (sphingomab) can bind and sequester S1P, thereby preventing its interaction with S1P receptors and its angiogenic/proliferative effects.
It has long been known that S1P can also act intracellularly to enhance cell proliferation and suppress apoptosis independently of S1P receptors (Van Brocklyn et al., 1998; Rosenfeldt et al., 2001). However, in contrast to the well-established actions of S1P through its receptors, identification of direct intracellular targets has remained elusive.
S1P1 is ubiquitously expressed and its genetic deletion in mice demonstrated that it has a key role in angiogenesis and vascular maturation (Liu et al., 2000b), as well as in regulation of immune cell trafficking (Matloubian et al., 2004), endothelial barrier function (Singleton et al., 2005), and vascular tone (Sanna et al., 2006). Additionally, binding of S1P to S1P1 can transactivate growth factor receptor tyrosine kinases (RTKs), such as VEGF receptor, EGF receptor, and PDGF receptor, through three not mutually exclusive mechanisms: 1) intracellular receptor cross talk and direct phosphorylation of the RTK by protein tyrosine kinases; 2) induced production and/or secretion of growth factors; and 3) participation of S1P1 and RTK in a signaling complex (signalplex), either by direct receptor/receptor interactions or by binding of both receptors to scaffolding proteins. Conversely, binding of these growth factors to their RTKs can also translocate and activate SphK1 (Pitson et al., 2005), leading to spatially restricted formation of S1P that in turn further activates inside-out signaling (Hobson et al., 2001). Additionally, some of these RTKs, such as PDGF receptor, can also increase S1P1 mRNA expression (Landeen et al., 2007), leading to further complex interplay between S1P receptors and RTKs.
S1P2, like S1P1 and S1P3 receptors, is also widely expressed and is the dominant receptor in the vascular smooth muscle cells (Waeber et al., 2004). Although S1P2-deficient mice are born with no apparent anatomical or physiological defects, the mice develop spontaneous, sporadic, and occasionally lethal seizures between 3 and 7 weeks of age. At the cellular level, loss of S1P2 leads to a large increase in the excitability of neocortical pyramidal neurons, demonstrating that S1P2 plays an essential and functionally important role in the development and/or mediation of neuronal excitability (MacLennan et al., 2001). Furthermore, because S1P2 is essential for proper functioning of the auditory and vestibular systems, its deficiency results in deafness (Herr et al., 2007; Kono et al., 2007).
S1P3 receptor is highly expressed in heart, lung, spleen, kidney, intestine, diaphragm, and certain cartilages, but genetic deletion of S1P3 does not result in an obvious phenotype (Ishii et al., 2001). However, this receptor fine-tunes several pulmonary functions in the adult. In S1P3 receptor-null mice, it was demonstrated that activation of S1P3 in alveolar epithelium through administration of S1P by the airway, but not by the vasculature, induces lung leakage, which is due to increased permeability resulting from tight junction opening and loss of ZO-1, an essential component of the cytoplasmic plaque associated with tight junctions (Gon et al., 2005). Thus, S1P1 and S1P3 have opposing effects on pulmonary epithelial and endothelial barriers.
S1P4 has a more restricted expression pattern and is detectable predominantly within immune compartments and leukocytes (Gräler et al., 1998, 1999). S1P4-deficient mice have not yet been described, but a role for this receptor in regulation of T-cell cytokine production has been proposed (Wang et al., 2005).
S1P5 is expressed primarily in the white matter tracts of the central nervous system with the highest levels in oligodendrocytes, the myelinating cells of the brain (Im et al., 2000; Terai et al., 2003). S1P5-deficient immature oligodendrocytes display reduced responses to S1P in vitro; however, no deficits in myelination were observed in S1P5-deficient mice (Jaillard et al., 2005). Therefore, the precise role of S1P5 in oligodendrocyte function remains to be clarified. Recently, it was found that S1P5 is also expressed in natural killer (NK) cells in mice and humans and that S1P5-deficient mice had aberrant NK cell homing during steady-state conditions and S1P5 was required for the mobilization of NK cells to inflamed organs (Walzer et al., 2007).
V. Sphingosine 1-Phosphate in the Blood
The concentration of S1P in plasma rages from 0.2 to 0.9 μM, where it is tightly associated with albumin and lipoproteins, particularly HDL (Murata et al., 2000), whereas in serum, it varies from 0.4 to 1.1 μM (Caligan et al., 2000; Berdyshev et al., 2005). This provides both a stable reservoir of S1P in extracellular fluids and efficient delivery to S1P receptors. Ample evidence indicates that HDL-bound S1P is biologically active (Nofer et al., 2004; Theilmeier et al., 2006; Matsuo et al., 2007), suggesting that S1P may rapidly associate and dissociate from HDL. By comparison, tissue S1P levels are low, ranging from 0.5 to 75 pmol/mg (Edsall and Spiegel, 1999; Schwab et al., 2005). Therefore, a significant concentration gradient of S1P exists between blood and interstitial fluids.
The source of S1P in blood has only recently begun to be identified. Originally, platelets, which possess highly active SphK1 and lack the lyase that irreversibly degrades S1P, were proposed to be the major source (Yatomi et al., 1997, 2001). Indeed, platelets produce S1P during activation and thrombotic processes (English et al., 2000). Another proposed source of extracellular S1P is secretion of SphK1 from vascular endothelial cells that then can act as an ectokinase to phosphorylate circulating sphingosine (Ancellin et al., 2002). The notion that the platelets are the major contributor to plasma S1P levels has recently been challenged by the observation that transcription factor nuclear factor-E2-deficient mice, which virtually lack circulating platelets, have normal plasma S1P concentrations (Pappu et al., 2007). Furthermore, recent data showed that erythrocytes, which also lack both S1P-degrading enzymes, S1P lyase and S1P phosphohydrolases (Ito et al., 2007), appear to be a major contributor to the storage and supply of plasma S1P (Hänel et al., 2007; Pappu et al., 2007). Studies with conditional SphK1/2-double-knockout mice confirmed that plasma S1P is mainly erythrocytic in origin and also showed that lymph S1P is from a distinct radiation-resistant source, presumably lymphatic endothelium (Pappu et al., 2007).
VI. Sphingosine 1-Phosphate in Human Diseases and Sphingosine 1-Phosphate-Targeted Therapies
As mentioned above, SphK1 generates S1P that regulates proliferation, survival, movement, and invasion of cancer cells. Indeed, fibroblasts overexpressing SphK1 acquire a transformed phenotype and the capability to form tumors in nude mice, leading to the suggestion that Sphk1 may be an oncogene (Xia et al., 2000). Furthermore, SphK1 has been shown to be significantly overexpressed in multiple types of cancers (brain, breast, colon, lung, ovary, stomach, uterus, kidney, rectum, and small intestine) compared with their healthy tissue counterparts (French et al., 2003; Johnson et al., 2005; Van Brocklyn et al., 2005). Enforced expression of SphK1 in MCF7 breast cancer cells, protected against cell death induced by the anthracycline doxorubicin (Nava et al., 2002) and against etoposide-induced apoptosis of HL-60 acute myeloid leukemia cells (Bonhoure et al., 2006). These findings are consistent with results in orthotopic nude mice models in which overexpression of SphK1 in MCF7 cells enhanced estradiol-dependent tumor formation (Nava et al., 2002) and in PC-3 prostate cancer cells which developed larger tumors that were resistant to docetaxel (Pchejetski et al., 2005). These animal studies suggest that SphK1 is a potential candidate therapeutic target for cancer.
There have been mixed results in regard to the effects of S1P on the cardiovascular system. Activation of S1P3 receptor lowers heart rate under most but not all conditions (Alewijnse et al., 2004; Czyborra et al., 2006) and predominantly causes vasoconstriction with some reported evidence of vasodilation (Hemmings, 2006). S1P also can cause coronary vasoconstriction because of coronary artery smooth muscle cell contraction via S1P2 (Sugiyama et al., 2000; Ohmori et al., 2003; Hemmings, 2006). On the other hand, exogenous S1P reduces mortality of hypoxic cardiac myocytes and is cardioprotective in mouse and rat models of cardiac ischemia/reperfusion injury (Karliner, 2004). Moreover, genetic deletion of SphK1 sensitizes the myocardium to ischemia/reperfusion injury and appears to impair the protective effect of ischemic preconditioning, suggesting that the SphK1-S1P axis plays an important role in protection against ischemia/reperfusion injury (Jin et al., 2007).
S1P also plays an important role in allergic reactions in the respiratory system. S1P levels are increased in bronchoalveolar lavage fluid of asthmatics after antigen challenge and correlate with increased eosinophils (Ammit et al., 2001). S1P is known to promote endothelial adherence junction assembly (Lee et al., 1999), which is critical for endothelial barrier maintenance to avoid hyperpermeability resulting in pulmonary edema (Bhattacharya, 2004). Indeed, administration of a new selective S1P1 competitive antagonist (3-amino-4-(3-hexylphenylamino)-4-oxobutylphosphonic acid) to mice induced disruption of barrier integrity in pulmonary endothelium (Sanna et al., 2006; Rosen et al., 2007). Intriguingly, a single intravenous injection of S1P showed protective effects against lung injury caused by high-volume mechanical ventilation and intratracheal endotoxin instillation in an animal model (McVerry and Garcia, 2004) that was attributed to expression of S1P1 by endothelial cells (Rosen and Goetzl, 2005).
Multiple sclerosis (MS) is the most common nontraumatic cause of neurological disability in young adults. It is generally believed to be an autoimmune disorder in which autoreactive T-cells migrate across the blood-brain barrier and attack myelin sheaths, leading to demyelination and axonal damage. Most patients have a relapse after varying lengths of symptom-free periods with worsening of symptoms that usually last several weeks and may or may not resolve completely. The therapeutic goal of currently approved immunomodulating treatments for MS (interferon-β and glatiramer acetate) is to reduce the frequency, severity, and duration of these relapses. However, these medications afford only modest benefits (approximately 30% reduction of relapse rates), despite administration of both. Moreover, interferon-β is associated with systemic reactions in more than 60% of patients, with implications for adherence to treatment. Recently, it has been reported that this combination therapy may even lead to the development of progressive multifocal leukoencephalopathy, a potentially life-threatening opportunistic infection (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005). Safer and more effective therapy options with more specific targets of action are needed. The S1P1 receptor has been shown to regulate the recirculation of lymphocytes (Brinkmann et al., 2002; Mandala et al., 2002; Rosen et al., 2003a,b; Xie et al., 2003) and regulates their egress from secondary lymphatic organs (Matloubian et al., 2004). Therefore, targeting S1P1 to reduce circulating T cells might be an effective treatment for multiple sclerosis and other autoimmune disorders. Currently, FTY720 is undergoing clinical evaluation for this purpose.
VII. FTY720 (Fingolimod)
A. Mechanism of Action
FTY720 (Novartis, Basel, Switzerland) was first synthesized in 1992 by structural modification of myriocin (ISP-1), a fungal metabolite with immunosuppressive properties isolated from Isaria sinclairii culture broth (Fujita et al., 1994). It is now known that FTY720 is a prodrug that is phosphorylated in vivo by SphK2, but not SphK1 (Allende et al., 2004; Kharel et al., 2005; Zemann et al., 2006) to biologically active FTY720-phosphate (FTY720-P), a structural analog of S1P.
FTY720-P binds to four of the five known S1P receptors, but not to S1P2. Although it is an S1P1 receptor agonist, FTY720-P induces internalization and degradation of the S1P1 receptor that results in prolonged receptor down-regulation (Matloubian et al., 2004), thereby depriving thymocytes and lymphocytes of an S1P signal necessary for their egress from secondary lymphoid tissues (Gräler and Goetzl, 2004; Matloubian et al., 2004; Cyster, 2005) (Fig. 5). The majority of circulating lymphocytes are thus sequestered in lymph nodes, reducing peripheral lymphocyte counts and the recirculation of lymphocytes to the central nervous system (Brinkmann et al., 2002, 2004; Mandala et al., 2002; Matloubian et al., 2004). Lymphocytes in secondary lymphoid organs and those remaining in blood continue to be functional. Interestingly, oral administration of FTY720 resulted in a significantly lower blood lymphocyte content compared with intravenous administration (Kovarik et al., 2007), which suggests that at least part of the phosphorylation of FTY720 may occur in the liver, where there is abundant expression of SphK2 (Liu et al., 2000a).
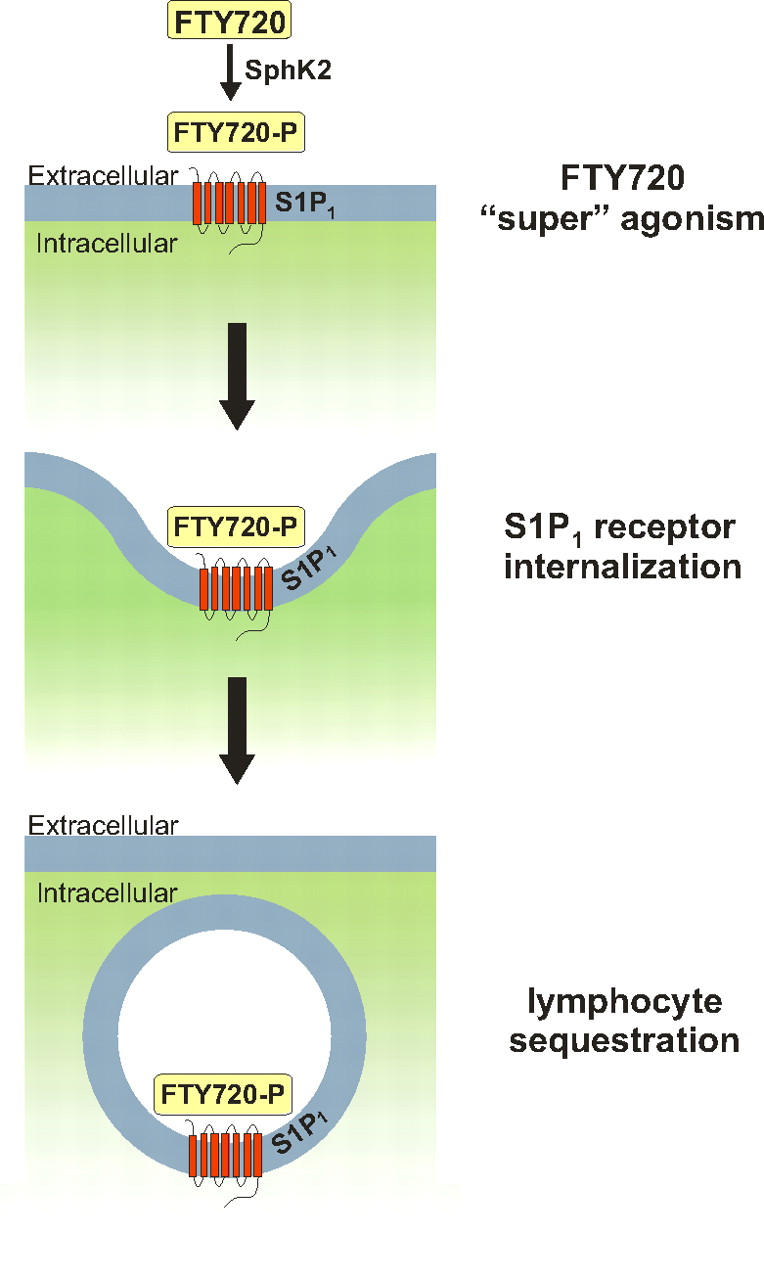
Internalization of S1P1 by FTY720 after phosphorylation by SphK2. FTY720 (fingolimod) is a sphingosine analog, which, after phosphorylation by SphK2, acts as a “ super” agonist of S1P1, leading to sustained S1P1 receptor internalization and lymphocyte sequestration.
Lymphocyte subsets including CD3+ (T cells), CD4+ (T helper cells), CD8+ (B cells), CD45RA+ (T naive cells), and CD45R0+ (T memory cells) are reduced by FTY720, whereas the numbers of peripheral blood granulocytes, monocytes, eosinophils, erythrocytes, and platelets remain unchanged (Kahan et al., 2003). T-cell counts are decreased more than B-cell counts, and CD4+ cells are affected even more than CD8+ cells (Budde et al., 2003). This relative sequestration of T cells may be particularly advantageous in treating MS, because CD4+ T-cells are considered to be major contributors to the pathophysiology. Indeed, in animal models of MS, FTY720 prevents the onset of disease and reduces established neurological deficits (Brinkmann et al., 2002; Fujino et al., 2003; Webb et al., 2004). FTY720 has the theoretical advantage of retarding lymphocyte mobilization to sites of inflammation without inducing a generalized state of immunosuppression.
B. Clinical Trials
There has been much optimism that FTY720 could become a large asset for suppressing transplant rejection with less toxicity, because of its actions that retard lymphocyte mobilization without inducing generalized immunosuppression (Brinkmann et al., 2002). It was expected that FTY720 would decrease the required doses of conventional immunosuppressive agents such as cyclosporine, particularly for renal transplantation, which is a known nephrotoxin. Unfortunately, recent results of multiple large clinical trails were all quite disappointing, failing to demonstrate a large advantage of FTY720 over standard care (Tedesco-Silva et al., 2005, 2006; Mulgaonkar et al., 2006; Salvadori et al., 2006).
Two phase 2 clinical trials evaluating the efficacy of FTY720 for immunosuppression in renal transplant recipients have been published (Tedesco-Silva et al., 2005; Mulgaonkar et al., 2006). The first study was a multicenter, open-label, dose-finding study comparing FTY720 with mycophenolate mofetil (MMF), in combination with cyclosporine and corticosteroids (Tedesco-Silva et al., 2005). Two hundred and eight patients were randomized, and the main differences in tolerability were mild and transient reductions in heart rate and decreases in peripheral lymphocytes that were reversible after cessation of FTY720 administration. The second phase 2 study compared low-dose FTY720 plus full-dose cyclosporine, low- or high-dose FTY720 plus reduced-dose cyclosporine, and MMF plus full-dose cyclosporine, in renal transplant patients, who were randomized into groups of 76, 74, 72, and 39, respectively (Mulgaonkar et al., 2006). This study design was based on the notion that addition of FTY720 might allow lower cyclosporine trough concentrations, while providing equivalent efficacy. However, low-dose FTY720 plus reduced-dose cyclosporine treatment group was discontinued early because of unusually high rejection rates. Furthermore, enrollment in this study was stopped prematurely because of a serious adverse event (cardiac arrest in the FTY720 group), and as a result, the study lacked adequate power for statistical analysis.
Two phase 3 clinical trials evaluating the efficacy of FTY720 in combination with reduced-dose cyclosporine in renal transplant recipients have also been published (Salvadori et al., 2006; Tedesco-Silva et al., 2006). These 1-year multicenter studies with randomized 668 and 696 de novo renal transplant recipients, respectively, compared low-dose FTY720 with reduced-dose cyclosporine, high-dose FTY720 with full-dose cyclosporine, and MMF with full-dose cyclosporine. Disappointingly, the study with patients randomized to high-dose FTY720 plus reduced-dose cyclosporine was discontinued early because of unusually high rejection rates. These phase 3 trials not only failed to show an advantage of FTY720 over standard of care but also demonstrated lower creatinine clearances and increased risks of macular edema in the FTY720 group. Therefore, further investigation of the use of FTY720 in renal transplantation was terminated. Furthermore, all phase 2 and 3 clinical trials of FTY720 described a transient but significant bradycardia after the first dose (Budde et al., 2002; Tedesco-Silva et al., 2005; Kappos et al., 2006). This bradycardia is probably due to the agonistic activity of FTY720-P on cardiac S1P3 receptors, because FTY720 failed to induce bradycardia in S1P3 knockout mice (Sanna et al., 2004). The specific mechanism leading to bradycardia is believed to involve the atrial muscarinic-gated potassium channel, which is a key element in vagal regulation of heart rate (Koyrakh et al., 2005). As expected, lymphopenia occurred throughout the duration of FTY720 treatment. The incidence of malignancies appeared to be low and comparable with the incidence seen with other immunosuppressive therapies. Diarrhea and nausea were also associated with FTY720 therapy during the first 6 months. However, no notable differences were seen by 12 months. Asymptomatic elevation of serum transaminases was also detected in half of the patients regardless of dose.
FTY720 is considered to be particularly promising for treatment of MS, and current therapies cannot avoid affecting other regulatory cytokines, which prevent long-term benefits and cause adverse effects. The results of a phase 2, double-blind, randomized, placebo-controlled clinical trial evaluating the efficacy and safety of FTY720 for treating relapsing MS has been published (Kappos et al., 2006). Two hundred and eighty-one patients were randomized to receive oral FTY720 or a placebo. The annualized relapse rate. in the FTY720 group was significantly lower than that in the placebo group. Adverse events included nasopharyngitis and a significant reduction in forced expiratory volume in 1 s, and some patients developed dyspnea, headache, diarrhea, and nausea. Asymptomatic elevations of alanine aminotransferase levels, symptomatic bradycardia, transient second-degree Wenckebach atrioventricular block, and posterior reversible encephalopathy syndrome were observed in FTY720 group. However, all episodes of bradycardia and atrioventricular block occurred only on the first day of therapy. Overall, the results of this phase 2 proof-of-concept study are very promising for the treatment of MS. The apparent higher rate of adverse events, coupled with the lack of difference in efficacy at higher doses of FTY720 warrants the exploration of the optimal dose for treating MS. Currently, a phase 3 study, FTY720 Research Evaluating Effects of Daily Oral therapy in Multiple Sclerosis (FREEDOMS), to further evaluate the safety and efficacy of FTY720, is underway.
VIII. KRP-203
Recently, another immunosuppressant, KRP-203 (Kyorin Pharmaceutical, Tokyo, Japan) was discovered that was found to be a S1P1 receptor-selective agonist (Shimizu et al., 2005), in contrast to FTY720. KRP-203 prolonged graft survival and attenuated chronic rejection in rat heart allograft models, suggesting that it not only regulates T-cell responses but also those of B cells, in contrast to FTY720, which does not affect B cells (Mizushima et al., 2004). KRP-203 also markedly improved allogeneic immune responses in the heart transplantation model when used in combination with low-dose cyclosporine or MMF (Takahashi et al., 2005; Suzuki et al., 2006). A similar positive effect was also noted in a rat renal allograft model in which KRP-203 in combination with a subtherapeutic dose of cyclosporine significantly prolonged skin and renal allograft survival and markedly improved graft kidney function (Fujishiro et al., 2006). These results are clinically relevant because despite the fact that cyclosporine is a powerful immunosuppressant and is regularly used for renal transplant recipient patients, it is also known to be a nephrotoxin. Furthermore, KRP-203 was highly effective in the treatment of lymphocyte-mediated hepatic injury in mice (Kaneko et al., 2006) and in experimental autoimmune myocarditis (Ogawa et al., 2007). KRP-203 was also found to significantly inhibit ongoing Crohn's-like enterocolitis in an interleukin-10-deficient mouse model (Song et al., 2008). As a selective S1P1 receptor agonist on lymphocytes, KRP-203 accelerated sequestration of circulating lymphocytes into Peyer's patches and mesenteric lymph nodes, resulting in a reduction of CD4+ T cells at the inflammatory site. Furthermore, KRP-203 inhibited T helper 1-type proinflammatory cytokine release in the lamina propria of the bowel, thus ameliorating the ongoing lesions of colitis.
Use of KRP-203 with its apparent selectivity for S1P1 would help avoid the clinical adverse effects associated with use of FTY720, which can induce transient but significant bradycardia because of its effect on cardiac S1P3 receptors. KRP-203 showed less of a tendency to cause bradycardia than FTY720 in Hartley guinea pigs (Shimizu et al., 2005). Ten-fold higher doses of phosphorylated KRP-203 than of FTY720-P were required to induce transient bradycardia (Fujishiro et al., 2006). Thus, with the given results, KRP-203 is expected to be useful for immunointervention in inflammatory and/or autoimmune diseases and graft rejection and potentially more advantageous for long-term usage with less adverse effects than FTY720. The results of human clinical trials are awaited.
IX. Other Sphingosine 1-Phosphate Receptor Agonists
The S1P1 agonist SEW2871 (Maybridge, Tintagel, Cornwall, UK) was originally identified by high-throughput screening of commercial chemical libraries with a FLIPR calcium flux assay and was found to induce lymphopenia in mice via a S1P1-dependent mechanism (Sanna et al., 2004). Although it is a S1P1-selective agonist, it is structurally unrelated to S1P and its phosphorylation is not required for binding to the receptor. SEW2871, like S1P, induces S1P1 internalization and recycling, in contrast to FTY720-P, which mediates S1P1 internalization and degradation (Jo et al., 2005). As SEW2871 does not activate S1P3, it also does not cause bradycardia (Brinkmann et al., 2004). SEW2871 was equivalent to S1P, although less potent, in guanosine 5′-O-(3-thio)triphosphate binding, calcium flux assays, ERK activation, and cell migration (Sanna et al., 2004). SEW2871 protects the kidneys against ischemia/reperfusion injury in animals (Awad et al., 2006) and preserves renal function, reduces neutrophil and macrophage infiltrates, and decreases the severity of acute tubular necrosis (Lien et al., 2006).
JTE 013, a specific S1P2 receptor antagonist (Kawasaki et al., 2001), is a pyrazolopyridine derivative originally generated at the Central Pharmaceutical Research Institute, Japan Tobacco Corporation (Osaka, Japan), which is now available from Tocris Cookson, Inc. (Ellisville, MO). The specificity of JTE-013 was established by showing that it inhibited specific binding of radiolabeled S1P to membranes from Chinese hamster ovary cells stably transfected with human S1P2 and rat S1P2 with IC50 values of 17 ± 6 and 22 ± 9 nM, respectively. In contrast, JTE-013 at concentrations up to 10 μM did not significantly decrease binding of S1P to human S1P1 or S1P3 (Ohmori et al., 2003). Only a few animal studies have been carried out to date with this S1P2 antagonist. Of note, S1P injection in diabetic mice significantly accelerated cutaneous wound healing, which was enhanced by coadministration of JTE-013 (Kawanabe et al., 2007). However, because the inhibitory effects of JTE-013 on vascular contraction were also present in basilar arteries from S1P2 knockout mice, it has been suggested that some of the effects of JTE-013 may be unrelated to S1P2 receptor antagonism (Salomone et al., 2008).
VPC 23019 is an aryl amide-containing S1P analog that acts as an unselective competitive antagonist at both S1P1 and S1P3 receptors (Davis et al., 2005) and is available from Avanti Polar Lipids (Alabaster, AL). VPC23019 has been shown to inhibit S1P-induced migration of thyroid cancer cells (Balthasar et al., 2006), ovarian cancer cells (Park et al., 2007), and neural stem cells (Kimura et al., 2007).
X. Sphingosine Kinase Inhibitors
Because the dynamic balance between the cellular levels of ceramide and S1P can determine cell fate, the enzymes that regulate this balance are potential targets for the development of new anticancer drugs that decrease S1P and/or increase ceramide (Spiegel and Milstien, 2003). In particular, there is much interest in isozyme-specific inhibitors of SphK1 because it is overexpressed in human cancers and plays a pivotal role in enhancing cancer cell proliferation and promotion of tumorigenesis in animal models. SphK1 expression is elevated in a variety of solid human tumors, such as breast, colorectal, stomach, lung, ovary, uterus, and kidney and may be involved in tumor progression and metastasis (French et al., 2003; Johnson et al., 2005; Kawamori et al., 2006). Increased SphK1 expression in tumor biopsies has been shown to correlate with a significant decrease in survival rate in patients with glioblastoma multiforme (Van Brocklyn et al., 2005).
One of the first compounds to be identified as a competitive inhibitor of sphingosine kinase activity was DHS (Saphingol) (Buehrer and Bell, 1992). It was later found that DMS, a N-methylated metabolite of sphingosine, inhibited sphingosine kinase activity in a concentration-dependent manner and was more potent than DHS (Yatomi et al., 1996). Since this finding, DMS has been one of the most commonly used inhibitors of sphingosine kinase activity in both intact cells, intact tissues, and cell-free systems (Edsall et al., 1998; Kohama et al., 1998; Pitson et al., 2000; Lee et al., 2007). However, neither DHS nor DMS can be considered as specific sphingosine kinase inhibitors, because they inhibit both SphK1 and SphK2 equally, inhibit ceramide kinase (Sugiura et al., 2002), and can also inhibit protein kinase C (Igarashi et al., 1989), as well as sphingosine-dependent protein kinase (Megidish et al., 1995), 3-phosphoinositide-dependent kinase (King et al., 2000), and casein kinase II (McDonald et al., 1991).
Both DHS and DMS have been used in several cancer preclinical trials in animal models. DMS dose dependently inhibited the growth of lung and gastric cancer cells in athymic mice (Endo et al., 1991) and decreased lung metastasis of melanoma cells (Okoshi et al., 1991). Safingol has been evaluated in a phase I clinical trial, which showed that it was tolerated well when combined with doxorubicin and did not alter the pharmacokinetics of the anticancer drug (Schwartz et al., 1997). Although sphingosine derivatives can effectively inhibit tumor growth in vivo, they also have been reported to cause significant hepatotoxicity and hemolysis at higher doses (Kedderis et al., 1995).
Screening assays identified several natural product inhibitors of SphK (Kono et al., 2000a, 2001). B-5354c, isolated from a novel marine bacterium, blocked the production of S1P and caused sphingosine to accumulate in sheep erythrocytes (Ochi et al., 2004). F-12509a, a sesquiterpene quinone isolated from a culture broth of the discomycete, Trichopezizella barbata SANK 25395, was shown to be a competitive inhibitor of SphK1 (Kono et al., 2000b; Kim et al., 2005). Although these natural products are moderately potent SphK inhibitors, the possibility of large-scale production remains unknown, and their synthesis seems impractical because of their complex structures.
A medium-throughput assay for recombinant human sphingosine kinase fused to glutathione S-transferase was developed, validated, and used to screen a library of synthetic compounds. Five nonlipid selective inhibitors toward human sphingosine kinase were identified (compounds SKI I–V) with nanomolar to low micromolar potencies, which were not competitive inhibitors at the ATP binding site of sphingosine kinase and also induced apoptosis of cancer cells (French et al., 2003, 2006). Among them, 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole was the most selective compound isolated (IC50, 0.5 mM for SphK activity), and demonstrated strong cytotoxicity toward T24 bladder carcinoma cells (IC50, 4.6 mM) and MCF-7 breast cancer cells (IC50, 1.2 mM). Interestingly, this inhibitor was almost as effective in MCF-7 cells overexpressing the multidrug resistance protein MRP1 (also known as ABCC1 and drug transporter P-glycoprotein or multidrug resistance gene 1, or ABCB1) (French et al., 2003). This result is intriguing as it suggests that inhibition of SphK1 may actually be effective against cancers that develop resistance to many chemotherapeutic drugs. The underlying mechanism behind this observation, however, is yet to be clarified. More recently, 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole was shown to kill both androgen-sensitive (LNCaP) and hormone-resistant human prostate cancer cells (PC-3), regardless of their p53 status, notably by tilting the ceramide/S1P biostat toward ceramide (Pchejetski et al., 2005). These compounds are now commercially available (Calbiochem or ChemBridge Corp, San Diego, CA), and they have been used in several other studies, including studies in murine bone marrow-derived dendritic cells (Jung et al., 2007a,b), murine cardiac cells (Pchejetski et al., 2007), human prostatic adenocarcinoma cells (Leroux et al., 2007), and a rat hemorrhagic shock model (Lee et al., 2004).
Consistent with the pharmacological effects of SphK1 inhibitors, specific knockdown of SphK1 by small interference RNA triggers apoptosis in multiple types of tumor cells in vitro, including leukemic (Taha et al., 2004; Bonhoure et al., 2006), melanoma (Bektas et al., 2005), breast (Taha et al., 2006b), glioblastoma (Van Brocklyn et al., 2005), and prostate (Pchejetski et al., 2005). Small interference RNA targeted to SphK1 induced activation of effector caspases and cytochrome c release (Bonhoure et al., 2006; Taha et al., 2006b) and significantly increased ceramide and sphingosine levels (Taha et al., 2006b).
XI. Anti-Sphingosine 1-Phosphate Monoclonal Antibody
Recently, Sabbadini and colleagues developed a highly specific monoclonal antibody against S1P (anti-S1P mAb; sphingomab) as a molecular sponge to selectively absorb and neutralize S1P and examined its effect on tumorigenesis (Visentin et al., 2006) (Fig. 4). Although this is the first report using this mAb and the models were all xenografts and allografts in nude mice, the results are very promising. The anti-S1P mAb substantially reduced tumor progression, and in some cases eliminated measurable tumors. Sphingomab blocked endothelial cell migration and resulting capillary formation, inhibited blood vessel formation induced by VEGF and basic fibroblast growth factor, and arrested tumor-associated angiogenesis. It also neutralized S1P-induced proliferation, decreased release of proangiogenic cytokines, and blocked the ability of S1P to protect tumor cells from apoptosis in multiple tumor cell lines: human lung carcinoma (A549), human breast adenocarcinoma (MCF-7, MDA MB-231, and MDA MB-468), and human ovarian carcinoma (SKOV3).
This study suggests that the general effectiveness of anti-S1P mAb in inhibiting xenograft tumor growth of many types of human cancers is probably due to prevention of the proangiogenic effects of the blood-borne lipid mediator S1P. The molecular sponge approach to neutralize S1P, coupled with the favorable pharmacokinetics and high therapeutic indices of sphingomab, suggest that a humanized anti-S1P mAb might provide therapeutic benefits in treating a broad range of human cancers. Clinical trials with a humanized version of this mAb will be important to demonstrate whether it will be of therapeutic use in humans.
XII. Conclusions
S1P is a bioactive lipid mediator that is now recognized as a bona fide regulator of many important cellular and biological processes, including proliferation, survival, migration, angiogenesis, and allergic responses. SphK1 is a key regulator that determines cell fate. S1P signaling has been implicated in many serious medical conditions, such as cancer, atherosclerosis, inflammation, and autoimmune diseases such as MS. Currently, numerous studies are underway to investigate the potential of new therapies targeting S1P signaling, including FTY720 (fingolimod), S1P receptor agonists (KRP-203 and others), sphingosine kinase inhibitors, and anti-S1P monoclonal antibody. The positive results of numerous cell culture and animal studies hold promise for successful development of a new generation of S1P targeted therapeutic agents.
Acknowledgments
This work was supported by National Research Service Award Training Grant 5T32GM008695-08 (K.T.) and in part by Virginia Commonwealth University Building Interdisciplinary Research Careers in Women's Health, National Institutes of Health 1K12HD055881-01 (K.T.), National Institute of Allergy and Infectious Diseases Training Grant T32AI007407 (S.W.P.) and Grants R01AI50094 (S.S.), R01CA61774 (S.S.), and R37GM043880 (S.S.). S.M. was supported by the Intramural Research Program of the National Institute of Mental Health.
Footnotes
-
↵1 Abbreviations: S1P, sphingosine 1-phosphate; GPCR, G protein-coupled receptor; FTY7220, 2-amino-2-propane-1,3-diol hydrochloride, fingolimod; ER, endoplasmic reticulum; GlcCer, glucosylceramide; SphK, sphingosine kinase; SPP, S1P phosphatase; PDGF, platelet-derived growth factor; VEGF, vascular endothelium growth factor; EGF, epidermal growth factor; ERK, extracellular signal regulated kinase; DMS, N,N-dimethylsphingosine; DHS, dl-threo-dihydrosphingosine; EDG, endothelium differentiation gene; ABC, ATP binding cassette; RTK, receptor tyrosine kinase; NK, natural killer; HDL, high-density lipoprotein; MS, multiple sclerosis; FTY720-P, FTY720 phosphate; MMF, mycophenolate mofetil; KRP-203, 2-amino-2-{2-{4-(3-benzyloxyphenylthio)-2-chlorophenyl}ethyl}-1,3-propanediol hydrochloride; SEW2871, 5-(4-phenyl-5-trifluoromethylthiophen-2-yl)--(3-trifluoromethylphenyl)-(1,2,4)-oxadiazole; JTE 013, 1-[1,3-dimethyl-4-(2-methylethyl)-1H-pyrazolo[3,4-b]pyridin-6-yl]-4-(3,5-dichloro-4-pyridinyl)-semicarbazide; VPC 23019, (R)-Phosphoric acid mono-[2-amino-2-(3-octyl-phenylcarbamoyl)-ethyl] ester; mAb, monoclonal antibody.
-
This article is available online at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.107.07113.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Biosynthesis and Metabolism of Sphingosine 1-Phosphate
- III. Mechanisms of Sphingosine Kinase Activation
- IV. Sphingosine 1-Phosphate Receptors
- V. Sphingosine 1-Phosphate in the Blood
- VI. Sphingosine 1-Phosphate in Human Diseases and Sphingosine 1-Phosphate-Targeted Therapies
- VII. FTY720 (Fingolimod)
- VIII. KRP-203
- IX. Other Sphingosine 1-Phosphate Receptor Agonists
- X. Sphingosine Kinase Inhibitors
- XI. Anti-Sphingosine 1-Phosphate Monoclonal Antibody
- XII. Conclusions
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters