Article Text

Abstract
The genetic aetiology of autism remains elusive. Occasionally, individuals with Cowden syndrome (a cancer syndrome) and other related hamartoma disorders such as Bannayan-Riley-Ruvalcaba syndrome, Proteus syndrome, and Proteus-like conditions, are characterised by germline PTEN mutations, and may have neurobehavioural features resembling autism as well as overgrowth and macrocephaly. Therefore, we undertook PTEN gene mutation analysis in 18 subjects mainly prospectively ascertained with autism spectrum disorder and macrocephaly. Of these 18 autistic subjects (13 males and five females; ages 3.1–18.4 years) with a head circumference range from 2.5 to 8.0 standard deviations above the mean, three males (17%) carried germline PTEN mutations. These three probands had previously undescribed PTEN mutations: H93R (exon 4), D252G (exon 7), and F241S (exon 7). They had the larger head circumference measurements amongst all our study subjects. The three residues altered in our patients were highly evolutionarily conserved. We suggest that PTEN gene testing be considered for patients with autistic behaviour and extreme macrocephaly. The gene findings may impact on recurrence risks as well as medical management for the patient.
- ADI-R, Autism Diagnostic Interview-Revised
- ASD, autism spectrum disorders
- BRRS, Bannayan-Riley-Ruvalcaba syndrome
- CS, Cowden syndrome
- HC, head circumference
- PDD-NOS, pervasive developmental disorder-not otherwise specified
- SD, standard deviations
- autism
- macrocephaly
- novel mutation
- PTEN
Statistics from Altmetric.com
- ADI-R, Autism Diagnostic Interview-Revised
- ASD, autism spectrum disorders
- BRRS, Bannayan-Riley-Ruvalcaba syndrome
- CS, Cowden syndrome
- HC, head circumference
- PDD-NOS, pervasive developmental disorder-not otherwise specified
- SD, standard deviations
Autism is a common neurodevelopmental disorder with a prevalence of 4–10 per 10 000 individuals with a three- to fourfold higher incidence in males than in females.1–3 Classical autism is an early onset disorder with developmental difficulties noted by 3 years of age and belongs to a group of heterogeneous conditions known as autism spectrum disorders (ASD). Asperger syndrome and pervasive developmental disorder-not otherwise specified (PDD-NOS) are also included in this group. Diagnostic features for classical autism include severe impairment in the development of social interactions, a marked and sustained impairment of both verbal and non-verbal communication, and restricted, repetitive, or stereotyped behaviours and interests.4–6 Macrocephaly is also seen in 20% of patients with autism.7
Several genome-wide scans have searched for autism susceptibility loci using DNA markers in multiplex families. Strong evidence for linkage has been reported for chromosomes 5, 7, 8, 16, 19, and X and nominal evidence for chromosomes 2, 3, 4, 10, 11, 12, 15, 18, and 20.8 The inheritance of autism is complex with more than 15 loci involved.
Multiple interacting genetic factors underlie the cause of the majority of cases of ASD.9 However, as many as 10% of cases are associated with a number of distinct genetic conditions including fragile X, tuberous sclerosis, phenylketonuria, Rett syndrome, and chromosomal anomalies.4,8,10,11 One of the most frequent cytogenetic abnormalities (that is, partial duplications, deletions, and inversions) includes the 15q11–q13 region and accounts for 1–4% of cases of autism.12,13 However, most cases of autism have no known aetiology. Several potential candidate genes have been identified in both autosomes and X chromosomes including the tuberous sclerosis genes (TSC1 and TSC2) on chromosomes 9 and 16, respectively; serotonin transporter (SERT) on chromosome 17; gamma-aminobutyric acid receptor-beta 3 (GABRB3) on chromosome 15; neuroligins (NLGN3 and NLGN4) on the X chromosome; and possibly PTEN on chromosome 10.
PTEN (phosphatase and tensin homologue with sequence homology to chicken tensin, bovine auxilin, and a protein tyrosine-phosphatase domain) is a tumour suppressor gene localised to chromosome band 10q23.14 It encodes a dual specificity phosphatase effecting G1 cell cycle arrest and/or apoptosis.15PTEN represents the first phosphatase gene implicated in an inherited cancer syndrome (Cowden syndrome). Germline mutations in PTEN have been found in patients with four hamartoma syndromes: Bannayan-Riley-Ruvalcaba syndrome (BRRS), Cowden syndrome (CS), Proteus syndrome, and Proteus-like conditions.15 Somatic mutations in PTEN have been reported to varying degrees in brain, colorectal, breast, kidney, uterine, thyroid, and haematological malignancies (reviewed in Eng15).
CS is an autosomal dominant disorder with a high risk of breast, thyroid, and endometrial cancer. In addition, Hanssen and Fryns reported mental retardation in 12% of CS subjects.16 Hallmark signs of BRRS include macrocephaly and multiple lipomas, Hashimoto’s thyroiditis, vascular malformations, pigmented macules of the glans penis, mental retardation, and delayed motor development. Interestingly, autistic behaviours have been observed in separate patients carrying germline PTEN mutations15,17–19 including a patient with VATER association and macrocephaly, a CS family with a boy with autistic behaviour and mental retardation, and a boy with BRRS and autism but with CS in the mother. Because of these reports and macrocephaly seen in patients with autism, we hypothesised an association between progressive macrocephaly and autistic behaviour and PTEN gene mutations.
METHODS
To address our hypothesis, we accrued subjects with the predetermined inclusion criteria of ASD and macrocephaly (>2.5 standard deviations (SD) above the mean), and the exclusion criteria of known CS or BRRS; ASD without macrocephaly was also a predetermined exclusionary criterion. Accordingly, we accrued 18 subjects ascertained with ASD and macrocephaly including 15 with classical autism and three with PDD-NOS (13 males and five females; 16 white and two black with an age range of 3.1–18.4 years). Twelve subjects were ascertained longitudinally or prospectively in the clinical setting and referred to either general genetics or autism clinics for diagnosis, medical management, and/or genetic testing. DNA was obtained from six subjects from the Autism Genetic Resource Exchange (information available at www.agre.org) and the patients selected based on the diagnosis of classical autism and having macrocephaly. The diagnosis was established with the use of the Autism Diagnostic Interview-Revised (ADI-R)5 and by clinical genetics evaluation. Psycho-behavioural and clinical assessments and diagnostic examinations were performed by neurologists, clinical geneticists, and other knowledgeable experts in autism (for example, developmental paediatricians, clinical psychologists) for 12 subjects. In addition, family and medical histories were reviewed and psychological/autism testing (for example, ADI-R) performed if not previously available to establish a diagnosis. Head circumference (HC) ranged from +2.5 to +8.0 SD for age and sex with an average HC of +4.0 SD (six subjects had HC greater than +4.0 SD). Chromosome analysis and fragile X gene testing were normal. Genomic DNA was extracted from peripheral blood for PTEN mutation analysis of the promoter and exons 1–9 using a combination of denaturing gradient gel electrophoresis and direct DNA sequencing as previously described.14
RESULTS AND DISCUSSION
Mutation survey in ASD subjects
Of our 18 ASD subjects, three males (17%) ranging in age from 2 to 4 years and with head circumferences ranging from +4.5 to +8 SD, carried germline PTEN mutations: D252G (exon 7), F241S (exon 7), and H93R (exon 4). The three males were identified from the group of autistic subjects with macrocephaly ascertained prospectively in the clinical setting. The mutations were all different and novel compared to those reported in the literature. There were no features suggestive of CS or BRRS except for pigmented macules on the glans penis in one mutation positive individual (subject three, see below). There was no family history suggestive of CS or BRRS in any of the three mutation positive children. The parents of the child (subject one) with the F241S mutation were mutation negative and non-paternity was excluded by microsatellite testing at three other loci. The three missense mutations were absent in >400 control chromosomes.
Clinical phenotypes of the three ASD individuals with PTEN mutations
Subject one (H93R)
Subject one was a 4 year old white male born to a 32 year old G2 P2 mother and a 35 year old father. He presented with macrocephaly, macrosomia, severe speech delay, and autistic behaviour. He had no history of seizures, hypoglycaemia, or hypotonia but did have an adenoidectomy and history of recurrent otitis media. There was a history of gestational diabetes, and a birth weight of 4.1 kg (90th centile). He was delivered by caesarian section. His family history was significant for an older brother with trisomy 21 and macrocephaly in the father (head circumference of 61.5 cm (+4 SD)). No consanguinity was noted. The physical examination at 4 years of age showed macrosomia with a weight of 27.4 kg (+4 SD), height of 109.8 cm (90th centile), and head circumference of 62 cm (+8 SD). Mild bilateral epicanthal folds and a flattened nasal bridge were observed. Severe speech delay and autistic behaviours were recognised during the toddler period and documented by developmental assessments. Clinical chemistry, haemogram, and urine mucopolysaccharides were all within normal range. A bone x ray survey and renal ultrasound were normal. A brain MRI showed marked dilatation of the perivascular spaces. Plasma acylcarnitine and urinary organic acid profiles were normal. Routine chromosome studies and fragile X molecular genetic testing were normal. PTEN gene mutation analysis was performed on genomic DNA from the patient and his parents. A novel germline PTEN mutation in exon 4 at codon 93 (CAT>CGT, H93R) was identified in the patient. Neither parent carried this mutation and non-paternity was excluded with microsatellite genotyping.
Subject two (D252G)
Subject two was a 3.5 year old white male seen for genetic evaluation due to developmental delay, speech apraxia, sensory integration problems, a short attention span, and macrocephaly (head circumference 61.1 cm (+7 SD)). He was born vaginally at approximately 18 days post-term, weighed 5 kg (+3 SD), and had a large appearing head. No other pregnancy problems were noted. The mother was 29 years old at the time of delivery and the father was 33 years old. There was no consanguinity. He has three siblings (two older sisters and one younger brother, all reported to be in good health). The mother’s head circumference was 56.5 cm (80th centile) and the father’s head circumference was 61.2 cm (+4 SD). Because of macrocephaly, sensory integration problems, mild hypotonia, and developmental delay, CT and MRI scans were each performed previously on two separate occasions and reported as normal. An EEG was also performed and interpreted as normal. Growth hormone testing was undertaken due to his large size and was found to be normal. Developmental testing at a chronological age of 28 months showed delayed early motor milestones (27th centile for overall fine motor ranking with the Peabody Developmental Motor Skills-II, birth to 72 months) which was thought to be due to his macrocephaly. His grasp rating was at 20 months and his visual motor integration was at 23 months. He walked between 12 and 18 months of age per history and demonstrated significant speech and language delay. The diagnosis of verbal apraxia was made (he currently has 15–20 words but can use sign language). He receives speech therapy. By history, his language development regressed at approximately 26 months of age. Sensory integration problems were noted as well as a short attention span. He was thought to have a pervasive developmental disorder. By exam he has one posterior hair whorl with coarse hair, joint laxity, and soft, loose skin. He has bilateral plantar creases and a flat appearing mid face with a prominent forehead (fig 1). He had a large appearing penis without freckles and with bilateral hydroceles. Routine chromosome studies and molecular genetic testing for the fragile X syndrome were normal. Clinical chemistry, organic and amino acid levels, and haematology testing were normal. Because of macrocephaly, developmental delay, and pervasive developmental disorder, PTEN gene mutation analysis was performed. A germline PTEN mutation was identified in exon 7 at codon 252 (GAC>GGC, D252G). The mother was PTEN mutation negative, but the father was not available for mutation testing.
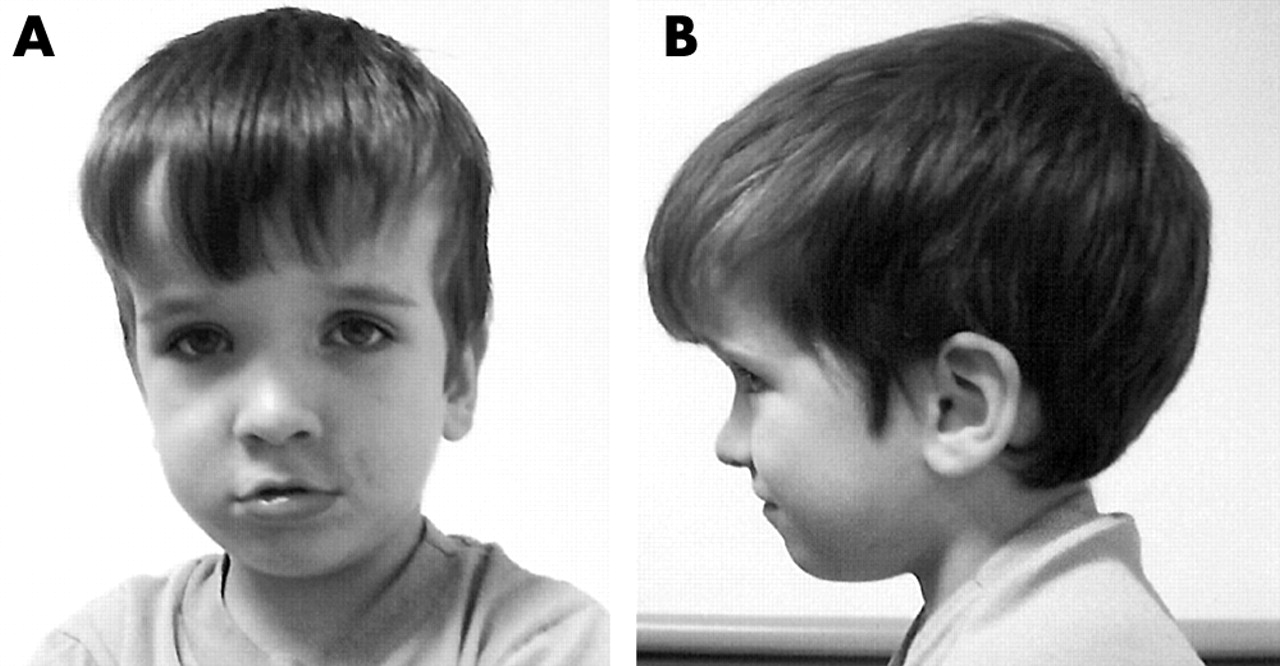
Frontal and profile views of subject two with a PTEN mutation (D252G) at 3.5 years of age showing macrocephaly. (Photograph reproduced with permission.)
Subject three (F241S)
Subject three was a 2.5 year old biracial male referred because of developmental delay, autistic behaviour, and overgrowth. Birth weight was 3.4 kg (97th centile) and birth length was 53.8 cm (95th centile). The historian (adoptive mother) reported alcohol and illicit drug use during the pregnancy. The family history was remarkable for mental health problems in the mother. At 3 months of age, questions were raised about his vision and later loss of skills was noted during infancy. Because of possible seizure activity, a video EEG and cranial CT and MRI scans were performed. The EEG and brain imaging scans were unremarkable. Urine organic acid levels were normal as were routine chromosome studies and fragile X DNA testing. At 2.5 years of age, his length was 96 cm (97th centile) and head circumference was 56.5 cm (+4.5 SD). His anterior fontanelle was closed, but he had a narrow appearing but prominent forehead. His hair was curly, light brown, and with normal texture. The craniofacial examination revealed a high arched palate. The ears were 5 cm long (+4 SD) but normally shaped and placed. The cardiovascular and abdominal examinations were normal without heart murmur, organomegaly, or hernias. The genital examination showed freckles on the glans penis, but both testes were of normal size. The hands and feet were of normal size and appearance. There were no cutaneous pigmentary or vascular abnormalities. The gait was wide based. Both Sotos syndrome and BRRS were under consideration and PTEN mutation testing was done. He has been followed in the autism treatment centre. He was found to have a germline mutation in exon 7 at codon 241 (TTT>TCT, F241S).
Predicted pathogenicity of missense PTEN mutations
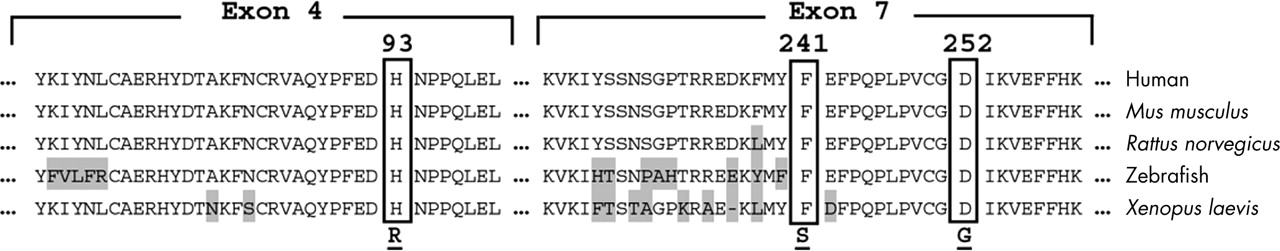
The alignment of the human PTEN gene with four homologous genes from other species indicated that the three residues altered in our patients are highly evolutionarily conserved down through zebrafish and Xenopus laevis, implying an important functional role for these residues in the PTEN protein (fig 2). Point mutations have been previously reported for two other conserved sequences surrounding these residues, in patients with CS (P96Q)20 and BRRS (P246L).21 No missense mutations have been previously reported to alter either residue 241 or 252. Missense mutations have been identified in other residues of exon 7 of the PTEN gene. A histidine to tyrosine substitution in codon 93 (H93Y) was reported in a 46 year old Japanese male with CS.22 This patient had CS and macrocephaly. It is not surprising that different substitutions at the same residue may result in different phenotypes. In vitro studies have indicated that point mutations at the same codon might produce different effects on the activity of PTEN. For example, the missense mutation, G129E, identified in at least two unrelated CS families, was found to have full protein phosphatase activity but absent lipid phosphatase activity.23,24 However, a mutation in tumour cells of the same residue to a different amino acid (G129R) resulted in a significant reduction in protein phosphatase activity of PTEN.23 Of note, all three subjects in this study had missense mutations which contrast markedly with the CS/BRRS mutational spectra where only ∼20% (of >13515) of mutations are missense (p = 0.009, Fisher’s two tailed exact test).

{kind=link}
{kind=link}
Multiple alignment of PTEN and homologous genes in other organisms (that is, Mus musculus, Rattus norvegicus, zebrafish, and Xenopus laevis) showing the extent of sequence conservation in exons 4 and 7. The three residues (H93, F241, and D252) implicated in our patients are conserved among other gene families (shown in boxes). Amino acids that are not conserved are shaded in grey. Amino acid substitutions are shown below the alignment.
The ideal method to assess the significance of missense mutations would include functional analysis of the translated protein. However, in the absence of functional data, we evaluated the impact of the observed missense mutations in our subjects on the secondary structure of the PTEN protein using protein profiling software. The preliminary protein analysis suggested that all three mutations resulted in a detectable change of the surface accessibility of the protein in a region surrounding the altered residues using MacVector software version 7.0 (ACCELRYS, San Diego, CA). This programme will predict hydrophilicity, hydrophobicity, surface probability, and antigenicity. Surface probability predicts the region of a protein most likely to lie on the surface based on the hydrophilicity and/or hydrophobicity of the amino acids (for example, a value is calculated between 0 and 1 with 0 representing an amino acid definitely buried in the interior of the protein while 1 represents an amino acid definitely exposed at the surface). Changing the H to R at position 93 (H93R) in exon 4 in one of our patients did increase hydrophilicity and reduced hydrophobicity which resulted in a higher surface probability (from 0.59 to 0.66) and antigenicity. Indeed H93Y in a CS individual abrogated phosphatase activity.15 Similar predicted changes were observed for the F241S substitution in exon 7 (surface probability of 0.39 to 0.44) in subject three, while the third mutation in exon 7 (D252G) had an opposite impact on the protein profile (0.35 to 0.27). These mutations affect the C2 domain and likely will impact on phospholipid binding, both substrate and membrane. The germline mutation G251C occurs at a residue next to the D252G, the latter of which is phosphatase null (reviewed by Waite and Eng14). Similarly, an existing mutation, F341V, in proximity to F241S in one of our patients, also has been shown to abrogate phosphatase activity.14 Therefore, taking together the informatics profiling and the functionality of the known mutations, these missense mutations would be pathogenic. It is also tempting to hypothesise that missense mutations can act as dominant negatives and hence result in a severe phenotype. Supporting this postulate is the observation that missense mutations in CS are associated with multi-organ involvement.25
Our findings suggest that molecular testing for PTEN gene mutations in patients with autistic behaviour and extreme macrocephaly, even in the absence of other CS/BRRS related clinical features, should be considered. The autistic subjects in our study with macrocephaly also had brain imaging performed (for example, CT or MRI scans), but no hydrocephaly or other brain pathology was found. Interestingly, two of the three subjects with PTEN mutations had the larger head circumference measurements (for example, +8 and +7 SD, respectively) amongst all our study subjects. These two subjects had no other additional clinical findings classically associated with CS or BRRS (for example, pigmentary abnormalities). Implications for the patient’s care and management as well as recurrence risks for other family members are evident if gene mutations are found.
Two of the fathers of our subjects with PTEN mutations were found to have macrocephaly. One was PTEN mutation negative and had an unremarkable history and physical examination, although no head imaging was performed. The other father was not available for either examination or testing (for example, PTEN gene mutation analysis or diagnostic evaluations such as head imaging). Therefore, other causes of macrocephaly in the fathers could not be ruled out (for example, familial macrocephaly or hydrocephaly).
Autism is genetically heterogeneous with previous genetic linkage analysis identifying a region on chromosome 10; PTEN gene may be in this region. A positive yield for PTEN gene mutations should be anticipated particularly in autistic subjects with extreme macrocephaly with or without features of BRRS or CS. Finally, presentation of an autism spectrum disorder in an individual should now be considered as another indication for PTEN gene mutation screening particularly in those with extreme macrocephaly (that is, HC greater than +4 SD) as observed in our study.
Acknowledgments
We thank the autism families presenting for genetic services and the Autism Genetic Resource Exchange. We also thank Cindy Holmberg and Deborah Moore for expert preparation of the manuscript and Molly Lund, M.S., and Holly Welsh, M.S., for assistance in obtaining family data and samples.
REFERENCES
Footnotes
-
Partial funding for this study was made possible through CMH Special Gift Funds (MGB), a CMH Physician Scientist Award (MGB), the Hall Foundation (MGB), and the American Cancer Society (RSG-02-151-01CCE) (CE). CE is a recipient of the Doris Duke Distinguished Clinical Scientist Award.
-
Competing interests: none declared