

Abstract
The loss of motor neurons (MNs) is a hallmark of the neuromuscular disease spinal muscular atrophy (SMA); however, it is unclear whether this phenotype autonomously originates within the MN. To address this question, we developed an inducible mouse model of severe SMA that has perinatal lethality, decreased motor function, motor unit pathology, and hyperexcitable MNs. Using an Hb9-Cre allele, we increased Smn levels autonomously within MNs and demonstrate that MN rescue significantly improves all phenotypes and pathologies commonly described in SMA mice. MN rescue also corrects hyperexcitability in SMA motor neurons and prevents sensory-motor synaptic stripping. Survival in MN-rescued SMA mice is extended by only 5 d, due in part to failed autonomic innervation of the heart. Collectively, this work demonstrates that the SMA phenotype autonomously originates in MNs and that sensory-motor synapse loss is a consequence, not a cause, of MN dysfunction.
Introduction
The survival motor neuron 1 (SMN1) gene encodes a protein that is essential for spliceosome assembly (Schrank et al., 1997; Pellizzoni et al., 1998). While complete loss of SMN is lethal to all cells, low levels of SMN cause the neuromuscular disease spinal muscular atrophy (SMA) (Lefebvre et al., 1995; Crawford and Pardo, 1996; Coovert et al., 1997; Lefebvre et al., 1997). The pathology of SMA is paradoxical because SMN is ubiquitously expressed in all cells, yet motor neurons (MNs) preferentially degenerate.
SMA has been modeled in several organisms to address SMN′s cellular requirements. In zebrafish and Caenorhabditis elegans, restoring SMN in MNs rescues neuromuscular junction (NMJ) pathology, while in Drosophila, both MN and muscle rescue are required to correct pathology (Chang et al., 2008; Boon et al., 2009; Briese et al., 2009). Survival analysis in these models is complicated, since they rely on either maternal transcripts or temporally discordant drivers to accomplish SMN rescue.
Conflicting evidence exists regarding whether neuromuscular dysfunction in SMA mice originates in MNs. Gavrilina et al. (2008) demonstrated that CNS-specific SMN restoration rescued the phenotype of a severe SMA mouse model, albeit with leaky expression in skeletal muscle. Importantly, muscle specific rescue where there was no leakiness in the CNS had no survival benefit.
In contrast, mice with disease-relevant levels of SMN only within MNs and oligodendrocytes do not develop severe SMA, as one would expect from a cell-autonomous disease, but instead develop an adult form of SMA (Park et al., 2010). This suggests an additive involvement of muscle, Schwann cells, and/or peripheral factors in defining disease severity. Alternatively, these results could be due to inefficient Olig2-Cre-mediated recombination, or an unknown disease modifying role of Olig2+ oligodendrocytes (Park et al., 2010). To date, genetic experiments in mice have failed to exclusively target motor neurons, making it difficult to draw conclusions pertaining to MN-autonomous disease mechanisms.
An important recent finding is that a loss of the central synapses, particularly the proprioceptive inputs onto MN somata and proximal dendrites, both parallels and precedes the pattern of MN loss in SMA mice (Ling et al., 2010; Park et al., 2010; Mentis et al., 2011). Mechanistically, this could indicate that defects within afferent neurons play a causative role in disease that is external to the MN (Mentis et al., 2011). If true, a simplistic interpretation would be that MNs are not affected by low Smn levels and would not benefit from autonomously increased Smn dosage; rather MNs are relatively healthy cells, whose dysfunction and loss is attributed to aberrant synaptic input.
Here, we directly address the question of MN specificity by generating a Cre-inducible mouse model of severe SMA. Using an Hb9-Cre allele, we demonstrate that MN rescue dramatically improves all neuromuscular phenotypes and pathologies observed in severe SMA model mice; however, survival is only extended 5 d due in part to failed autonomic innervation of the heart. This work demonstrates that the neuromuscular phenotype in SMA mice autonomously originates in MNs, while their lethality is multifactorial.
Materials and Methods
Ethics statement.
All studies performed on mice were in accordance with the Institutional Animal Care and Use Committee regulations in place at Children's Memorial Research Center and Northwestern University's Animal Care and Use Committee guidelines, and were specifically approved under Protocols 2006-22 and 2008-03.
Animal models.
Animals were kept in a controlled vivarium at 25°C and 50% humidity with a 12 h light/dark photoperiod and monitored for health. The official strain designations for the alleles used in this manuscript are as follows: SMN2, FVB.Cg-Tg(SMN2)89Ahmb Smntm1Msd; Smn2B-Neo, B6.127-Smn1tm1Cdid; Smn2B, B6.127-Smn1tm1.1Cdid; Hb9-Cre, B6;129S-Hlxb9tm4(cre)Tmj; ZGFP, B6.Cg-Tg(ACTB-Bgeo/GFP)21Lbe (Arber et al., 1999; Monani et al., 2000; Novak et al., 2000; Hammond et al., 2010). The official name for the severe inducible model reported here [(SMN2)89Ahmb+/−, Smn2B-Neo/2B-Neo] is Tg(SMN2)89Ahmb/J, Smn1tm1Cdid/ tm1Cdid.
Severe inducible SMA pups were generated by crossing a breeder stock of SMN2+/+; Smn2B-Neo/+ SMA mice with a Smn2B-Neo/+ mouse to create SMN2+/−; Smn2B-Neo/2B-Neo SMA pups. For MN-rescue experiments, an Hb9-Cre+/− mouse was intercrossed with a Smn2B-Neo/+ heterozygote. F1 progeny that were positive for both the Smn2B-Neo and the Hb9-Cre alleles were backcrossed to the SMN2+/−; Smn2B-Neo/+ breeder stock to create SMA mice containing both with (MN-rescued SMA) and without (severe inducible SMA) Hb9-Cre. In all of the described experiments, both male and female pups were used in proportions approximated to be equal.
Genotyping.
The presence of the neomycin cassette was verified via PCR using a forward primer within neomycin (5′-gcattaaagcttggctggac) and a reverse primer in Smn intron 7 (5′-gagaccgaggcaggctaac) that amplifies a neomycin specific band of 353 bp (Smn2B-Neo). The addition of a third primer within intron 6 (5′-tcccaggcagttttagactca) allows for the detection of a WT band of 750bp (Smn+). When the neomycin cassette has been removed by Cre-recombinase, a third 850 bp band corresponds to the excised allele (Smn2B). The presence of the Hb9-Cre allele was detected using generic Cre primers (forward, 5′-atgtccaatttactgaccg; reverse, 5′-cgccgcataaccagtgaaac). Assessment of the (SMN2)89Ahmb allele was conducted as described previously (Gavrilina et al., 2008).
Phenotyping.
In all phenotypic assays, the researcher recording the score was blinded to the genotype of the litter being assessed. Tube test was conducted as described by Heier et al. (2010a). Righting reflex was monitored on odd days beginning at postnatal day 3 (P3) and defined as the time taken to turn over and place all four paws simultaneously on a hard benchtop after being placed in supine position. If righting did not occur in, 1 min the test was scored as a failure. Electrocardiography (ECG) was conducted as described by Heier et al. (2010b). The end stage was defined as 2 consecutive days of weight loss and an inability to right within 60 s in cage bedding.
Western blot analysis.
To quantify Smn levels, 5 μg of total protein per tissue was resolved on a NuPage Novex 4–12% Bis-Tris gel and transferred to nitrocellulose membrane. Smn was detected by Western blot using the Li-COR Odyssey system as described previously (Hammond et al., 2010).
Immunofluorescence.
NMJ staining was performed on whole-mount intercostals (ICs) and triangularis sterni (TS) using a combination of neurofilament [1:100; 2H3; Developmental Hybridoma Bank (DHB)], synaptic vesicle 2 (SV2; 1:10; DHB), and rhodamine conjugated α-bungarotoxin (αBTX; 1:250; Invitrogen). Briefly, whole-mount tissues were dissected and fixed in 2% paraformaldehyde overnight at 4°C. The following day, they were washed in PBS for 2 h at room temperature (RT). Primary antibodies (NF and SV2) were then added in blocking buffer (5% bovine serum, 2% Triton X-100, 0.5% glycine in PBT). Samples were incubated in primary for 48–72 h at RT on a rocker, washed three times for 10 min each in PBS, and incubated in secondary antibody (1:250, GAM-488 and 594-αBTX) for 24 h at RT. Samples were again washed three times for 10 min each in PBS and mounted on slides with Clear Mount mounting media (Invitrogen).
For MN staining, spinal cords were dissected, fixed in 2% PFA at 4°C overnight, cryoprotected in 30% sucrose for 12–24 h, and embedded in OCT. Serial sections (12 μm thick) were collected, and to avoid repeat counts, every 15th section (180 μm apart) was used to quantify MN number. Before ChAT and Islet-1/2 staining, one slide per series was stained with fluorescent Nissl (0.5 mg/ml cresyl violet) to assist in determining the exact location within the spinal cord. Following antigen retrieval, equivalent sections from an adjacent slide were stained in 5% donkey serum for ChAT (Millipore Bioscience Research Reagents; 1:100) and Islet-1/2 (DHB; 4D5, 1:100 plus 2H3, 1:100). Only double-positive cells localized in the ventral horn were classified as MNs. Tyrosine hydroxylase (TH), vesicular glutamate transporter 1 (vGlut1), and Smn staining, as well as Smn gem counts were conducted as described previously (DiDonato et al., 2003; Heier et al., 2010a; Ling et al., 2010; Park et al., 2010; Le et al., 2011).
NMJ innervation/maturity quantification.
All NMJs were imaged whole mount on a Leica spinning disc confocal microscope using Slidebook software. Z stacks were deconvolved in Slidebook, and the projection image was used for final analysis. Images were de-identified and randomized before quantification for blinding purposes. Blinded images were assessed for innervation and maturity using Adobe Photoshop, CS4 extended edition. Innervation status was defined visually as fully innervated (>80%), partially innervated (15–80%), or denervated (≤15%). Postsynaptic maturity was defined as junctions containing homogeneous plaques, folds, perforations, or secondary structure. Only after quantification was complete were the images reidentified for statistical analysis.
Electrophysiology.
Mice at P4–P5 were deeply anesthetized with isofluorane, decapitated, and eviscerated. The spinal cord was quickly removed and embedded in 2.5% w/v agar, and super glued to a stainless-steel platform, and 350 μm transverse slices were made using a Leica 1000 vibratome as described previously (Quinlan et al., 2011). During both spinal cord isolation and slicing, the spinal cord was immersed in 1–4°C high-osmolarity dissecting solution containing the following (in mm): 234 sucrose, 2.5 KCl, 0.1 CaCl2 · 2H2O, 4 MgSO4 · 7H2O, 15 HEPES, 11 glucose, and 1 Na2PO4, pH 7.4 when bubbled with 95% O2/5% CO2. After cutting, the slices were incubated for >1 h at 30°C in incubating solution containing the following (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2 · 2H2O, 2 MgCl2 · 6H2O, 26 NaHCO3, and 10 glucose, pH 7.4 when bubbled with 95% O2/5% CO2. During recording, slices were perfused at a rate of 2.5–3.0 ml/min with a modified Ringer's solution containing the following (in mm): 111 NaCl, 3.09 KCl, 25 NaHCO3, 1.10 KH2PO4, 1.26 MgSO4, 2.52 CaCl2, and 11.1 glucose. Whole-cell patch-clamp recording was performed using 1–4 MΩ glass electrodes containing the following (in mm): 150 Texan red dextran (3000 molecular weight; Invitrogen), 138 K-gluconate, 10 HEPES, 5 ATP-Mg, and 0.3 GTP-Li (all from Sigma). Electrodes were positioned using a Sutter Instrument MP-285 motorized micromanipulator. Whole-cell patch-clamp measurements were performed at room temperature using the Multiclamp 700B amplifier (Molecular Devices).
Only neurons maintaining a resting potential of −50 mV or lower, an action potential height of >0 mV, a series resistance of ≤25 MΩ, and an input resistance of ≤200 MΩ were included in this study. Recordings were performed in current- and voltage-clamp modes [more details available in the study by Quinlan et al. (2011)]. In voltage-clamp mode, holding potential was set to −90 mV, and neurons were subjected to a slow, depolarizing voltage ramp of 12 mV/s, bringing the cell to +6 mV in 8 s, and then back to the holding potential in the following 8 s. Persistent inward current (PIC) amplitudes were measured from leak-subtracted current traces. In current-clamp mode, depolarizing ramps were used for testing ION (the current level at firing onset), IOFF (the current level at cessation of firing), and the frequency–current relationship. Characteristics of the action potential, including overshoot (past 0 mV), duration at half peak, and rate of rise were measured from the first spike elicited from the current ramp. Threshold voltage was determined as the voltage at which the rate of rise of the membrane potential exceeded 10 mV/ms, indicating the beginning of the spike. Synaptic potentials were quantified as the number of sharp deflections in membrane potential (either depolarizing or hyperpolarizing) occurring over 5–10 s. Data was collected on Winfluor software (University of Strathclyde, Glasgow, Scotland) and analyzed using Spike2 software (Cambridge Electronic Design). Z stacks were obtained using a Bio-Rad 2100 MPD laser scanning microscope. The Coherent Chameleon Ultra II laser was used at 920 nm. Bio-Rad LaserSharp software was used to collect 1024 × 1024 pixel Z stacks, up to 200 μm depth, in 1.0 μm steps, and using a Kalman 4 filter. Image analysis was performed using ImageJ (NIH).
Statistical analysis.
Statistical analysis was analyzed using Graph Pad Prism software (version 4.0) and Microsoft Excel. Kaplan–Meier survival curves were compared using log-rank tests. Significance in weights, tube test scores, innervation status, and postsynaptic maturity was determined using either a one- or two-way ANOVA with a Bonferroni post hoc comparison or Student's t test as appropriate. Significance pertaining to MN counts and heart rate was analyzed with a Student's t test.
Results
Smn inducible model of severe SMA
Smn inducible SMA mice were generated by breeding the Smn1tm1Cdid allele (hereafter referred to as Smn2B-Neo) with the commonly used Tg(SMN2)89Ahmb allele (hereafter referred to as SMN2) (Monani et al., 2000; Hammond et al., 2010). Smn2B-Neo/2B-Neo embryos die by E9 due to low Smn protein levels (1–3%), but Smn dosage can be dramatically increased if the floxed neomycin cassette is removed by Cre-recombinase [discussed extensively in the study by Hammond et al. (2010)]. By breeding one copy of the SMN2 transgene to a Smn2B-Neo/2B-Neo background (SMN2+/−; Smn2B-Neo/2B-Neo), we increased Smn levels to 6% relative to SMN2+/+; Smn+/+ controls (Fig. 1A). Importantly, this rescued the lethality observed in Smn2B-Neo/2B-Neo embryos and allowed for postnatal survival.

Phenotypic characterization of severe inducible SMA mice. A, Western blot analysis of Smn levels. The homozygous presence of the neomycin cassette decreases Smn to 6% relative to littermate controls. This level is increased to an asymptomatic 37% if one neomycin-containing allele is replaced with a rescued allele. B, Photomicrograph. Severe inducible SMA mice were smaller and weaker in appearance than littermate controls and exhibited near complete paralysis by P5. The ruler is in centimeters. C, Kaplan–Meier survival curve. Median survival of severe inducible SMA mice is 7 d (control, n > 50; SMA, n = 15; p ≤ 0.001, log-rank test). D, SMA mice weighed significantly less than controls (P2 or later; p ≤ 0.05) and generally died after 2 consecutive days of weight loss (control, n = 16; SMA, n = 14; 2-way ANOVA, Bonferroni's post-test). E, Tube test scores in severe SMA mice were significantly diminished from the earliest detectable points (P2 or later; p ≤ 0.01; 1-way ANOVA, Bonferroni's post-test; control, n = 22; SMA, n = 15). F, SMA mice were bradycardic from the earliest point analyzed by ECG (P3; p ≤ 0.04; Student's t test; control, n = 10; SMA, n = 7). Values are shown as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Severe inducible SMA mice have a neuromuscular phenotype
SMN2+/−; Smn2B-Neo/2B-Neo (hereafter referred to as severe inducible SMA) pups exhibited phenotypes similar to established severe models of SMA, including reduced survival (median survival, 7 d; p = 0.001), reduced weight (P2 or later; p ≤ 0.05), and diminished motor function (as quantified by tube test; P2 or later; p ≤ 0.01) (Fig. 1B–E) (Hsieh-Li et al., 2000; Monani et al., 2000; Le et al., 2005). Severe inducible SMA pups had near complete paralysis by P5 and never developed the ability to right when placed on their back. To assess overall health, we used electrocardiography (ECG) and found that severe inducible SMA mouse heart rates were significantly bradycardic from the earliest time point assayed (P3, SMA, 351 ± 95 bpm vs control, 462 ± 118 bpm; p = 0.04) and declined thereafter (Fig. 1F).
To confirm that the minimal level of excision (one allele per cell) would increase Smn to asymptomatic levels, we used breeding to replace one of the inducible alleles (Smn2B-Neo) with a Smn1tm1.1Cdid allele (referred to hereafter as Smn2B) that had already been induced by a E2A germline Cre. Western blot analysis confirmed SMN2+/−; Smn2B/2B-Neo pups had significantly increased Smn levels from 6 to 37% in P5 spinal cord (Fig. 1A). Importantly, this whole body rescue approach resulted in mice that survived over one year and were phenotypically indistinguishable from control littermates (data not shown). These experiments demonstrate that neomycin excision in even one of the two alleles is enough to correct the severe SMA-like phenotype documented in this model.
Presynaptic and postsynaptic defects in severe inducible SMA mice
To determine the extent of motor unit pathology in end-stage (P6) severe inducible SMA mice, we examined the fast-synapsing intercostal (IC) and the delayed synapsing triangularis sterni (TS) muscles. These muscles were chosen due to their role in maintaining proper respiratory function, one of the most critical deficits in human SMA patients.
The ICs of severe inducible SMA mice had a significant decrease in the number of fully innervated motor endplates (−36 ± 9%; p = 0.007) (Fig. 2A,B,E,F). This correlated with significant increases in both the number of partially occupied and denervated endplates. In contrast, TS NMJs were largely protected and did not significantly differ from controls (Fig. 2C–E,G). Hence, in this SMA model and time point, we consider the IC to be a vulnerable muscle and the TS to be a resistant muscle (McGovern et al., 2008; Ling et al., 2010).

Severe inducible SMA mice have presynaptic and postsynaptic defects. A–D, Micrograph images depicting control (A, C) and mutant (B, D) IC and TS NMJs, respectively. E, Representative images of NMJs that have full innervation, have partial innervation, or are denervated. F, Severe inducible (n = 7) and delta-7 SMA mice (n = 3) had a decrease in the number of fully occupied motor endplates in the ICs (p = 0.007 and p = 0.02, respectively; 2-way ANOVA, Bonferroni's post-test). G, Severe inducible and delta-7 SMA mice showed no defects in innervation of the TS. H, Examples of postsynaptic maturity. I, J, Severe inducible (n = 7) and delta-7 SMA mice (n = 3) had immature motor endplates in the ICs and protection in the TS (n = 5, control). K, L, Severe inducible (n = 7) and delta-7 SMA mice (n = 3) had NF accumulation in the ICs and exhibited resistance to disease in the TS (n = 5, control). Greater than 50 motor endplates per biological replicate were assessed for innervation, postsynaptic maturity, and presynaptic defects. Scale bars: 50 μm. Graph values are shown as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01.
Additionally, we assessed the postsynaptic architecture of αBTX-stained motor endplates, which are known to have delayed maturation in SMA mice (Murray et al., 2008; Kong et al., 2009). Relative to control littermates, severe inducible SMA pups had a 13 ± 4% (p = 0.05) increase in IC homogeneous motor endplates, and a 7 ± 0% (p = 0.04) decrease in the number of endplates containing secondary structure (Fig. 2H,I). Consistent with the innervation results, a similar pattern of protection was observed in the TS (Fig. 2J).
Finally, we quantified two classic presynaptic hallmarks of neurodegeneration: neurofilament accumulation and delayed/failed axonal pruning (Fig. 2H). We observed an 18 ± 5% (p = 0.03) increase in these presynaptic defects in SMA motor axons within the ICs, while SMA motor axons in the TS did not significantly differ from controls (Fig. 2I).
To place the pathology of our model in context with a commonly used model of SMA, we included the delta-7 mouse in our analysis (Le et al., 2005). Consistent with the milder phenotype at P6, the decrease in fully occupied NMJs (−15 ± 2%; p = 0.02) and the increase in partially occupied NMJs (+20 ± 4%; p = 0.02) within the ICs were all less dramatic in delta-7 mice relative to severe inducible SMA mice. Delta-7 and severe inducible SMA mice also demonstrated a parallel pattern of delayed motor endplate maturation and presynaptic defects (Fig. 2K,L) (Murray et al., 2008; Kong et al., 2009).
Hb9-Cre-driven recombination increases Smn expression in spinal MNs
The exact role of MNs in SMA remains an enigmatic mechanistic question. To address this point using severe inducible SMA mice, we used a Cre allele driven by the Hb9 promoter (Jax strain 6600) (Arber et al., 1999). In this widely used allele, Cre becomes active in all MNs and in a rare population of interneuron progenitors at embryonic day 9.5 (E9.5). It is of note that the prion promoter used in previous murine rescue experiments had a comparable temporal expression pattern, with expression being detected at E7.5 (Tremblay et al., 2007). Hb9-Cre is notably absent in a small percentage of lumbar MNs in the lateral motor column (LMC), but has robust expression in the cervical and thoracic spinal cord (Kramer et al., 2006). Importantly, it is not expressed in skeletal muscle, although minor amounts of excision were observed in kidney and stomach via PCR analysis (Fig. 3A). P5 mice doubly transgenic for Hb9-Cre and a ZGFP reporter allele were used to verify MN specificity, as only ChAT-positive MNs stained positive for GFP (with the rare exception of the Hb9 interneurons) (Fig. 3B) (Novak et al., 2000; Kramer et al., 2006).

Specificity and efficiency of Hb9-Cre-driven recombination in vivo. A, A 3-primer PCR assay detects the presence or absence of the neomycin cassette in P5 MN-rescued SMA mice. Excision was observed in the spinal cord, with only minor amounts of promiscuous Cre activity in the stomach and kidney. TA, Tibialis anterior; LG, lateral gastrocnemius; Bicep, biceps brachii. A tamoxifen responsive Cre allele was used to induce severe inducible SMA pups as a positive control for excision. B, SMN2+/−, Smn2B-Neo/2B-Neo, Hb9-Cre+/−, (Cg)-Tg(CAG-Bgeo/GFP) mice show strong GFP expression in ChAT-positive cells, indicating efficient recombination specific to spinal MNs in vivo. C, Western blot. MN-rescued SMA mice showed a visible increase in Smn within the spinal cord and no increase within the ICs, kidney, or brain. D, ChAT-positive SMA motor neurons demonstrated a significant increase in the number of MNs containing nuclear gems (SMA, 17 ± 3% vs MN-rescued SMA, 51 ± 2%; p = 0.003), as well as the average number of gems per MN, confirming that Smn dosage was increased in MNs. White arrows denote gems (Table 1). Scale bars: B, 50 μm; D, 100 μm.
We next bred the Hb9-Cre allele onto our severe inducible SMA background (hereafter referred to as MN-rescued SMA) and analyzed Smn levels in a variety of tissues via Western blot. In agreement with the expression pattern of Hb9-Cre, Smn levels increased slightly in the spinal cord (a dramatic increase would not be expected given the relatively small cellular contribution of MNs) (Fig. 3C). Importantly, there was no increase in Smn levels in the skeletal muscle, kidneys, or brain. To confirm Smn was increased in SMA motor neurons, we quantified nuclear gems, and found a significant increase in the average number of gems per MN in MN-rescued SMA mice (0.63 ± 0.11 gems; p ≤ 0.05) relative to severe inducible SMA mice (0.19 ± 0.02 gems) (Table 1). These results demonstrate that Hb9-Cre significantly increases Smn levels in MNs, while leaving peripheral tissues with SMA relevant Smn protein dosage.
Spinal motor neuron gem counts
Hb9-Cre significantly improves phenotype, but minimally improves survival
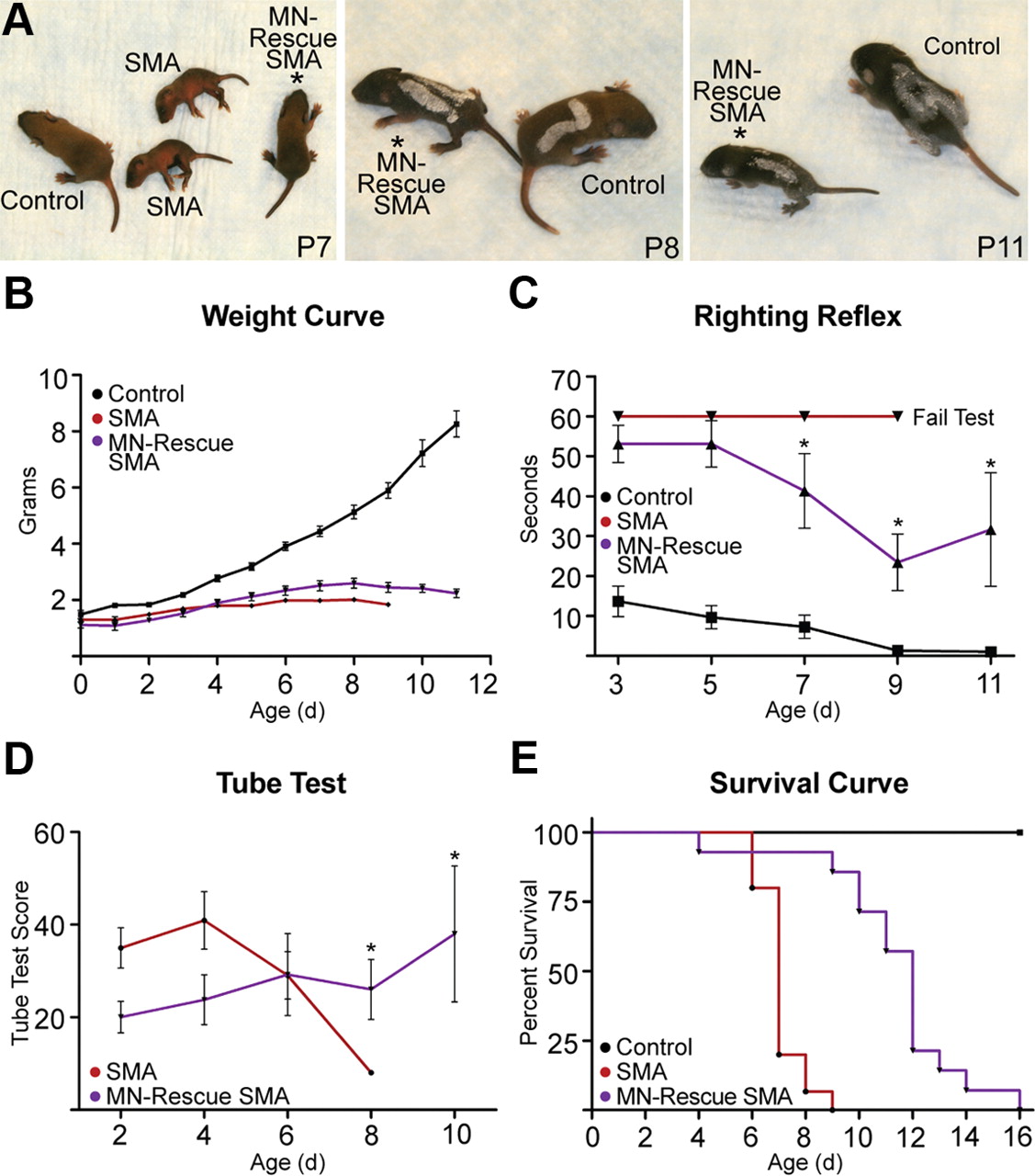
Having validated that Hb9-Cre was increasing Smn dosage exclusively in MNs, we assessed the phenotype of MN-rescued SMA mice. MN rescue dramatically improved the appearance and motor function of severe inducible SMA mice. While still runted, MN-rescued SMA pups had significantly improved righting reflex times (P7–P11; p ≤ 0.01) and were ambulatory throughout their entire life, a feature not seen in other severe SMA models (Fig. 4A–C). MN-rescued SMA mice also had significantly improved tube test scores (P8–P10; p ≤ 0.05) that improved steadily with age (Fig. 4D). Strangely, MN-rescued SMA mice exhibited a decreased level of consciousness or lethargy early in life that was not observed in severe inducible SMA mice or in control littermates, with or without Hb9-Cre. In these instances, MN-rescued SMA mice refused to attempt the righting reflex and tube tests, despite having the functional ability to successfully complete each test. The pups outgrew this passive behavior after P6; however, its presence resulted in poor motor function scores early in life (Fig. 4C,D).

MN-rescued SMA mice have improved motor function, but minimally improved survival. A, Gross phenotype of severe inducible SMA mice is improved by Hb9-Cre. Pups are numbered using a white paint pen. B, Despite improved motor function, MN-rescued SMA mice were runted and did not show significantly improved weights (control, n > 50; SMA, n = 13; MN-rescued SMA, n = 10). C, D, MN-rescued SMA mice demonstrated a decreased level of consciousness (lethargy) early in life, resulting in poor motor function scores. This phenotype dissipates between P6 and P8, and righting reflex and tube test scores significantly improved (p ≤ 0.05 and p ≤ 0.05, respectively; 2-way ANOVA, Bonferroni's post-test; control, n ≥ 21; SMA, n = 9; MN-rescued SMA, n = 7). E, Kaplan–Meier survival curve. Despite the dramatic improvement in motor function, median survival was only increased 5 d in MN-rescued SMA mice (p ≤ 0.001, log-rank test) (control, n > 50; SMA, n = 13; MN-rescued SMA, n = 12). Values are shown as mean ± SEM. *p ≤ 0.05.
Despite the improved motor function, median survival of MN-rescued SMA mice increased by only 5 d (p = 0.001) (Fig. 4E). At the end stage, MN-rescued SMA mice were ambulatory and did not have significant paralysis, demonstrating only lateral instability of their hind limbs. This is likely attributed to a loss of MNs within the lumbar LMC, which control muscles responsible for stability and demonstrate weak Hb9-Cre expression (discussed further in next section).
MN rescue improves SMA motor unit pathology
To determine whether MN rescue corrected the presynaptic and postsynaptic motor unit pathology observed in severe inducible SMA pups, we examined NMJs in the ICs and TS of control and end-stage MN-rescued SMA mutants (the end stage was phenotypically defined as 2 consecutive days of weight loss). After blinded quantification, analysis revealed no significant differences in innervation status, endplate maturity, or presynaptic defects in the ICs of control and MN-rescued SMA mice (Fig. 5A–C) (data not shown). The TS also had no difference in innervation status and demonstrated only a significant decrease in junctions containing secondary structure (−34 ± 6%; p = 0.04) (Fig. 5D,E).

Hb9-Cre rescues motor unit pathology in SMA mice. A–C, At end stage, MN-rescued SMA NMJs within the ICs were indistinguishable from controls in innervation status and endplate maturity (n = 6 per genotype, >50 NMJs per mouse). D, E, NMJs within the TS of MN-rescued SMA mice showed no difference in innervation and demonstrated only a variable delay in postsynaptic maturity (n = 3 per genotype, >50 NMJs per mouse; p = 0.04; 2-way ANOVA, Bonferroni's post-test). F, G, Severe inducible SMA mice had a significant reduction in the average number of ChAT and Islet-1 double-positive MNs per ventral horn within cervical (C4–C8), thoracic (T8–T11), and lumbar (L2–L5) spinal cord (p = 0.01, p = 0.04, and p = 0.05, respectively). In contrast, MN-rescued SMA mice showed no loss in MNs in cervical and thoracic spinal segments, and had only a significant loss at the lumbar level, where Hb9-Cre expression is incomplete. Black, Control; red, SMA; purple, MN-rescue SMA. H, I, MN-rescued SMA mice did not differ from controls in average number of lumbar MMC neurons and showed a significant 25 ± 9% (p = 0.04) reduction in LMC neurons. A significant decrease in both MMC (−47 ± 12%; p = 0.05) and LMC (−29 ± 10%; p = 0.04) neurons was observed in severe inducible SMA mice (Student's t test). J, Confocal images of P5 L2–L5 motor neurons. vGlut1 labels the synapses (green), and the postsynaptic MN is marked by ChAT (red; control, n = 3; SMA, n = 3; MN-rescued SMA, n = 6; average of 44 MN per mouse). K, SMA mice had significantly fewer synapses juxtaposed to MNs per 100 μm of soma (2.74 ± 0.05, p = 0.04) relative to control littermates (3.67 ± 0.32). MN-rescued SMA mice were significantly improved relative to SMA mice (3.92 ± 0.19; p = 0.003; 1-way ANOVA, Bonferroni's post-test). Scale bars: 50 μm. Values are shown as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01.
To discount the possibility that MNs outside of those that innervate the TS and ICs were affected by disease, we quantified ChAT and Islet-1/2 double-positive MNs in the cervical, thoracic, and lumbar spinal cords of control, severe inducible, and MN-rescued SMA pups at end stage (Fig. 3F). In severe inducible SMA mice, a significant loss in average MNs per ventral horn of 4.4 ± 0.2, 4.1 ± 0.3, and 4.6 ± 1.3 was observed in cervical, thoracic, and lumbar spinal cords (p = 0.01, p = 0.04, and p = 0.05), respectively (Fig. 5G). In contrast, MN-rescued SMA spinal cords did not differ significantly from controls in the cervical and thoracic regions. A significant 5 ± 1 MN decrease was observed in L3–L5 spinal cord sections (p = 0.03) (Fig. 5G). This was not unanticipated, as the Hb9-Cre allele used here is known to have incomplete expression in the lumbar LMC (Kramer et al., 2006). A blinded postanalysis confirmed that the LMC in MN-rescued SMA mice presented with a 25 ± 9% reduction in MNs per ventral horn (p = 0.04), while the medial motor column (MMC) showed no significant decrease (Fig. 5H,I). These results support the conclusion that MN rescue is sufficient to prevent the neuromuscular pathology in our SMA model mice, since only MNs that appeared to be lost were those known to lack Hb9-Cre expression.
Hb9-Cre rescues loss of sensory-motor synapses
To determine the effect of MN rescue on glutamatergic excitatory synapses, which are known to be lost in delta-7 SMA mice, we quantified vGlut1-positive puncta juxtaposed to ChAT-positive MNs (Ling et al., 2010; Park et al., 2010; Mentis et al., 2011). End-stage (P5) severe inducible SMA mice had a significant reduction in the number of vGlut1-positive synapses per 100 μm perimeter when compared to control littermates (control, 3.67 ± 0.32 vs SMA, 2.74 ± 0.05; p = 0.04) (Fig. 5J,K). Conversely, MN-rescued SMA mice had significantly restored vGlut1 synapse numbers (3.92 ± 0.19; p ≤ 0.003) and were largely indistinguishable from controls (Fig. 5J,K). These results demonstrate that the loss of afferent synapses in SMA mice can be rescued by increasing Smn exclusively in MNs.
Hb9-Cre corrects SMA motor neuron hyperexcitability and excess synaptic activity
To confirm that SMA motor neurons rescued with Hb9-Cre were functionally normal, we performed whole-cell patch-clamp measurements on spinal MNs from P4–P5 control, SMA, and MN-rescued SMA mice. MNs from all three groups were indistinguishable in appearance, and stable recordings were readily achieved (Fig. 6A–C). There were no significant differences between the control, SMA, or MN-rescued SMA motor neurons in the resting membrane potential, whole-cell capacitance, input resistance, or action potential characteristics (all parameter values are provided in Table 2).

Whole-cell patch clamp on medial MNs. A–C, Images representative of control, SMA, and MN-rescued SMA motor neurons. MNs were filled with Texas Red during recording. D, The current command shows the protocol of hyperpolarizing and depolarizing pulses. E–G, Representative traces of fluctuations in membrane potential in control, SMA, and MN-rescued SMA motor neurons in response to voltage changes. E, F, While some postsynaptic potentials are common in the slice preparation when no blockers are present, there was an overt increase in synaptic activity in the SMA motor neurons. The frequency of postsynaptic potentials per second was quantified as significantly higher in SMA motor neurons (12.4 ± 5.1) relative to control motor neurons (3.3 ± 0.3; p = 0.05) (Table 2). G, MN-rescued SMA motor neurons had complete correction of aberrant postsynaptic potentials (2.5 ± 0.6), suggesting that this phenotype originates in MNs. Boxed insets are magnified from the boxed regions of each trace. Scale bars: C, 100 μm; D, 0.5 nA. Calibration: E–G, vertical, 20 mV; horizontal, 1 s; insets, vertical, 15 mV; horizontal, 1 s.
Electrophysiological properties of motor neurons
Interestingly, the threshold voltage in SMA motor neurons was significantly lower (more hyperpolarized) relative to controls, and this phenotype was corrected in MN-rescued SMA motor neurons (Table 2). The amplitude of the persistent inward current (PIC) was significantly larger in SMA motor neurons and was restored to control levels by Hb9-Cre. Together, the changed PIC amplitude and threshold voltage parameters would suggest that increased excitability (hyperexcitability) is a property of SMA motor neurons that can be corrected by rescuing Smn levels exclusively within MNs.
Normally, it is not necessary to add blockers of synaptic activity to this preparation since there is minimal spontaneous synaptic activity in the quiescent preparation, though much of the circuitry remains intact. However, there was an unusually high frequency of postsynaptic potentials (PSPs) in SMA motor neurons, as shown in the voltage traces in Figure 6D–G. This was quantified as a significant increase in the number of potentials per second in severe inducible SMA mice (12.4 ± 5.1; p = 0.05) relative to control littermates (3.3 ± 0.3). Importantly, MN-rescued SMA motor neurons did not show increased PSPs relative to control littermates (2.5 ± 0.6) (Table 2). These results highlight the fact that alterations in motor circuitry are concomitant with alterations in the intrinsic properties of SMA motor neurons, since Hb9-Cre abolished the increased frequency of postsynaptic potentials.
MN-rescued SMA mice have autonomic defects
The degree of improvement observed in motor function, pathology, and electrophysiology makes it unlikely that NMJ dysfunction is the underlying cause of lethality in MN-rescued SMA pups. In recent years, a number of non-neuromuscular defects have been observed in SMA mice that could additively contribute to lethality in the absence of paralysis, one of which is cardiac defects (Bevan et al., 2010; Heier et al., 2010a; Shababi et al., 2010). Since we observed bradyarrhythmia in our severe inducible SMA mice and have previously documented similar findings in delta-7 SMA mice (Heier et al., 2010a), we assessed its presence as a contributing factor to lethality in MN-rescued SMA mutants. Unlike SMA mice without induction, MN-rescued SMA pups did not present with cardiac defects quantifiable by ECG early in life. However, by P9, mutants began to exhibit significant and progressive bradycardia (MN-rescued SMA, 503 ± 32 bpm vs control, 688 ± 9 bpm; p = 0.001) (Fig. 7A,B). Similar to what has been reported in delta-7 mice, end-stage MN-rescued SMA mice had signs of terminal heart block that manifested as skipped or dropped beats (Fig. 7B) (Heier et al., 2010a).

MN-rescued SMA mice have deficits in autonomic control of the heart. A, B, The heart rate of MN-rescued SMA mice (n = 7) did not differ from controls (n = 14) early in life. However, at P9, MN-rescued SMA mice became significantly bradycardic (p ≤ 0.001) and began to show hallmark signs of terminal heart block, which manifested as dropped beats and prolonged PR interval (SMA, n = 6; Student's t test). C, Schematic of the relevant components of ECG. SA, sinoatrial node; AV, atrioventricular node. D, E, MN-rescued SMA mice demonstrated deficits in aspects of ECG that are attributable to problems in sympathetic innervation of the heart, such as SA-AV signal transmission (P9–P11; p ≤ 0.01) and duration from depolarization to repolarization of the ventricles (P9–P11; p ≤ 0.05) (Student's t test). F, MN-rescued SMA mice did not differ from controls in duration of the QRS complex, suggesting that the ability of the myocardium to contract once depolarized was intact. G, H, Sympathetic innervation of the heart was visualized with tyrosine hydroxylase staining at P5. A significant loss in major visible branches was quantified in both SMA (n = 3) and MN-rescued SMA mouse hearts (n = 4) relative to controls (n = 4), suggesting that this phenotype is independent of motor unit dysfunction (Student's t test). Values are shown as mean ± SEM. Scale bar, 1 mm. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
We further analyzed the components of ECG, such as the PR interval, QT interval, and QRS interval (Fig. 7C). Severe inducible SMA mice had significantly longer PR, QT, and QRS intervals at all time points examined (P3–P7; p < 0.05) (Fig. 7D–F). In contrast, MN-rescued SMA mice did not differ from controls early in life, but did exhibit significantly longer PR and QT intervals from P9 to P11 (p < 0.01 and p < 0.05, respectively) (Fig. 7D,E). MN-rescued SMA mice did not display any alterations in QRS interval (Fig. 7F). These results indicate that the ability of the myocardium to contract in response to ventricular depolarization was unaffected by disease (QRS), while the cardiac nervous system demonstrated delays in electrical conductance (PR and QT).
Previously, we demonstrated reduced sympathetic neuron staining and axon branches in the hearts of delta-7 SMA mice; however, it was unclear whether this was a primary deficit or a secondary complication of muscle weakness (Heier et al., 2010a). To assess the cardiac autonomic innervation pattern in MN-rescued SMA mice, who do not demonstrate significant paralysis, we performed tyrosine hydroxylase (TH) staining on control, SMA, and MN-rescued SMA mice. After standardizing the size of control and mutant hearts digitally using Photoshop, an average of 14 ± 0.5 major visible branches was quantified in control mice. In contrast, a significant reduction to 10 ± 1 and 11 ± 1 visible branches was observed in SMA and MN-rescued SMA mice (p = 0.02 and p = 0.008), respectively (Fig. 7G,H). Importantly, these defects in autonomic innervation exist in the absence of significant paralysis and/or motor unit pathology. This suggests that they are primary defects that likely combine with other non-neuromuscular defects and additively contribute to the poor health of SMA mice, both in the presence and absence of motor unit dysfunction.
Discussion
Here we describe a novel mouse model of SMA with the ability to increase Smn dosage to asymptomatic levels after exposure to Cre-recombinase. Our model presents with severe SMA-like symptoms and pathology that are most similar to the severe model described by Monani et al. (2000) (Jax strain 5024). The experiments described in this manuscript demonstrate that MN rescue with Hb9-Cre can significantly improve motor function and motor unit pathology, which are commonly deficient in SMA mice. Furthermore, electrophysiology demonstrates that increasing Smn specifically within MNs is sufficient to correct hyperexcitability in SMA MNs, as well as abolish aberrant synaptic input to MNs.
Involvement of pathology outside the CNS
Despite a lack of significant paralysis, median survival is only extended 5 d in MN-rescued SMA mice. Our results, combined with those of Park et al. (2010), indicate that defects external to the MN exasperate the SMA phenotype by compromising the overall health of the mouse, but future experiments are required to definitively confirm this. One potential caveat to this interpretation is that inefficient Cre excision could be the root of lethality in our model and the cause of recovery in the Olig2-Cre SMA mice (Park et al., 2010). While unlikely, such a result would further emphasize the importance of MNs in the SMA phenotype, since a dramatic change in phenotype would be observed despite inefficient recombination. We attribute lethality in MN-rescued SMA mice, in part, to autonomic deficits in cardiac innervation and resulting heart failure. However, the severe phenotype in this model makes it probable that additional, non-neuromuscular factors also contribute to the observed lethality. Interestingly, there is mounting evidence to suggest that other cell types such as hepatocytes, osteoclasts, and myofibers may be affected in SMA mice, although it is unclear whether these defects are primary or secondary (Guettier-Sigrist et al., 2002; Araujo Ade et al., 2009; Shanmugarajan et al., 2009; Bevan et al., 2010; Heier et al., 2010a; Shababi et al., 2010; Wishart et al., 2010; Hua et al., 2011; Mutsaers et al., 2011). Despite the presence of non-neuromuscular phenotypes in severe SMA mice, it remains clear that long-term survival can be achieved with predominantly CNS-specific SMN rescue (Gavrilina et al., 2008; Foust et al., 2010; Passini et al., 2010, 2011; Hua et al., 2011; Osman et al., 2011).
MN pathology supports defects in central spinal circuitry
Several recent reports indicate that loss of the primary afferents that synapse onto MNs is an early hallmark and potentially significant contributor or mechanistic root of the SMA phenotype in mice (Ling et al., 2010; Park et al., 2010; Mentis et al., 2011). As has been noted in other SMA mouse models, the level of MN electrophysiological dysfunction quantified in our model is not sufficient to explain the significant reduction in motor function that exists (Zhang et al., 2010; Mentis et al., 2011). Given the high safety factor for neuromuscular transmission, the synaptic transmission defects that have been described in delta-7 SMA mice are also unlikely, on their own, to account for the observed phenotype (Kong et al., 2009; Ling et al., 2010; Ruiz et al., 2010). Hence, in our model the presence of hyperexcitable MNs, which experience an increased frequency of postsynaptic events and a loss of glutamatergic synapses, supports the conclusion that defects in spinal circuitry above the NMJ, such as sensory-motor synapses, play a significant role in driving the progression of a SMA-like disease in mice and potentially human SMA patients.
Here, we show an increased frequency of postsynaptic events in SMA MNs, whereas a previous study demonstrated decreased afferent input (Mentis et al., 2011). On the surface, these results appear to conflict; however, they are not mutually exclusive. Since synaptic formation and maintenance is altered between proprioceptive, vGlut1-expressing neurons and MNs, the formation and maintenance of synaptic contacts with other neurons, for example, local spinal interneurons that do not express vGlut1, could be altered as well. Therefore, deficits in afferent input are potentially part of the same altered circuitry that is responsible for the increased spontaneous synaptic activity.
In addition to Hb9-positive MNs, another group of neurons could be involved in the rescued phenotype: the Hb9 interneurons. This small population of glutamatergic interneurons (∼40 per segment) is present in limited thoracic and lumbar spinal cord segments, T13–L3 (Hinckley et al., 2005; Wilson et al., 2005; Kwan et al., 2009). They receive monosynaptic input from sensory afferents (Hinckley et al., 2010) and have sparse synaptic contacts with MNs (Hinckley et al., 2005; Wilson et al., 2005). Since Hb9 interneurons are also rescued, it could be argued that loss of Smn protein renders them hyperexcitable, and their subsequent rescue explains the absence of aberrant synaptic activity. However, this is highly improbable, since contacts between Hb9 interneurons and MNs are rare (Wilson et al., 2005), and based on their low levels of activity during activation of the locomotor network, the possibility that these interneurons drive another population of premotor interneurons in quiescent cord appears somewhat remote (Kwan et al., 2009).
Pathology of MNs
Some of the findings here are not identical to those of previous studies. In this SMA model, we did not observe changes in input resistance or action potential height, as was reported in Mentis et al. (2011). This may be due to the different SMA models or the preparations used (whole isolated spinal cord vs spinal cord slice). However, our observations are similar to previous studies in that the threshold voltage of SMA MNs is more hyperpolarized, which would allow them to fire with less depolarizing input (Zhang et al., 2010; Mentis et al., 2011). Increased excitability, including an increased persistent inward current, has been observed in other conditions of spinal MN dysfunction, such as spinal cord injury and MN disease (Li and Bennett, 2003; Kuo et al., 2005; Harvey et al., 2006; Meehan et al., 2010; Quinlan et al., 2011). This might indicate that MN excitability is a common thread in SMA and, more broadly, in MN pathology.
Defects in sensory-motor synapses are specific to MNs
Aberrant synaptic activity in SMA can be explained by altered synapse development and maintenance. Since synaptic integrity requires faithful and reciprocal presynaptic and postsynaptic signaling, the loss of sensory-MN interaction could be due to a failure of primary afferents to deliver appropriate neurotransmitters across the synapse. Alternatively, the inability of the postsynaptic MN to maintain normal levels of excitability could result in the loss of primary afferent synapses, while other synaptic contacts are upregulated. Results from this study would argue that the latter is the mechanism of afferent synapse loss in SMA, since our MN-rescued SMA mice, who express only asymptomatic levels of Smn in MNs, do not show a loss in vGlut1 bouton number or evidence of altered synaptic inputs. This finding is in agreement with the previous observation that sensory-motor synapses are lost in mice that have normal Smn dosage in all cells except MNs and oligodendrocytes (Park et al., 2010).
The existence of non-neuromuscular contributions to lethality makes it challenging to determine whether, with prolonged survival, aberrant synaptic activity in Hb9-Cre-rescued SMA motor neurons would develop; however, dissecting this will require milder inducible models of SMA than the one described here. Together, these findings suggest that the role of the postsynaptic cell should not be underestimated in maintenance of afferent synapses.
Conclusions
Collectively, our results and those of others indicate that deficits in the central circuit play a significant role in SMA disease progression and paralysis in severe SMA mice and potentially human SMA patients (Ling et al., 2010; Park et al., 2010; Mentis et al., 2011). Moreover, our results support the idea that loss of sensory-motor synapses is a consequence, and not a cause, of MN dysfunction. Finally, this work demonstrates that neuromuscular dysfunction in SMA mice is mechanistically due to deficits within MNs. These defects are enhanced by the presence of additional primary or secondary defects, all of which act additively to comprise the general health of the mouse. As such, this work does not rule out the potentially significant benefits of restoring Smn within the other components of the motor unit, such as skeletal muscle and/or Schwann cells, which may play important roles in defining disease severity. Future work determining how many rescued MNs are required to affect a phenotypic benefit will have important implications in developing effective therapies to treat SMA patients.
Footnotes
This work was supported by NIH Training Grant T32 AG000260, Drug Discovery Training in Age-Related Disorders (R.G.), Families of Spinal Muscular Atrophy Grant DiD0809 (C.J.D.), and NIH NINDS Grants 1ROIN5060926 (C.J.D.), 3RO1NS060926-02S3 (C.J.D.), NS034382 (C.J.H.), and NS071951 (C.J.H., K.A.Q.). K.A.Q. made use of equipment supported by NS054850. We acknowledge Herminio Cardona's and Michael Jorgensen's roles in overall colony maintenance, Carina Emery for experimental assistance, as well as the labs of Dr. Greg Cox and Dr. Robert Burgess for technical training rendered.
The authors declare no competing financial interests.
- Correspondence should be addressed to Christine J. DiDonato, Children's Memorial Research Center, 2300 Children's Plaza, Box 211, Chicago, Illinois 60614. c-didonato{at}northwestern.edu
References
- Araujo Ade et al., 2009.↵
- Arber et al., 1999.↵
- Bevan et al., 2010.↵
- Boon et al., 2009.↵
- Briese et al., 2009.↵
- Chang et al., 2008.↵
- Coovert et al., 1997.↵
- Crawford and Pardo, 1996.↵
- DiDonato et al., 2003.↵
- Foust et al., 2010.↵
- Gavrilina et al., 2008.↵
- Guettier-Sigrist et al., 2002.↵
- Hammond et al., 2010.↵
- Harvey et al., 2006.↵
- Heier et al., 2010a.↵
- Heier et al., 2010b.↵
- Hinckley et al., 2005.↵
- Hinckley et al., 2010.↵
- Hsieh-Li et al., 2000.↵
- Hua et al., 2011.↵
- Kong et al., 2009.↵
- Kramer et al., 2006.↵
- Kuo et al., 2005.↵
- Kwan et al., 2009.↵
- Le et al., 2005.↵
- Le et al., 2011.↵
- Lefebvre et al., 1995.↵
- Lefebvre et al., 1997.↵
- Li and Bennett, 2003.↵
- Ling et al., 2010.↵
- McGovern et al., 2008.↵
- Meehan et al., 2010.↵
- Mentis et al., 2011.↵
- Monani et al., 2000.↵
- Murray et al., 2008.↵
- Mutsaers et al., 2011.↵
- Novak et al., 2000.↵
- Osman et al., 2011.↵
- Park et al., 2010.↵
- Passini et al., 2010.↵
- Passini et al., 2011.↵
- Pellizzoni et al., 1998.↵
- Quinlan et al., 2011.↵
- Ruiz et al., 2010.↵
- Schrank et al., 1997.↵
- Shababi et al., 2010.↵
- Shanmugarajan et al., 2009.↵
- Tremblay et al., 2007.↵
- Wilson et al., 2005.↵
- Wishart et al., 2010.↵
- Zhang et al., 2010.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}