

Abstract
The mammalian auditory sensory epithelium, the organ of Corti, is a highly ordered cellular structure that comprises two types of auditory hair cells and several types of nonsensory supporting cells. During embryogenesis, a stereotyped sequence of cellular and molecular events is required for its development. These processes are assumed to be regulated by multiple growth and transcription factors. However, the majority of these factors have not been identified. One potential regulator of cochlear development is the insulin-like growth factor (IGF) signaling family. To examine the roles of the IGF pathway in inner ear formation, cochleae from Igf1r mutant mice were analyzed. Deletion of Igf1r leads to several changes in inner ear development including a shortened cochlear duct, a decrease in the total number of cochlear hair cells, and defects in the formation of the semicircular canals. In addition, maturation of the cochlear sensory epithelium was delayed at the transition point between cellular proliferation and differentiation. To determine the molecular basis for these defects, inhibition of IGF signaling was replicated pharmacologically in vitro. Results indicated that IGF signaling regulates cochlear length and hair cell number as well as Atoh1 expression through the phosphatidylinositol 3-kinase/Akt signaling pathway. These results demonstrate novel roles for IGF signaling in inner ear development including regulation of vestibular formation, length of the cochlear duct, and the number of cochlear hair cells. The results also provide new insights regarding the pathological processes that underlie auditory defects in the absence of IGF signaling in both humans and mice.
Introduction
The auditory sensory epithelium of the mammalian inner ear, also referred to as the organ of Corti, is a complex structure consisting of two distinct types of hair cells (inner and outer) and at least six distinct types of associated supporting cells. All of these cell types are arranged in regular rows that extend along the length of the coiled cochlea to form a highly ordered mosaic. The formation of this structure requires a coordinated sequence of cellular and molecular processes that act to generate a pool of precursor cells, known as prosensory cells, and then to direct subsets of those prosensory cells toward one of the possible cell fates. At the same time, these cells must become organized into specific rows, some of which contain an alternating series of hair cells and particular supporting cells. While considerable progress has been made in the identification of the factors that mediate these events, our understanding of how this structure develops is still limited.
One potential regulator of some aspects of cochlear development is the insulin-like growth factor (IGF) signaling family. Woods et al. (1996) described severe hearing loss along with growth failure in a 15-year-old boy with a homozygous mutation in the IGF1 gene. In addition, mice that are deficient in Igf1 exhibit severe hearing loss as well as loss of spiral ganglion neurons (Cediel et al., 2006). A recent study by Sanchez-Calderon et al. (2010) demonstrated that Igf1 and Igf-I receptor (Igf1r) are both expressed in the developing mouse cochlea at embryonic day 15.5 (E15.5) and E18.5. Based on these observations, a role for the IGF signaling pathway in some aspect of cochlear development seems likely.
The IGF signaling pathway consists of two secreted ligands (IGF1 and IGF2), two receptors (IGF1R and IGF2R), and six soluble binding proteins (IGFBP1–6). IGF1R is a classic transmembrane tyrosine kinase receptor and is believed to mediate all cellular responses to the IGF ligands because IGF2R lacks an intracellular signaling domain (Jones and Clemmons, 1995). The specific roles of IGFBPs are not entirely clear, but they are believed to modulate pathway activity via inhibition or augmentation of bioavailability of the IGF ligands (Schneider et al., 2000). During embryonic development, IGF signaling plays an important role in determining body size by promoting cell proliferation and survival (Liu et al., 1993). In the nervous system, IGF signaling has been shown to regulate differentiation of oligodendrocytes and to promote growth cone formation and axon outgrowth (Hsieh et al., 2004; Laurino et al., 2005; Scolnick et al., 2008). However, a specific role(s) for IGF signaling in inner ear development has not been fully examined.
In this study, we describe the patterns of expression for IGF signaling components in the embryonic cochlea, and then examine the effect of genetic or pharmacological inhibition of IGF1R on cochlear development. Our results demonstrate important roles for IGF signaling in inner ear morphogenesis as well as in proliferation of prosensory cells and differentiation of hair cells and supporting cells.
Materials and Methods
Animals.
Heterozygous Igf1r mutants were generated by crossing Igf1r floxed mice (Dietrich et al., 2000) with Zp3-cre mice in which cre recombinase expression is controlled by regulatory sequence from the mouse zona pellucida 3 (Zp3) gene leading to specific expression in oocyte (Lewandoski et al., 1997). Resulting offspring were heterozygous for a somatic deletion of Igf1r. Heterozygous animals were crossed to generate embryos with a complete deletion of Igf1r (Igf1r−/−). Animals were maintained on a mixed 129/Bl6 background. Genotyping for Igf1r floxed or heterozygous Igf1r mutant mice was performed as previously described (Xuan et al., 2002). For in vitro experiments, timed-pregnant CD1 mice were purchased from Charles River Laboratories. The morning of the plug was designated as E0.5 and the day of birth as postnatal day 0 (P0), and animals of either sex were used in all the experiments in this study. All animals were maintained based on the standards outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In situ hybridization.
In situ hybridization for Igf1, Igf2, Igf1r, Igf2r, and Atoh1 was performed as described previously (Wu and Oh, 1996) on 12-μm-thick frozen sections of mouse inner ears that were isolated between E10.5 and P0.
Quantitative reverse transcription PCR.
Gene expression levels were determined by quantitative reverse transcription PCR (qPCR) as previously described (Yamamoto et al., 2011). Briefly, total RNA was extracted from six cochleae for each genotype using the RNaquous kit (Ambion) according to the manufacturer's protocol. Before extraction, spiral ganglion neurons and other surrounding tissue were physically dissected away from the cochlea, but some mesenchymal tissue was included. cDNA was synthesized using the SuperScript III cDNA Synthesis kit (Invitrogen). cDNA and primer sets were mixed with Power SYBR Green PCR Master Mix (Applied Biosystems), and real-time PCR quantification was performed using an ABI Prism 7000 Sequence Detection System (Applied Biosystems). The relative amount of mRNA was calculated by the standard curve method using mouse Arbp mRNA as an invariant control (Yabe et al., 2003). For the analysis of in vitro gene expression following drug treatments, cochlear explants were directly processed for RNA extraction at the end of designated culturing periods. Sequences of primer sets for Igf1, Igf2, Igf1r, Atoh1, Prox1, p27Kip1, Jag1, Sox2, and Arbp are available on request.
Paint fills of the labyrinth.
Gross morphology of the membranous labyrinth was visualized by filling the labyrinth with paint as previously described (Morsli et al., 1998).
Immunohistochemistry.
For whole mounts, cochleae were prepared as described previously (Jacques et al., 2007). For immunohistochemistry on frozen sections, cochleae were dissected in ice-cold PBS and fixed in 4% paraformaldehyde overnight. Samples were cryoprotected through a sucrose gradient, embedded in Tissue-Tek OCT Compound (Sakura), and sectioned on a cryostat at 12 μm thickness. The primary antibodies used in this study were as follows: rabbit anti-Myosin6 (Myo6) (1:1000) (Proteus BioSciences), rabbit anti-Prox1 (1:1000) (Covance), goat anti-Sox2 (1:250) (Santa Cruz Biotechnology), goat anti-Jag1 (1:300) (Santa Cruz Biotechnology), rabbit anti-p27Kip1 (1:300) (Thermo Fisher Scientific), mouse anti-Lhx3 (1:50) (Developmental Studies Hybridoma Bank), rabbit anti-Calbindin (1:1000) (Millipore), rabbit anti-cleaved Caspase 3 (1:100) (Cell Signaling Technology), mouse anti-phospho-Histone H3 (1:100) (Cell Signaling Technology), rabbit anti-Ki67 (1:200) (Thermo Fisher Scientific), mouse anti-5-bromo-2′-deoxyuridine (BrdU) (1:50) (BD Biosciences). For BrdU staining, specimens were pretreated with 2N HCl at 37°C for 20 min. Primary antibodies were visualized with Alexa Fluor 488 (AF488)-, AF555-, or AF633-conjugated secondary antibodies (1:500) (Invitrogen). AF488-, AF546-, or AF633-conjugated phalloidin (Invitrogen) was used for labeling filamentous actin. All fluorescent images were obtained on a Nikon E800 (Nikon) or an LSM 510 laser-scanning microscope (Carl Zeiss).
Quantification of cell number, cell density, length of sensory epithelium, and stereociliary bundle orientation.
To determine the total number of hair cells or supporting cells in the cochlear sensory epithelium, images of the entire cochlea labeled with anti-Myo6 or anti-Prox1 were taken with an LSM 510 laser-scanning microscope. The number of Myo6-positive hair cells or Prox1-positive supporting cells was counted in each whole cochlea from base to apex. The density of proliferating cells was determined on sections stained with anti-phospho-Histone H3 and anti-Sox2 using ImageJ software (http://rsbweb.nih.gov/ij/). The number of phospho-Histone H3-positive cells in Sox2-positive prosensory cells was divided by the area occupied by Sox2-positive cells. Using three nonadjacent sections in each ear, the density of proliferating cells was calculated as cell number per 10,000 μm2. Length of cochlear sensory epithelium was determined on whole-mount cochleae stained with Sox2 by measurement of the length of the Sox2-positive domain along the longitudinal axis of the cochlear duct using ImageJ software. Orientations for stereociliary bundles in the basal turn of cochlea at E18.5 were determined as previously described (Dabdoub et al., 2003; Montcouquiol et al., 2003).
Scanning electron microscopy.
The inner ears from E18.5 mice were fixed in 4% paraformaldehyde with 2.5% glutaraldehyde overnight at 4°C. The bony capsule of the cochlea was removed and the roof of the cochlear duct was opened to expose the lumenal surface of cochlear sensory epithelium. Specimens were postfixed with 1% osmium tetroxide, incubated twice in 1% tannic acid followed by 1% osmium tetroxide for conduction staining, and dehydrated through a graded ethanol series from 30 to 100%. Specimens were dried at the critical point and mounted on metal studs. Samples were examined on a Hitachi S-4800 field-emission scanning electron microscope (Hitachi).
Explant cultures of mouse embryonic cochlea.
Explant cultures of embryonic cochleae were established at indicated ages as described previously (Montcouquiol and Kelley, 2003) and maintained for a maximum of 4 d in vitro. For experiments with BrdU, explants were incubated with 3 μg/ml BrdU (Sigma-Aldrich) in culture media for 24 h.
Treatment of cochlear explants with an activator or an inhibitor for the IGF signaling pathway.
Cochlear explants were established at indicated time points and treated immediately with one of the following reagents. For activation of the IGF signaling pathway, cochlear explants were treated with 1 μg/ml recombinant mouse IGF1 (R&D Systems) plus PBS as a vehicle. For inhibition of IGF signaling, cochlear explants were treated with specific concentrations between 0.15 and 5 μm picropodophyllin (PPP) (EMD Chemicals), or between 5 and 30 μm IGF-1R inhibitor II [N-(5-chloro-2-methoxyphenyl)-N′-(2-methyl-4-quinolinyl)urea (PQ401)] (EMD Chemicals), plus 0.1% dimethyl sulfoxide (DMSO) as a vehicle. Phosphatidylinositol-3-kinase (PI3K) was inhibited by treating cochlear explants with specific concentrations between 10 and 40 μm 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) (EMD Chemicals), and MEK1/2 was inhibited by treatment with 10 μm 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) (EMD Chemicals) plus 0.1% DMSO as a vehicle. Control explants were treated with vehicle alone in each experiment.
Immunoblotting.
Protein extracts from four cochlear explants were prepared at 4°C using RIPA buffer (Sigma-Aldrich) with 300 μm NaCl, protease inhibitor mixture (Complete Mini; Roche), and phosphatase inhibitor mixture (Sigma-Aldrich). The lysates were denatured at 70°C for 10 min with NuPAGE LDS Sample Buffer (Invitrogen) and β-mercaptoethanol (Sigma-Aldrich). Total protein was fractionated by SDS-PAGE on a 4–12% gradient Bis-Tris gel (Invitrogen) and transferred onto nitrocellulose membranes (Invitrogen). The origins and dilutions of antibodies used in this study were as follow: mouse anti-β-actin (1:10,000) (Sigma-Aldrich), rabbit anti-phospho-Akt (1:2000) (Cell Signaling Technology), and rabbit anti-phospho-Erk (1:1000) (Cell Signaling Technology). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgGs were used as secondary antibodies (1:50,000) (GE Healthcare). The specific bands were visualized by chemiluminescence using GE Healthcare ECL plus Western Blotting Detection System (GE Healthcare) according to the manufacturer's instruction.
Statistics.
At least four samples for each experimental group were analyzed in all of the experiments performed in this study. All data are shown as mean ± SEM. An ANOVA or a Student's t test was used to determine significance of difference among experimental groups except that Kolmogorov–Smirnov test was used to analyze the deviations for hair bundle orientation. Values of p < 0.05 were considered to be statistically significant.
Results
Igf1, Igf2, and Igf1r are expressed in the developing mouse cochlea
To determine which cells within the developing cochlea might be regulated by IGF signaling, we performed in situ hybridization for Igf1, Igf2, and Igf1r on sections through developing mouse cochleae at specific ages between E10.5 and P0. Expression of Igf1 was not observed in the cochlea between E10.5 and E12.5, but on E13.5 and E14.5, Igf1 expression was observed in the extreme medial edge of the greater epithelial ridge (GER) and in the presumptive stria vascularis (Fig. 1A). This pattern of expression was largely maintained from E14.5 to P0 (Fig. 1B–D); however, a clear basal-to-apical gradient in expression is present at E16.5 (Fig. 1B). Expression of Igf2 overlaps with Igf1 in the medial region of the cochlear duct but is not expressed in the stria vascularis (Fig. 1E–H). In addition, Igf2 is expressed in the lateral edge of the cochlear duct and the surrounding mesenchyme (Fig. 1E, arrowhead, asterisks). In contrast with Igf1 and Igf2, Igf1r is already expressed in the ventral region of the otocyst at E10.5 in a broad and diffuse pattern (data not shown). At E13.5, Igf1r is broadly expressed within the cochlear duct including the prosensory domain, GER, and future Reissner's membrane (Fig. 1I), as well as the developing spiral ganglion (data not shown). At E16.5, Igf1r expression persisted in the floor of the cochlear duct and spiral ganglion but was downregulated in developing Reissner's membrane (Fig. 1J,K). At P0, expression was largely restricted to the floor of the cochlear duct and to the basal cell layer of the stria vascularis (Fig. 1L). Expression of Igf2r was not observed within the developing cochlea at any time point between E10.5 and P0 by in situ hybridization (data not shown).

Expression of Igfs and Igf1r in the embryonic cochlea. A–D, In situ hybridization for Igf1 in cross-sections through the embryonic cochlea at the indicated time points. At E13.5, Igf1 expression is present in two regions of the cochlear duct, the medial edge of the GER (A, arrowhead) and the developing stria vascularis (SV) (A, arrow). This pattern persists at E16.5 (B, C) and P0 (D). However, while the pattern of expression is unchanged, a clear base-to-apex gradient in the intensity of Igf1 expression is present at E16.5 (B, arrows). E–H, Similar views as for Igf1 but probed for expression of Igf2. At E13.5, Igf2 is expressed at the medial edge of the GER (E, arrow) and also in the lateral edge of the cochlear duct (arrowhead). At E16.5, lateral expression of Igf2 is downregulated, but expression in the GER, which is almost identical with expression for Igf1, persists. In addition, Igf2 is also broadly expressed in the surrounding mesenchyme (E, asterisks). By P0, expression of Igf2 is reduced in the mesenchyme and is more restricted within the GER (H, arrow). I–L, Similar views for expression of Igf1r. At E13.5 and E16.5, Igf1r is broadly expressed initially in the cochlear duct (I–K) and spiral ganglion (SG) (J, K, arrows). The white box indicates the region illustrated in the inset in J. The expression of Igf1r is still prominent in the spiral ganglion of the apical part at E16.5 (inset), while only weak expression of Igf1r is seen in the spiral ganglion of the basal turn (K, arrow). At P0, expression of Igf1r is largely restricted to the cochlear duct (L). M, qPCR data indicates a 180-fold increase in Igf1 expression in the cochlea between E13.5 and E16.5, and an additional 4.5-fold increase from E16.5 to P0. In contrast, expression of Igf2 and Igf1r was stable over the period between E13.5 and P0. Mean ± SE are shown. ANOVA followed by Tukey–Kramer's test, *p < 0.05. Scale bars: (in I, K, L) A, C–E, G–I, K, L, 50 μm; (in J) B, F, J, 200 μm; J, inset, 50 μm.
To examine changes in relative levels of expression of Igf1, Igf2, and Igf1r at E13.5, E16.5, and P0, qPCR was performed on cochlear tissue from each age. While the expression of Igf2 and Igf1r was constant across the three time points, expression of Igf1 increased significantly between E13.5 and E16.5 (180-fold) and continued to increase between E16.5 and P0 (an additional 4.5-fold) (Fig. 1M).
IGF signaling is required for normal morphogenesis of the inner ear
As elongation of the cochlear duct takes place between E12.5 and P0, and hair cell differentiation begins around E13.5, the expression data presented above suggest that the IGF pathway could be involved in one or more aspects of cochlear development or hair cell differentiation. To test this hypothesis, inner ear development was evaluated in Igf1r−/− mice. Igf1r−/− pups die immediately after birth as a result of respiratory failure; therefore, all analyses were made at embryonic stages. As previously reported, Igf1r−/− mice showed severe intrauterine growth deficiency only reaching one-half the size of heterozygous littermates (Liu et al., 1993). Consistent with the overall growth defect, the size of the bony labyrinth in Igf1r−/− mice was noticeably smaller by comparison with heterozygotes at E18.5 (Fig. 2A). In addition to the decreased size, the bony labyrinth in Igf1r−/− mice also appeared to be flattened, and the posterior canal was obscured.

Effects of deletion of Igf1r on inner ear morphogenesis. A, Gross morphology of the bony labyrinth in a control and an Igf1r−/− mutant at E18.5. The bony labyrinths in Igf1r−/− mutants show an overall growth deficiency as well as abnormal morphology including a flattened shape. The cochleae are at the top (brackets). B, C, Paint fills of the membranous labyrinth at E15.5 from control (B) and an Igf1r−/− mutant (C). Visualization of the membranous labyrinth indicates truncation of the lateral canal (asterisk), a hypomorphic posterior canal (arrowhead), and a shortened cochlear duct in the Igf1r−/− mutant. cd, Cochlear duct; Lc, lateral canal; Ac, anterior canal; Pc, posterior canal; ed, endolymphatic duct; sac, saccule; utr, utricle. D, E, Whole-mount preparations of the cochlear duct at E18.5 from a control (D) and an Igf1r−/− mutant (E) labeled with anti-Myosin6 (Myo6). The number of Myo6-positve hair cells is decreased in the Igf1r−/− cochlea. In addition, the length of sensory epithelium is shorter in the Igf1r−/− mutant. F, G, Whole-mount preparations of the cochlear duct at E18.5 from a control (F) and an Igf1r−/− mutant (G) labeled with anti-Prox1. The number of Prox1-positive supporting cells is decreased proportionally with the reduction in the total number of Myo6-positive hair cells. H, Confocal images of the surface of the sensory epithelium labeled with actin from basal, middle, and apical region of control and Igf1r−/− cochleae at E18.5. Cellular patterning is disrupted in the Igf1r−/− mutant, including misalignment of inner hair cells (arrows). A comparison between the morphological structure of the epithelium in the basal turn of the Igf1r−/− cochlea and the apical turn from a control suggests a possible developmental delay in the Igf1r−/− mutant. I, Cross-sections through the indicated maculae and cristae at E18.5 from controls and Igf1r−/− mutants labeled with Myo6 and actin. The development of hair cells in all the vestibular sensory epithelia of Igf1r−/− mutants is comparable with controls. Scale bars: A, 1 mm; B, C, 500 μm; D–G, 200 μm; H, 50 μm; I, 20 μm.
To confirm the vestibular phenotype in Igf1r−/− animals, the membranous labyrinth was visualized by paint fill at E15.5 (Fig. 2B,C). The utricle and saccule, as well as the anterior canal and crista, were present and appeared normal in Igf1r−/− mutants. In contrast, the lateral canal was truncated, although the lateral crista appeared to be nearly intact. Similarly, the posterior canal was hypomorphic in Igf1r−/− mutants, although the posterior crista appeared normal. The hypomorphic vestibular phenotypes for the lateral and posterior canals were fully penetrant in all mice examined in this study. Finally, the cochlear duct in Igf1r−/− mice appeared shortened and slightly wider by comparison with heterozygotes.
To examine the cochlear phenotype further, cochlear ducts were dissected from Igf1r−/− mutants and controls at E18.5, and hair cells were labeled by immunostaining for Myo6 (Fig. 2D,E). The overall length of the cochlear sensory epithelium was 33% shorter in Igf1r−/− mutants by comparison with heterozygous controls (4691 ± 157.7 μm in controls and 3148 ± 140.0 μm in Igf1r−/− mutants). Next, the total number of inner and outer hair cells was determined in whole-mount cochleae from controls and Igf1r−/− mutants. By comparison with controls, the number of both inner (634.0 ± 10.2 per control cochlea vs 462.6 ± 5.2 per Igf1r−/−) and outer (1763 ± 44.6 per control vs 1140 ± 40.7 per Igf1r−/−) hair cells was significantly decreased in Igf1r−/− mutants. The reduction in the overall number of hair cells correlated proportionally with the decrease in the length of the cochlear sensory epithelium, resulting in no significant change in the density of hair cells per unit length in Igf1r−/− mutants. To examine changes in supporting cell density, whole-mount cochleae were immunostained for Prox1, a marker for pillar cells and Deiters' cells (Bermingham-McDonogh et al., 2006), and total number of Prox1-positive cells in a whole cochleae was determined. By comparison with controls (3616 ± 88.2 Prox1-positive cells per cochlea), a significant decrease was observed in Igf1r−/− mutants (2745 ± 133 Prox1-positive per cochlea) (Fig. 2F,G).
Analysis of cellular patterning in Igf1r−/− mutants indicated a general decrease in organization that was present in all three turns of the cochlea (Fig. 2H). In particular, many examples of inner hair cells that were not aligned within a single row were observed. In addition, the lumenal surfaces of both inner and outer hair cells appeared smaller in Igf1r−/− mutants. These results are suggestive of a developmental delay in the Igf1r−/− cochlea, as illustrated by comparing cellular patterning in the base of the cochlea of an Igf1r−/− mutant with the apex of a control.
Finally, cross-sections through the vestibular organs were labeled with Myo6 and actin to examine the development of vestibular sensory epithelia (Fig. 2I). Although paint fill analysis of Igf1r−/− mutants indicated an absence of the lateral canal and hypoplasia of the posterior canal, all the vestibular sensory epithelia in Igf1r−/− mutants were present and showed no apparent defects by comparison with controls.
Deletion of Igf1r results in a delay in hair cell and stereociliary bundle development
To further characterize the defects/delays in hair cell development in Igf1r−/− mutants, we examined the expression of specific markers of hair cell development and the morphological development of stereociliary bundles. In controls, the prosensory marker Sox2 is downregulated in hair cells in the basal turn of the cochlea by E18.5 (Fig. 3A) (Dabdoub et al., 2008; Cotanche and Kaiser, 2010). However, in Igf1r−/− mutants, strong expression of Sox2 was still observed in hair cells at the same position along the duct (Fig. 3B). Conversely, Lhx3, a transcription factor that begins to be expressed in the nuclei of cochlear hair cells between E16.5 and P0 (Hertzano et al., 2007), was absent from hair cells in Igf1r−/− mutants at E18.5 (Fig. 3C,D). Finally, expression of the calcium-binding protein, Calbindin, begins in a gradient that extends along both the basal-to-apical and medial-to-lateral axes of the cochlea beginning around E17.5 (Buckiová and Syka, 2009). As a result, in controls at E18.5, both inner and outer hair cells in the base of the cochlea were strongly positive for Calbindin expression (Fig. 3E). In contrast, in Igf1r−/− mutants, Calbindin expression at a similar position within the cochlear duct was present in inner hair cells, but was only weakly expressed in outer hair cells (Fig. 3F).

Hair cell maturation is delayed in Igf1r−/− mutants. A, B, Cross-sections through the base of the cochlea at E18.5 labeled as indicated. Sox2 expression is normally downregulated in hair cell nuclei (A, arrows), but is still present in both inner and outer hair cells, shown as Myosin6 (Myo6)-positive, in Igf1r−/− mutants (B, arrows). C, D, In controls, Lhx3 is expressed in hair cell nuclei at E18.5 (C, arrows); however, no expression of Lhx3 is present in hair cells in the basal turn of Igf1r−/− mutants (D, arrows). E, F, By E18.5, both inner and outer hair cells in the base of the cochlea are strongly positive for Calbindin expression in controls (E, arrows). In contrast in Igf1r−/− mutants, only weak expression of Calbindin is observed in outer hair cells (F, arrows). G, H, Confocal images of the surface of the organ of Corti from the basal turn of control (G) and Igf1r−/− (H) cochleae at E18.5. The formation of kinocilia appears largely comparable, although hair bundles appear less organized in outer hair cells, and some variations in the position and orientation of stereociliary bundles are present in the third row of outer hair cells in the mutant (arrows). I, Quantification of stereociliary bundle orientation in the basal turn of control and Igf1r−/− cochleae plotted as average deviations from an alignment of 0° (see text for details). Average deviations are 4.01 ± 0.4 versus 9.32 ± 0.8 in inner hair cells (IHC), 3.91 ± 0.4 versus 7.99 ± 0.8 in the first row of outer hair cells (OHC1), 4.82 ± 0.5 versus 12.5 ± 1.2 in the second row of outer hair cells (OHC2), 10.4 ± 0.8 versus 17.6 ± 1.2 in the third row of outer hair cells (OHC3) (mean ± SE; controls vs Igf1r−/− mutants, respectively). The mean of variance in deviations for sterociliary bundle orientation is statistically significant in all cell types (Kolmogorov–Smirnov test, *p < 0.05). J–M, Scanning electron micrographs of IHC and OHC2 from the basal turn of the cochlea at E18.5 from control (J, L) and Igf1r−/− mutant (K, M). A defect in the lateral migration of developing kinocilia in both IHC and OHC2 is present in Igf1r−/− mutants. The resorption of the microvilli located inside the inverted v-shaped stereociliary bundle in OHC2 appears also delayed in Igf1r−/− mutants. Scale bars: A–F, 50 μm; G, H, 5 μm; J–M, 2 μm.
Next, the development of stereociliary bundles was studied using immunofluorescence and scanning electron microscopy (SEM) at E18.5. Visualization of developing kinocilia by immunostaining of acetylated α-tubulin (Piperno and Fuller, 1985) indicated that kinocilia formation was largely normal in Igf1r−/− mutants, although some variation was observed in lumenal position. In controls, kinocilia were uniformly located at the extreme lateral edge of each hair cell in a position that corresponded with the vertex of the stereociliary bundle. In contrast, in Igf1r−/− mutants, many kinocilia, while located at the circumference of a hair cell, were not positioned at the extreme lateral edge (Fig. 3G,H). Similar misorientations in stereociliary bundle orientation were also observed.
To determine the extent of variation in bundle orientation, average deviations were determined for hair cells located in the basal turn of the cochlea in each hair cell row. Significantly increased deviations were observed in Igf1r−/− cochleae for hair cells in each row (Fig. 3I). However, orientation of stereociliary bundles is a gradual process that progresses from inner hair cells toward the third row of outer hair cells (Dabdoub et al., 2003); therefore, the increased deviations in Igf1r−/− cochleae could be a result of a delay in maturation rather than a disruption in orientation.
Finally, SEM analysis confirmed defects in the lateral migration of developing kinocilia, the organization of the stereociliary bundle, and in the resorption of microvilli of outer hair cells, all of which are consistent with a defect in hair cell maturation (Fig. 3J–M).
Initial differentiation of hair cells is delayed in Igf1r−/− mutants
The results described above are consistent with the hypothesis that IGF signaling plays a significant role in the differentiation/development of cochlear hair cells. To determine the developmental time point at which disruption of hair cell development begins, we examined the expression of prosensory and hair cell differentiation markers at earlier time points in Igf1r−/− mutants. During normal development, the onset of Myo6 expression begins in the inner hair cells in the midbasal region of the cochlea around E14.5 (Xiang et al., 1998). Expression extends along both the basal-to-apical and medial-to-lateral axes such that, by E16.5, Myo6-positive inner hair cells are present along the length of cochlea and Myo6-positive outer hair cells are present along the basal half (Fig. 4A). In contrast, in Igf1r−/− mutants, expression of Myo6 was delayed in terms of both extension toward the apex of cochlea and the strength of expression in outer hair cells (Fig. 4B). The delay in the onset of expression of Myo6 was already evident 1 d earlier, at E15.5. At this point, inner hair cells in the basal and middle regions of the duct are already positive for Myo6 in controls (Fig. 4C), but no Myo6-positive cells were present in the cochlea of Igf1r−/− mutants (Fig. 4D). In contrast, the pattern of expression for the prosensory marker Sox2 appeared comparable in control and Igf1r−/− mutants.

Delayed development of cochlear hair cells and supporting cells in Igf1r−/− mutants. A, B, Whole-mount preparations of the cochlear duct from control (A) and Igf1r−/− mutant (B) labeled with Myosin6 (Myo6) at E16.5. Higher magnification images of the boxed regions in A and B are shown in A′ and B′, respectively. In control, Myo6-positive inner hair cells extend from the base to the midapical region and Myo6-positve outer hair cells are present along the basal half of cochleae. In contrast, the onset of expression of Myo6 in inner hair cells is delayed in the apical half of the cochlea (A, B), and the strength of Myo6 expression in outer hair cells is reduced in the basal part of Igf1r−/− mutants (A′, B′). C, D, Cross-sections through comparable regions of the basal turn of the cochlea from a control and an Igf1r−/− mutant at E15.5. By comparison with control (C, arrow), the onset of Myo6 expression is delayed in the inner hair cell of the Igf1r−/− mutant (D). In contrast, expression of Sox2 appears similar in both cross-sections, although downregulation in developing hair cells appears to be delayed. E, F, The expression of Prox1 was restricted to the nuclei of supporting cells in the basal turn of a control cochlea at E15.5 (E); however, the expression of Prox1 was still diffuse and expressed in both hair cell and supporting cell layers in an Igf1r−/− mutant (F). Higher magnification images of the prosensory region are shown in the insets in E and F. In contrast, the expression pattern for Jag1 in Igf1r−/− mutants appears comparable with controls. G, H, The extent and relative position of expression for p27Kip1 at E15.5 appears unchanged between control (G) and Igf1r−/− mutant (H). I, J, In situ hybridization for Atoh1 in cross-sections at E15.5. In control, expression of Atoh1 is restricted to the developing hair cells in the base of the cochlea (I), but in an Igf1r−/− mutant, Atoh1 is expressed diffusely within the prosensory region at E15.5 (J). Scale bars: A, B, 200 μm; A′, B′, 50 μm; C–J, 50 μm; E, F, insets, 10 μm.
Next, the onset of the late prosensory/supporting cell marker Prox1 was examined (Bermingham-McDonogh et al., 2006). In controls, a group of Prox1-positive supporting cells was already present in the base of the cochlea at E14.5 (data not shown), and expression of Prox1 was clearly restricted to the nuclei of pillar cells and Deiters' cells at E15.5 (Fig. 4E). But in a similar cross-section from an Igf1r−/− mutant at E15.5, Prox1 expression was weak and diffuse in the prosensory area with some Prox1 expression located in the hair cell layer (Fig. 4F). These results are consistent with a developmental delay and could also suggest a possible additional role for IGF signaling in supporting cell development. In contrast, the patterns of expression for the early prosensory markers Jag1 and p27Kip1 appeared unchanged between controls and Igf1r−/− mutants (Fig. 4E–H).
Finally, expression of mRNA for Atoh1 was examined by in situ hybridization. In controls at E15.5, expression of Atoh1 was restricted to developing hair cells in the base of the cochlea (Fig. 4I). But in Igf1r−/− mutants at a comparable developmental time point and location within the duct, Atoh1 was expressed diffusely within the prosensory region including progenitor cells within the supporting cell layer (Fig. 4J). This result, along with the delay in the restriction of Prox1 to supporting cells, suggests that there is a delay in the early sorting of progenitor cells into hair cells and supporting cells.
IGF signaling regulates hair cell, but not prosensory cell, development
The results presented above indicate a disruption in the development of hair cells, and possibly supporting cells, in Igf1r−/− mutants. However, formation of the prosensory domain appeared to be relatively normal. To confirm this, expression of the prosensory domain markers Sox2, Jag1, and p27Kip1 was examined at E13.5 in control and Igf1r−/− cochleae. Sox2 and Jag1 are expressed in the otocyst as early as E10.5 and play key roles in the establishment of the prosensory cell population (Kiernan et al., 2005, 2006; Brooker et al., 2006). In contrast, p27Kip1 is initiated in the apex of the cochlea at E12.5 and proceeds toward the base in a gradient that reaches the base of the cochlea between E13.5 and E14.5 (Chen and Segil, 1999; Löwenheim et al., 1999; Lee et al., 2006). Both whole-mount preparations and cross-sections indicated comparable expression of Sox2 throughout the entire sensory epithelium (Fig. 5A–D). Similarly, the extent and relative positioning of expression of Jag1 and p27Kip1 were unchanged between controls and Igf1r−/− mutants (Fig. 5E–H); however, it is important to note that the timing of the onset of these factors was not examined.

The prosensory domain is relatively normal in Igf1r−/− mutants. A, B, Whole-mount immunostaining for Sox2 in control (A) and Igf1r−/− cochleae (B) at E13.5. Despite the shortening of the cochlea in the Igf1r−/− mutant, Sox2 is expressed throughout the entire length of the cochlear prosensory domain. C–H, Cross-sections through the base of the cochlear duct at E13.5 labeled with prosensory markers. As was observed in the whole mounts, expression of Sox2 appears comparable between control (C) and Igf1r−/− mutant (D). Similarly, the extent and relative positioning of the expression domains for Jag1 and p27Kip1 also appear comparable between controls (E, G) and Igf1r−/− mutants (F, H). I, J, In situ hybridization for Atoh1 in cross-sections through the base of the cochlea at E13.5. Despite the normal formation of prosensory cells, the onset of expression of Atoh1 is delayed in the Igf1r−/− cochlea. While a broad and diffuse pattern of Atoh1 is observed in control (I, bracket), no expression of Atoh1 is observed in Igf1r−/− mutant (J). K, L, Analysis of expression of Atoh1 and prosensory markers at E13.5 by qPCR. Results indicate a significant decrease in expression of Atoh1 in Igf1r−/− mutants at E13.5. In contrast, expression of the prosensory markers, Sox2, p27Kip1, and Jag1, remained unchanged or increased in Igf1r−/− mutants (K). Similarly, expression levels of Atoh1 and Prox1 are significantly decreased in Igf1r−/− mutants at E14.5, while expression of Sox2 remains increased (L). Shown are mean ± SE. Unpaired t test, *p < 0.05. Scale bars: A, B, 200 μm; C–J, 50 μm.
Next, to examine the early specification of hair cells, the onset of expression of Atoh1 was examined by in situ hybridization. While a broad and diffuse pattern of Atoh1 expression was observed in the basal part of E13.5 controls, no expression of Atoh1 was present in Igf1r−/− mutants at the same time point (Fig. 5I,J).
Finally, to confirm the results of immunohistochemistry and in situ hybridization, expression of prosensory markers was examined by qPCR. Results indicated a significant decrease in Atoh1 expression in the cochlear sensory epithelium of Igf1r−/− mutants at E13.5, a significant increase in expression of Sox2, but no change in Jag1, or p27Kip1 (Fig. 5K). Similarly, at E14.5 expression of Atoh1 was still reduced in Igf1r−/− mutants, as was Prox1, but expression of Sox2 remained increased (Fig. 5L). The basis for this increase in unclear, but could reflect a delay in the downregulation of Sox2 that normally occurs in developing hair cells. These results suggest that prosensory formation is relatively normal in Igf1r−/− mutants, but subsequent development of hair cells and supporting cells is delayed and/or disrupted.
Deletion of Igf1r leads to a decrease in prosensory cell proliferation
Prosensory cells are known to exit the cell cycle in an apical-to-basal gradient that is essentially complete by E14.5 (Ruben, 1967). In addition, precise regulation of the initial number of cells within the prosensory domain has been shown to play a key role in the appropriate patterning of the organ of Corti (Chen and Segil, 1999; Löwenheim et al., 1999; Lee et al., 2006). Considering the decrease in the number of hair cells observed in Igf1r−/− mutants at E18.5, cellular proliferation and apoptosis within the Sox2-positive prosensory domain were assayed through immunohistochemistry for cleaved Caspase3, phospho-Histone H3, and Ki67 at E12.5 and E13.5. Results indicated no significant alteration in the number of cleaved Caspase 3-positive apoptotic cells between controls and Igf1r−/− mutants (data not shown). In contrast, quantification of the density of proliferating cells labeled with phospho-Histone H3 within the Sox2-positive domain indicated a significant decrease in Igf1r−/− mutants at both E12.5 and E13.5 (Fig. 6A–D). The density of proliferating cells in Igf1r−/− mutants was reduced to 69 and 54% of controls at E12.5 and E13.5, respectively (Fig. 6E). Similarly, strong expression of Ki67 was observed in the GER and medial edge of the Sox2-positive domain in the basal region of control cochleae at E13.5, whereas only weak expression of Ki67 was observed in the corresponding area in Igf1r−/− mutants (Fig. 6F,G), suggesting that the number of prosensory cells in the cell cycle is decreased in Igf1r−/− mutants. These data indicate a role for IGF signaling in prosensory cell proliferation and suggest that the decrease in the number of hair cells in Igf1r−/− mutants can be attributed, at least in part, to a reduction in the proliferation rate of prosensory cells.

The number of proliferating prosensory cells is decreased in the absence of IGF signaling. A–D, Cross-sections through the basal region of the cochlear duct at the indicated time points. Proliferating cells are labeled with anti-phospho-Histone H3 (pHH3), a specific marker for M-phase cells, and the prosensory domain is marked by expression of Sox2. Numerous pHH3-positive cells are present in the control cochlea at E12.5 (A). In contrast, the number of proliferating cells is reduced in the Igf1r−/− cochleae (B). At E13.5, proliferating cells are restricted to the medial edge of Sox2-positive domain in both control (C, arrows) and Igf1r−/− mutant (D, arrow), but the number of labeled cells is remarkably reduced in the mutant. E, Density of proliferating cells within the prosensory domain at E12.5 and E13.5 (for details, see Materials and Methods). At E12.5, mean density of proliferating cells in the prosensory domain is 4.54 ± 0.45 cells/10,000 μm2 in control, whereas in Igf1r−/− mutants the density is 3.13 ± 0.19. A similar decrease is also present at E13.5 (1.43 ± 0.16 and 0.77 ± 0.20 in controls and Igf1r−/− mutants, respectively). Shown are mean ± SE. Unpaired t test, *p < 0.05. F, G, Immunostaining for Ki67 in cross-sections of the basal region of the cochlea from control (F) and Igf1r−/− cochleae (G) at E13.5. Weakly Sox2-positive cells at the medial edge of the prosensory domain are strongly positive for Ki67 in the control cochlea (bracket), indicating active proliferation in this region. By comparison, Ki67 expression appears weaker in the same region of the duct in the Igf1r−/− mutant (bracket). Scale bars: A–D, F, G, 50 μm.
Pharmacological inhibition of IGF1R in vitro phenocopies the Igf1r−/− mutant
To examine the signaling pathways that are activated through IGF signaling, we used an in vitro system that would allow more specific control of the IGF pathway during cochlear development (Dabdoub et al., 2003). Two IGF1R antagonists with different mechanisms of action, PPP, which blocks phosphorylation of Y1136 within the IGF1R activation loop (Vasilcanu et al., 2004), and PQ401, which is thought to inhibit binding of ATP to IGF1R (Gable et al., 2006), were used. Cochlear explants were established from embryos at E13.5, treated with one of the IGF1R antagonists for 4 d, and then fixed and labeled with Sox2 and Myo6 to determine the effect on the size of the sensory domain (defined as the Sox2-positive area) and the number of hair cells (defined as the number of Myo6-positive cells).
Treatment with PPP resulted in a disorganization in hair cell patterning and overall cochlear development that was comparable with that observed in Igf1r−/− mutants (Fig. 7A,B). Exposure to PPP also caused a dose-dependent shortening in the length of the sensory domain and a reduction in the number of hair cells. In control explants, the average length of the sensory domain was 1417 ± 83.1 μm and the average number of hair cells was 1271 ± 69.7 (Fig. 7C,D). By contrast, at the maximal dose of PPP tested (5 μm), the length of the sensory domain was reduced to 503.9 ± 37.1 μm and the number of hair cells in the explants was reduced to 16.8 ± 2.7. We found that exposure to PPP at concentrations >5 μm led to a general toxicity because explants became detached from the underlying substrate. Significant reductions in the length of the sensory epithelium were observed at PPP concentrations of 500 nm or greater, and significant reductions in hair cell number were observed at PPP concentrations of 1 μm or greater.

Pharmacological inhibition of IGF1R in vitro phenocopies Igf1r−/− mutants. A, B, The IGF1R inhibitor PPP alters cochlear length and hair cell number. Explants of embryonic cochleae were established at E13.5 and maintained for 4 d, and hair cells were immunostained with Myosin6 (Myo6). Treatment with PPP causes a shortened length of the sensory epithelium and a loss in the number of hair cells. A′, B′, Confocal images in the boxes in A and B, respectively, showing an alteration in cellular patterning. In the basal part of a control explant, the typical one row of inner hair cells and three to four rows of outer hair cells were observed, whereas a PPP-treated explant showed misaligned inner hair cells and dispersed outer hair cells. C, D, Quantification of the length of the cochlear sensory epithelium and the total number of Myo6-positive hair cells in control (Ctrl) and PPP-treated explants. Treatment with PPP caused a dose-dependent decrease in both the length of the sensory epithelium (C) and the total number of hair cells (D). Shown are mean ± SE. ANOVA followed by Dunnett's test, *p < 0.05. E, F, Cochlear explants from E13.5 were cultured with IGF-1R inhibitor II (PQ401) for 4 d. E′ and F′ are confocal images of the regions boxed in E and F. Similar decreases in sensory length and total number of hair cells (F), as well as changes in cellular patterning (F′) were observed. G, H, Average length of the cochlear sensory epithelium and hair cell number in explants treated with PQ401 at the indicated concentrations. Shown are mean ± SE. ANOVA followed by Dunnett's test, *p < 0.05. I, J, Pharmacological inhibition of IGF1R affects development of supporting cells. The number of Prox1-positive supporting cells is decreased in a PPP-treated explant. I′, J′, Higher-magnification images taken from boxed region in I and J, showing that cellular patterning is altered. K, Expression of sensory markers is altered following 24 h treatment with PPP. Cochlear explants were treated with PPP for 24 h beginning on the indicated embryonic day, and levels of expression for the indicated genes were determined by qPCR. While expression of Sox2 did not change between controls (Ctrl) and PPP-treated explants at E14.5, expression of both Atoh1 and Prox1 was significantly decreased. Similar PPP treatments beginning at E16.5 did not cause significant changes in Atoh1 expression, although expression of Prox1 was still decreased. Shown are mean ± SE. Unpaired t test, *p < 0.05. Scale bars: A, B, E, F, I, J, 200 μm; A′, B′, E′, F′, I′, J′, 50 μm.
Explants treated with PQ401 (Fig. 7E,F) appeared similar to ones treated with PPP, and reductions in the length of the sensory domain and in the number of hair cells were comparable with the reductions observed in explants treated with PPP (Fig. 7G,H).
To determine whether treatment with PPP also affected supporting cell development, as was observed in Igf1r−/− mutants, the number of Prox1-positive cells was determined in explants treated with 1 μm PPP (Fig. 7I,J). In control explants, the average number of supporting cells was 2187 ± 127.5, while in PPP-treated explants the average number of supporting cells was significantly reduced to 1362 ± 130, suggesting that the development of supporting cells is also affected by pharmacological inhibition of IGF1R in vitro.
To further examine the results obtained by immunostaining, expression levels for Atoh1, Sox2, and Prox1 were obtained by qPCR for cochlear explants established at E14.5 and treated with PPP for 24 h. Results indicated significant decreases in the level of expression for Atoh1 and Prox1 but no change in the level of expression of Sox2 (Fig. 7K). Interestingly, similar PPP treatments beginning 2 d later, at E16.5, still resulted in a decrease in expression of Prox1, but expression of Atoh1, as well as Sox2, was unchanged. Expression of Atoh1 in cochlear explants was decreased to 9, 25, 50, and 81% of control by PPP treatment for 24 h beginning at E13.5, E14.5, E15.5, and E16.5, respectively. These results suggest that the role of IGF signaling in Atoh1 expression is limited to the early initiation phase. In contrast, the role of IGF signaling in supporting cell development, or at least in the expression of Prox1, appears to be more prolonged.
Finally, to determine whether the reduction in the number of hair cells and supporting cells in PPP-treated explants was attributable to cell death, the number of cleaved-Caspase 3-positive apoptotic cells within the Sox2-positive domain was quantified in control explants and explants treated with 1 μm PPP beginning on E13.5. No significant difference in the number of cleaved-Caspase 3-positive cells was observed after 1, 2, or 3 d in vitro (47.3 ± 2.3 vs 50.5 ± 2.7, at day 1; 67.0 ± 6.6 vs 76.5 ± 5.3, at day 2; 75.0 ± 6.0 vs 77.7 ± 3.5, at day 3; control vs PPP-treated, respectively). We also examined whether inhibition of IGF1R leads to any change in the rate of cellular proliferation within the prosensory domain. Addition of 1 μm PPP for 24 h beginning on E13.5 led to a significant decrease in the number of BrdU-positive cells within the Sox2-positive prosensory domain (114 ± 7.3 cells per control explants vs 61.2 ± 5.7 cells per PPP-treated explants). These results are consistent with results from the Igf1r−/− mutants and indicate an effect of PPP treatment on prosensory cell proliferation, but not on cell survival. However, since levels of Sox2 expression were unchanged between control and PPP-treated explants at E14.5, the decrease in proliferation is probably independent from the subsequent role of IGF signaling on cellular differentiation.
IGF signaling regulates the length of the cochlear epithelium and hair cell differentiation independently
Inhibition or inactivation of the IGF signaling pathway leads to both a decrease in the length of the sensory domain, probably as a result of decreased prosensory cell proliferation, and a decrease in the number of hair cells. Previous results have demonstrated that the number of cochlear prosensory cells is directly related to the termination of cellular proliferation by E14.5 (Ruben, 1967; Chen and Segil, 1999; Löwenheim et al., 1999; Lee et al., 2006), while hair cell specification and differentiation begin around E14 (Driver and Kelley, 2009; Puligilla and Kelley, 2009). Based on the effects of IGF signaling on prosensory proliferation, it seemed possible that the changes in hair cell number could be a secondary outcome of the decreased number of progenitor cells. To determine whether IGF signaling plays a direct role in both of these processes, IGF1R inhibition was initiated at different developmental time points in vitro. Explants were established at E13.5 (defined as day 0) and maintained for 2 d before addition of 5 μm PPP. Explants were maintained for another 2 d and then fixed. Delayed addition of PPP resulted in no obvious change in the length of the sensory epithelium; however, hair cell development in the apical half of the explant was disrupted (Fig. 8A,B). In addition, marked disruptions in cellular patterning, as seen in the failed alignment of hair cell rows, were evident in basal regions of the explants in which hair cells were present (Fig. 8A′,B′). Quantification indicated a significant decrease in hair cell number despite no change in the length of the sensory epithelium. The overall length of the cochlear sensory epithelium was 1330 ± 88.9 μm in controls and 1208 ± 77.6 in explants treated with PPP from day 2 (PPP-delayed). The average number of hair cells was 1106 ± 40.0 in controls, while in PPP-delayed explants the average number of hair cells was significantly reduced to 416.6 ± 60.8. Since IGF1R inhibition was not initiated until the equivalent of E15.5, well after formation and terminal mitosis within the prosensory cell population, these results indicate that IGF1R plays a direct role in hair cell differentiation, independent of its earlier role in prosensory cell proliferation.

Independent roles for IGF signaling in cochlear length and hair cell differentiation. A, B, Cochlear explants from E13.5 were maintained for 2 d before addition of the IGF1R inhibitor (PPP), and then maintained for another 2 d with PPP. Higher-magnification images taken from the boxed region in A and B are shown in A′ and B′, respectively. Delayed addition of PPP results in no change in the length of the sensory epithelium by comparison with control (A, B), but development of hair cells that express Myosin6 (Myo6) in the apical half of the explant is substantially inhibited. In addition, cellular patterning in the basal half of the explant is disorganized (A′, B′). C, D, Cochlear explants from E13.5 were cultured with PPP for 24 h followed by washout and a 72 h recovery period. C′ and D′ are higher-magnification images taken from the boxed region in E and F. Compared with controls (C), PPP (washed) explants (D) have a shorter sensory epithelium and a decreased number of hair cells. However, cellular patterning appears relatively normal (C′, D′). Scale bars: A–D, 200 μm; A′–D′, 50 μm.
To determine whether inhibition of IGF1R at the onset of sensory differentiation leads to permanent changes in hair cell development, explants were treated with 5 μm PPP for 24 h beginning on E13.5. PPP was then washed out, and explants were maintained for an additional 3 d in normal culture medium. Overall development of the sensory epithelium appeared relatively normal in these explants, including the formation of a normal pattern of hair cells and supporting cells (Fig. 8C,D). However, quantification indicated significant decreases in both the length of the sensory epithelium and in hair cell number. The average length of the cochlear sensory epithelium was 1432 ± 81.1 μm in controls, whereas the cochlear length was reduced to 882.0 ± 34.6 in explants treated with PPP just for the first 24 h (PPP-washed). Similarly, the average number of hair cells was reduced from 1241 ± 100 in controls to 669.4 ± 38.4 in PPP-washed explants. The decreases were not as pronounced as in explants that were treated with 5 μm PPP continuously (compare hair cell numbers above with Fig. 7C,D), suggesting that the transient inhibition of IGF1R can be compensated for to some degree during later developmental processes.
Together with the findings on both PPP-delayed and PPP-washed cochlear explants, these results are consistent with the hypothesis that IGF signaling directly regulates both the length of the sensory epithelium and the differentiation and patterning of hair cells independently.
PI3 kinase is a downstream target of IGF1R in the developing cochlea
The IGF signaling pathway has been shown to activate two major signaling cascades, the PI3K/Akt pathway and the MEK/Erk pathway (Annenkov, 2009). To determine which pathway is activated by IGF1R during cochlear development, we examined changes in the phosphorylation levels of Akt, a direct downstream target of PI3K, and Erk, a direct downstream target of MEK, in serum-starved cochlear cultures treated with 1 μg/ml IGF1 for 20 min. Addition of IGF1 induced a rapid increase in phospho-Akt (pAkt), indicating activation of PI3K, but no change was observed in the level of phospho-Erk (pErk1/2) (Fig. 9A). Similarly, inhibition of IGF1R by treatment with 5 μm PPP for 1 h led to a decrease in the level of pAkt but did not change the amount of pErk1/2 (Fig. 9B).

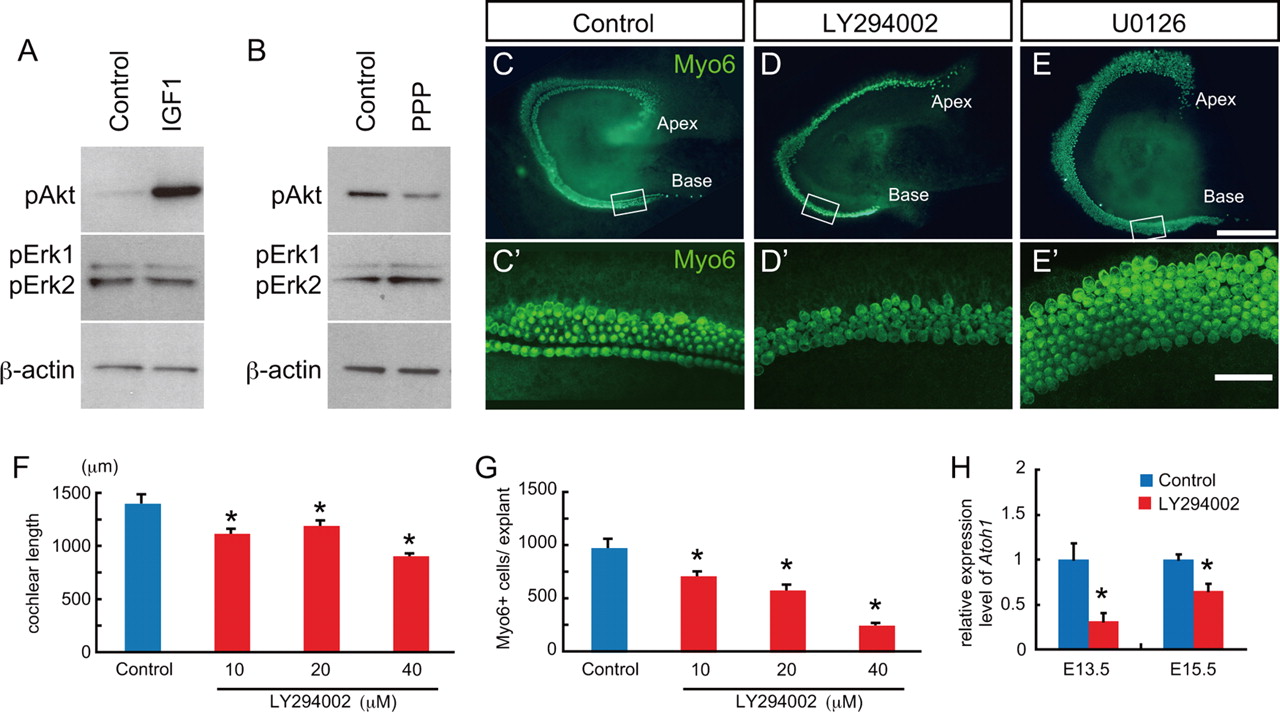
PI3-kinase is a downstream target of IGF1R in the developing cochlea. A, B, Western blots for potential downstream targets of IGF1R. Explants from E14.5 were treated with IGF1 at 1 μg/ml for 20 min and then probed with antibodies against phospho-Akt (pAkt) or phospho-Erk1/2 (pErk1/2) (A). The amount of pAkt is markedly increased in the presence of IGF1. In contrast, pErk1/2 was unchanged by IGF1. Similarly, Western blots from cochlear explants treated with or without 5 μm the IGF1R inhibitor (PPP) for 1 h indicate a decrease in pAkt in the presence of PPP but no change in the levels of pERK1/2. Anti-β-actin was used as a loading control for each experiment. C–E, Pharmacological inhibition of the PI3K/Akt pathway recapitulates the phenotype of Igf1r−/− cochleae. C′–E′ are confocal micrographs corresponding to the areas boxed in C–E. Cochlear explants from E13.5 were cultured with a specific inhibitor for either PI3K, LY294002, or MEK1/2, U0126, for 4 d. Control explants show an organized pattern of inner and outer hair cells labeled with Myosin6 (Myo6) (C, C′), while LY294002-treated explants demonstrate shortened cochlear sensory epithelia and disorganized patterning of hair cells (D, D′). In contrast, in cochlear explants treated with 10 μm U0126, the gap between inner and outer hair cells is missing, but the length of sensory epithelium is comparable with control, and the number of hair cell appears to be slightly increased (E, E′). F, G, Quantification of cochlear length (F) and number of hair cells (G) in response to different dosages of LY294002. Both were significantly reduced at all concentrations tested. Shown are mean ± SE. ANOVA followed by Dunnett's test, *p < 0.05. H, qPCR for Atoh1 expression in cochlear explants treated with 40 μm LY294002 for 24 h beginning at E13.5 or E15.5. Results indicate a significant downregulation of Atoh1 in explants treated with LY294002 at both time points. Shown are mean ± SE. Unpaired t test, *p < 0.05. Scale bars: C–E, 200 μm; C′–E′, 50 μm.
Next, cochlear explants established on E13.5 were exposed to a specific inhibitor for either PI3K, LY294002, or MEK, U0126 (Montcouquiol and Corwin, 2001), and changes in length of the sensory epithelium and hair cell numbers were assayed as described. Treatment with LY294002 resulted in mild shortening of the sensory epithelium, as well as a noticeable decrease in the number of hair cells (Fig. 9C,D). In contrast, cochlear explants exposed to U0126 showed no significant change in either the length of the sensory epithelium or hair cell number, although there was a trend toward increased hair cells (Fig. 9E). In addition, the gap between inner and outer hair cells was absent, suggesting a disruption in the development of the pillar cells. This phenotype is consistent with previous reports demonstrating a role for Fgfr3, a known activator of the MEK/Erk pathway, in both pillar cell development and inhibition of hair cell formation (Mueller et al., 2002; Hayashi et al., 2007; Jacques et al., 2007; Puligilla et al., 2007).
Quantification of the effects of LY294002 at different concentrations indicated a mild, but significant, effect on the overall length of the sensory epithelium as well as a more potent effect on the number of cells that developed as hair cells (Fig. 9F,G). These results were comparable with those observed in the presence of the IGF1R inhibitors. The mean length of the sensory epithelium was 1212 ± 21.1 μm in controls and 1328 ± 52.0 in U0126-treated explants, the average number of total Myo6-positive hair cells was 1123 ± 51.6 in controls and 1417 ± 137 in U0126-treated explants. Although quantification of the effects of U0126 demonstrated increases in both the cochlear length and the total number of Myo6-positive hair cells in U0126-treated explants, the differences were not significant (p = 0.108 in the cochlear length, and p = 0.119 in the number of hair cells).
Finally, qPCR data demonstrated a significant downregulation in expression of Atoh1 when explants established at either E13.5 or E15.5 were treated with LY294002 for 24 h (Fig. 9H). These results are consistent with the hypothesis that IGF signaling acts via the PI3K/Akt pathway to regulate several aspects of cochlear development, including expression of Atoh1.
Discussion
Previous studies had demonstrated a role for Igf1 in hearing and in the postnatal development of the auditory system, with particular roles in the survival of spiral ganglion neurons, maturation of the tectorial membrane, and increases in cochlear volume (Camarero et al., 2001). However, most of these changes were reported to occur after P5. Here, we demonstrate a much more profound role for IGF signaling with alterations in development evident as early as E13. Phenotypes were more widespread and included changes in both vestibular and cochlear structures. The underlying bases for the more extensive phenotypes are not entirely clear, but are most likely related to the deletion of Igf1r in this study as opposed to deletion of Igf1 in the previous work. As discussed, expression of Igf2 in the cochlea overlaps with Igf1, suggesting possible functional compensation in Igf1−/− mutants, or alternatively, unique roles for IGF2. Existing data have demonstrated specific roles for IGF2, in particular in regulation of cellular proliferation during placental growth and development of the heart (Constância et al., 2002; Li et al., 2011).
IGF signaling during initial cochlear development
Initial studies on the effects of activation of the IGF signaling pathway focused on promotion of cellular proliferation (Baker et al., 1993; Liu et al., 1993). However, more recent findings have linked IGF signaling with pleiotropic effects including cellular differentiation, axon outgrowth and polarity, cell migration and patterning, and cell survival (Fukudome et al., 2003; Vicario-Abejón et al., 2003; Ye and D'Ercole, 2006; Scolnick et al., 2008). The results presented here are consistent with a broader role for IGF signaling as many of the changes observed in the inner ears of Igf1r−/− mutants are not consistent with a decrease in proliferation.
A particularly intriguing aspect of the effect of the deletion of Igf1r was the disruption in the transition between uncommitted prosensory cells and differentiating hair cells and supporting cells. Recent work has demonstrated that an important step in cochlear development is the transition of proliferative Sox2-positive prosensory cells to a nonmitotic progenitor state in which the cells are poised to differentiate (Lee et al., 2006; Kwan et al., 2009). Two key factors in this transition are the cell cycle inhibitor p27Kip1, which regulates the temporal and spatial pattern of terminal mitoses (Chen and Segil, 1999; Löwenheim et al., 1999), and the transcription factor Atoh1, which regulates the initiation of hair cell differentiation (Bermingham et al., 1999; Woods et al., 2004). The precise mechanisms underlying the temporal separation between cell cycle exit and cell fate decision during cochlear development have not been identified, but, as will be discussed below, the results presented here suggest that IGF signaling could play a role in this process.
As IGF signaling is known to regulate cellular proliferation during development (D'Ercole et al., 1996; Aberg et al., 2000) and in some forms of cancer (Fürstenberger and Senn, 2002; Bruchim et al., 2009), the decrease in cellular proliferation observed in cochleae from Igf1r−/− mutants was not unexpected. Considering the role of cellular proliferation in growth, this decrease may account, at least in part, for the observed shortening of the cochlear duct. However, the presence of shortened sensory epithelia in explants treated with an IGF1R antagonist beginning at E13.5, only a half day before terminal mitoses (Ruben, 1967; Lee et al., 2006), suggests that changes in proliferation alone may not be sufficient to explain the shortened phenotype. Despite the decrease in the rate of cellular proliferation, the timing and pattern of expression for p27Kip1 appeared relatively normal in Igf1r−/− mutants, suggesting that the timing of cell cycle exit is normal. These results are consistent with a role for IGF signaling in the rate of proliferation, but not in the timing of terminal mitosis.
While expression of prosensory markers, as well as p27Kip1, was not altered in Igf1r−/− mutants, expression of Atoh1 and the supporting cell marker Prox1 were significantly downregulated. These results suggest that IGF signaling does not act as a nonspecific regulator of the overall pace of development. Instead, IGF signaling seems to act specifically to regulate the timing of developmental events that occur after the terminal mitosis of prosensory cells. In particular, the onset of Atoh1, which regulates hair cell specification and differentiation and the restriction of Prox1 to support cells, are altered in the absence of IGF signaling. Changes were also observed in the expression of Lhx3 and Calbindin, which could be a result of a direct effect of IGF signaling on these factors, suggesting a general role in the pace of development after prosensory cell formation, a result that is supported by the defects observed in the base of cochlear explants in which IGF signaling was normal until the equivalent of E15.5 (Fig. 8B).
Regulation of Atoh1 expression in the developing cochlea
As discussed, Atoh1 is a major determinant of hair cell specification and acts very early in the molecular cascade that leads to hair cell formation. Within the cochlea, the onset of Atoh1 expression follows a stereotypical pattern that initiates near the base of the cochlea and extends rapidly toward the apex. Considering the importance of Atoh1 expression in hair cell development and regeneration, significant effort has been applied toward the identification of factors that can positively or negatively regulate Atoh1. Previously, Sox2, Notch signaling, and FGF signaling have been shown to positively regulate expression of Atoh1 (Pirvola et al., 2002; Kiernan et al., 2005, 2006; Hayashi et al., 2008). Moreover, a recent study (Shi et al., 2010) demonstrated that β-catenin signaling upregulates Atoh1 expression in neural progenitor cells possibly through an interaction with Notch signaling. The results presented here add IGF signaling, and most likely PI3K/Akt signaling, to the list of known Atoh1 regulators. While Atoh1 was still expressed and many hair cells formed in Igf1r−/− mutants, the timing and overall level of Atoh1 was retarded. Neither IGF nor PI3K/Akt signaling has been implicated in the direct regulation of Atoh1; however, PI3K/Akt signaling has been shown to regulate expression of other basic helix-loop-helix molecules, including Ngn1 (Vojtek et al., 2003), suggesting a possible regulatory pathway. Alternatively, IGF and/or PI3K/Akt signaling could indirectly regulate Atoh1 expression through one of its known targets, such as GSK3β, a known modulator of canonical Wnt signaling, which has been implicated in development of the cochlea (Sienknecht and Fekete, 2008).
The role of IGF signaling in auditory function and inner ear morphogenesis
As discussed, defects in IGF signaling have been shown to lead to hearing deficits in humans and mice (Woods et al., 1996; Bonapace et al., 2003; Cediel et al., 2006). In mouse models, Camarero et al. (2001) reported a decrease in the number of spiral ganglion neurons in Igf1−/− mutant mice as a result of apoptotic cell death beginning after P20. The basis for the loss of spiral ganglion neurons was unclear since the overall structure of the organ of Corti appeared largely normal in Igf1−/− mutants. However, considering the delay in hair cell development and the overall decrease in total number of hair cells in Igf1r−/− mutants, it seems possible that defects in hair cell development, such as insufficient production of neurotrophins, could lead to the subsequent loss of spiral ganglion neurons in Igf1−/− mutants. It should be noted that a recent examination of changes in gene expression between wild-type and Igf1−/− cochleae did not note changes in neurotrophin expression, but the time points analyzed were before the onset of neuronal degeneration (Sanchez-Calderon et al., 2010). Unfortunately, Igf1r−/− mutants die at birth, preventing a subsequent analysis of inner ear; however, conditional deletion of Igf1r using an inner ear-specific driver could be used to analyze postnatal cochlear development and hearing function in the absence of Igf1r.
In addition to defects in cochlear development, inner ears from Igf1r−/− mutants also had a phenotype consistent with Mondini dysplasia, one of the most common congenital anomalies of the human inner ear. Mondini dysplasia is a complex malformation that includes a flattened and shortened cochlear duct, dilated vestibular aqueduct, and, in some cases, other vestibular malformations. It has been suggested that Mondini dysplasia should be regarded as arising as a result of fetal growth retardation (Barrenäs et al., 2005), which is consistent with some of the known effects of IGF1R deficiency, but the fact that similar phenotypes have been reported in mice with mutations in FoxG1, Six1, or Eya1 (Zheng et al., 2003; Pauley et al., 2006) suggests that the Mondini dysplasia reflects a specific pathological phenotype and that IGF signaling could interact in a common pathway with FoxG1, Six1, and Eya1.
In summary, the results presented here demonstrate novel roles for IGF signaling in the development of the inner ear. In Igf1r−/− mutants, the cochlear duct is shortened, possibly as a result of a decreased rate of proliferation, but prosensory formation appears to proceed normally through expression of p27Kip1. In contrast, significant defects occur at the transition between uncommitted prosensory cell and differentiating precursor. The onset of Atoh1 expression is delayed in Igf1r−/− mutants, which results in defects in hair cell formation and subsequent cellular patterning. In vitro experiments demonstrate that these effects are independently regulated through IGF signaling and that the PI3K/Akt pathway acts as a primary mediator of IGF1R within the cochlea. The results provide new insights regarding the pathological processes that underlie auditory defects in the absence of IGF signaling in both humans and mice.
Footnotes
This work was supported by funds from the NIH Intramural program (M.W.K.) and by a grant from the Japan Society for the Promotion of Science Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH (T.O.). We thank Drs. Doris Wu and Lisa Cunningham for helpful comments on this manuscript. We are also grateful to Dr. Weise Chang for his excellent technical assistance. We thank Dr. Helen May-Simera for assistance with scanning electron microscopy and Dr. Junko Okano for critical discussion.
The authors declare no competing financial interests.
- Correspondence should be addressed to Matthew W. Kelley, Laboratory of Cochlear Development, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Porter Neuroscience Research Center, 35 Convent Drive, 2A-100, Bethesda, MD 20892-3729. kelleymt{at}nidcd.nih.gov





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}