

Abstract
Here we demonstrate that interleukin-2-inducible T-cell kinase (Itk) signaling in cluster of differentiation 4-positive (CD4+) T cells promotes experimental autoimmune encephalomyelitis (EAE), the animal model of multiple sclerosis (MS). We show that Itk−/− mice exhibit reduced disease severity, and transfer of Itk−/− CD4+ T cells into T cell-deficient recipients results in lower disease severity. We observed a significant reduction of CD4+ T cells in the CNS of Itk−/− mice or recipients of Itk−/− CD4+ T cells during EAE, which is consistent with attenuated disease. Itk−/− CD4+ T cells exhibit defective response to myelin antigen stimulation attributable to displacement of filamentous actin from the CD4+ coreceptor. This results in inadequate transmigration of Itk−/− CD4+ T cells into the CNS and across brain endothelial barriers in vitro. Finally, Itk−/− CD4+ T cells show significant reduction in production of T-helper 1 (Th1) and Th17 cytokines and exhibit skewed T effector/T regulatory cell ratios. These results indicate that signaling by Itk promotes autoimmunity and CNS inflammation, suggesting that it may be a viable target for treatment of MS.
Introduction
Tyrosine kinase expressed in hepatocellular carcinoma (TEC) family non-receptor tyrosine kinases are critical for the regulation of intracellular signaling in lymphocytes for proper immune responses. The Tec kinase interleukin-2-inducible T-cell kinase (Itk) regulates signaling via the T-cell receptor (TCR) and has been shown to be involved in the activation of intracellular calcium signaling pathways, MAPK pathways, and polarization of actin cytoskeleton, supporting an integral role for Itk in T-cell activation and function (Gomez-Rodriguez et al., 2009; Andreotti et al., 2010). The absence of Itk results in development of cluster of differentiation 8-positive (CD8+) T cells with innate function (Atherly et al., 2006; Broussard et al., 2006; Hu et al., 2007; Huang et al., 2013, 2014b; Prince et al., 2014b), reduced development of invariant natural killer T (NKT) cells and ability to produce cytokines (Gadue and Stein, 2002; Au-Yeung and Fowell, 2007; Felices and Berg, 2008; Qi et al., 2011a,b, 2012), and expansion of a unique population of γδ NKT-like and promyelocytic leukemia zinc finger-positive innate like cells (Felices et al., 2009; Qi et al., 2009; Yin et al., 2013; Prince et al., 2014a). Mice deficient in Itk also exhibit significant alteration in T-helper (Th) cell development and function, including Th2 and Th17, as well as T-regulatory cell development (Fowell et al., 1999; Gomez-Rodriguez et al., 2009, 2014; Kannan et al., 2013; Huang et al., 2014a). This has made Itk a promising target for the development of drugs that target Th cytokine-mediated diseases (Sahu and August, 2009; August and Ragin, 2012).
Multiple sclerosis (MS) is a multifaceted neuroinflammatory disease influenced by environmental factors such as infection, vitamin D deficiency, and gonadal hormones (Whitacre, 2001; Cantorna, 2006; Ascherio and Munger, 2007). Although the etiology of MS is unknown, it is evident that aberrations in the immune response compartment can either trigger its onset or exacerbate its pathogenicity (Navikas and Link, 1996). Thus, imbalance in factors that induce and/or prolong inflammation versus those that resolve and/or suppress inflammation affects disease outcome. MS is characterized by infiltration of inflammatory immune cells into the CNS (Hafler and Weiner, 1989). CD4+ and CD8+ T cells play critical roles in the disease pathogenesis (Keegan and Noseworthy, 2002). CD4+ T cells in MS lesions have been determined to be primarily of the Th1 and Th17 lineages (El-behi et al., 2010). The murine model of MS, experimental autoimmune encephalomyelitis (EAE), is induced in susceptible mouse strains after immunization with myelin components, such as myelin oligodendrocyte glycoprotein (MOG), or by passive transfer of myelin antigen-specific T cells (Wekerle et al., 1994). Like MS, the neuroinflammatory response in EAE is mediated mainly by effector Th1 and Th17 cells that migrate to the CNS in which they attack the myelin sheath, resulting in demyelination and subsequent paralysis (Jäger et al., 2009). These pathogenic effector Th cells can be controlled by T regulatory (Treg) cells, which suppress inflammatory responses (Kohm et al., 2002).
We investigated the role of Itk in the development of EAE and found that Itk−/− mice are significantly protected from EAE and have diminished frequency of immune cells in their CNS. Similarly, in a transfer model of EAE, recipients lacking CD4+ T cells reconstituted with Itk−/− CD4+ T cells develop attenuated EAE compared with recipients reconstituted with wild-type (WT) CD4+ T cells. We also found that Itk−/− CD4+ T cells are defective in their ability to migrate across an in vitro blood–brain barrier (BBB). Itk−/− CD4+ T cells exhibit defects in actin cytoskeleton reorganization in response to myelin antigen stimulation, resulting in diminished ability to proliferate, and also show defective Th1 and Th17 cytokine production. Based on these findings, we conclude that Itk promotes CD4+ T cell migration into the CNS and contribute to neuroinflammation. We propose that inhibitors of Itk signaling have strong potential as therapeutics against MS.
Materials and Methods
Mice.
WT mice were obtained from The Jackson Laboratory, and Itk−/− mice were as described previously (Liao and Littman, 1995). All mice were on the C57BL/6 background and were used when they were 6–8 weeks of age. Mice of both sexes were used and were maintained in specific pathogen-free environment. All experiments were approved by the Institutional Animal Care and Use Committee of the Office of Research Protection at Cornell University.
Flow cytometry and intracellular cytokine staining.
To stain for intracellular cytokines and transcription factors, cells were stimulated with phorbol 12-myristate 13-acetate (PMA)/ionomycin (50 ng/1 μg/ml; Sigma) and Brefeldin A (Sigma) for 5 h. To examine antigen-specific recall responses, single-cell suspensions of splenocytes or lymph node cells were cultured with the indicated concentration of MOG peptide for 72 h and restimulated with either 10 μg/ml peptide from MOG35–55 or with PMA/ionomycin (50 ng/1 μg/ml; Sigma) in the presence of Brefeldin A (Sigma) for 5 h. Cells were then fixed/permeabilized using the forkhead box p3 (Foxp3) fixation/permeabilization kit and stained with the indicated antibodies (or isotype controls) against surface/intracellular proteins. Data were acquired and analyzed using LSRII (BD Biosciences) and FlowJo (TreeStar), respectively. Cells were identified by gating on forward scatter versus side scatter, gating on the lymphocyte population, followed by gating on TcRβ+CD4+ T cells and analysis of intracellular cytokine or Foxp3 expression as shown in Figure 3A. T cells were isolated from brains and spinal cords as follows: after homogenization and pelleting, cells were resuspended in the 30% Percoll. Thereafter, 70% of Percoll was layered at the bottom of 30% Percoll layer. Samples were spun down at 600 rpm for 20 min without acceleration and brake. The cell layer was collected and washed with media and used for additional analysis.
ELISA.
The protein concentrations of the cytokines IFNγ and IL-17A in cell culture supernatants were quantified using commercially available kits for two-site ELISA (eBioscience) according to the instructions of the manufacturer.
Carboxyfluorescein succinimidyl ester labeling and H3-thymidine incorporation assay.
Splenocytes from immunized WT and Itk−/− mice were incubated at 37°C for 10 min in PBS containing 5 mm carboxyfluorescein succinimidyl ester (CFSE; Invitrogen). Subsequently, the cells were washed twice with complete RPMI 1640 and cultured in the presence of the MOG peptide for 72 h, with cells cultured in media alone serving as controls. For analysis of H3-thymidine incorporation, cells were seeded at 2 × 106 cells/ml and left unstimulated or stimulated with MOG peptide for 72 h with H3-thymidine (1 μCi) added for the last 18 h, after which cells were washed and pelleted and the radioactivity was quantified and expressed as fold change over media-only controls.
EAE induction, scoring, and in vivo CD25+ cell depletion.
EAE was induced as described previously (Mills et al., 2012). Briefly, a 1:1 emulsion of MOG peptide (3 mg/ml in PBS; Anaspec) and complete Freund's adjuvant (CFA; Sigma) was injected subcutaneously (50 μl) into each flank (50 μg each flank = 100 μg). Pertussis toxin (200 ng in 200 μl of PBS; Biological Laboratories) was given intravenously at the time of immunization and again 2 d later. Mice were scored daily for EAE based on disease symptom severity: 0, no disease; 0.5, weak tail (cannot curl tail completely); 1.0, limp tail (complete inability to move tail); 2, limp tail and partial hindlimb paralysis; 3, total hindlimb paralysis; 4, both hindlimb and forelimb paralysis; and 5, death. Mice with a score of 4 were killed. For in vivo depletion of CD25+ cells, we administered either vehicle (PBS) control or 200 μg of α-CD25 (PC61 mAb) antibody every 4 d. Quantification of CD4+ T-cell infiltration was performed on anatomically similar brain (i.e., cerebellum and hippocampus, choroid plexus) and spinal cord immunohistochemically stained sections. Ten randomly selected fields from each mouse brain per group of mice (n = 4) were visualized at 10× magnification. Fields were then averaged to determine mean cell infiltration per brain.
Actin cytoskeleton analysis.
CD4+ T cells were isolated from MOG peptide–CFA-immunized WT or Itk−/− mice and stimulated with the MOG peptide or α-CD3 antibodies for 24 or 72 h. Cells were then fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100. Filamentous actin (F-actin) was stained with Alexa Fluor 568-conjugated phalloidin (1:200; Invitrogen), and CD4 was detected using allophycocyanin (APC)-conjugated α-CD4 primary antibody (1:100; BD Biosciences) for 30 min. The image was visualized by a Leica SP5 confocal microscope, and the localization of F-actin and CD4 was analyzed by a Leica suite image analysis program.
T-cell purification, differentiation, and transmigration assay.
Naive CD44loCD62Lhi CD4+ T cells were isolated from spleens and lymph nodes using the Miltenyi Naive CD4+ T cell isolation kit according to the instructions of the manufacturer. For transmigration assays, a mouse brain endothelial cell line (bEnd3; American Type Culture Collection) was cultured on an 8 μm porous membrane insert (BD Biosciences) as an in vitro model of the BBB (Wilhelm et al., 2011). CD4+ T cells (2.5 × 105) isolated from MOG and CFA-immunized WT and Itk−/− mice were placed in the cell containing insert, with media at the bottom well. The media at the bottom well were collected at 1 and 24 h after treatment, and the number of cells that crossed the barrier was counted. For the Latrunculin B (LatB)-induced transmigration recovery assay, cells were pretreated with DMSO or LatB (1 μm) for 1 h, washed with media, and loaded on the in vitro BBB insert. Cells were collected at 1 and 24 h after treatment, and the number of cells that crossed the barrier was counted. For the transmigration assay of differentiated T cells, CD4+ T cells were isolated from MOG–TCR transgenic mice (2D2-TCR-Tg) and differentiated into Th1, Th2, and Th17 cells as described previously (Jäger et al., 2009). In brief, purified naive CD44loCD62Lhi CD4+ T cells were cultured in 2 ml of Bruff's media and stimulated with plate-bound mouse anti-CD3 (3 μg/ml, clone 2C11; BD Biosciences) and anti-CD28 (1 μg/ml, clone 37.51; BD Biosciences) under neutral or polarizing conditions: Th1 [IL-2 (25 U/ml), IL-12 (10 ng/ml; eBioscience), and anti-IL-4 (10 μg/ml)], Th2 [IL-4 (20 ng/ml), anti-IFNγ and anti-IL-12 (10 μg/ml)], and Th17 [IL-2 (25 U/ml), IL-6 (20 ng/ml; eBioscience), TGF-β (3 ng/ml; R&D Systems), anti-IL-4 (10 μg/ml), anti-IL-12 (10 μg/ml, clone JES6–1A12; BD Biosciences), and anti-IFNγ (10 μg/ml) supplemented with IL-23]. On day 3 after stimulation, cells were expanded for an additional 4 d in fresh media containing 25 U/ml mouse IL-2. Th0, Th1, Th2, or Th17 CD4+ cells at 5 × 105 were pretreated with DMSO or Itk inhibitor (1 μm) for 2 h and loaded onto mouse brain endothelial cells cultured on porous membrane insert containing DMSO or Itk inhibitor (1 μm) with media at the bottom well. Cells at the bottom well were collected at 1 and 24 h after treatment and enumerated.
Statistical analysis.
Results are expressed as means ± SEMs, and statistical significance between groups determined by either unpaired Student's t test or two-way ANOVA using GraphPad Prism version 5.00 for Windows (GraphPad Software). Values with a probability of p ≤ 0.05 are considered statistically significant.
Results
Itk promotes development of EAE
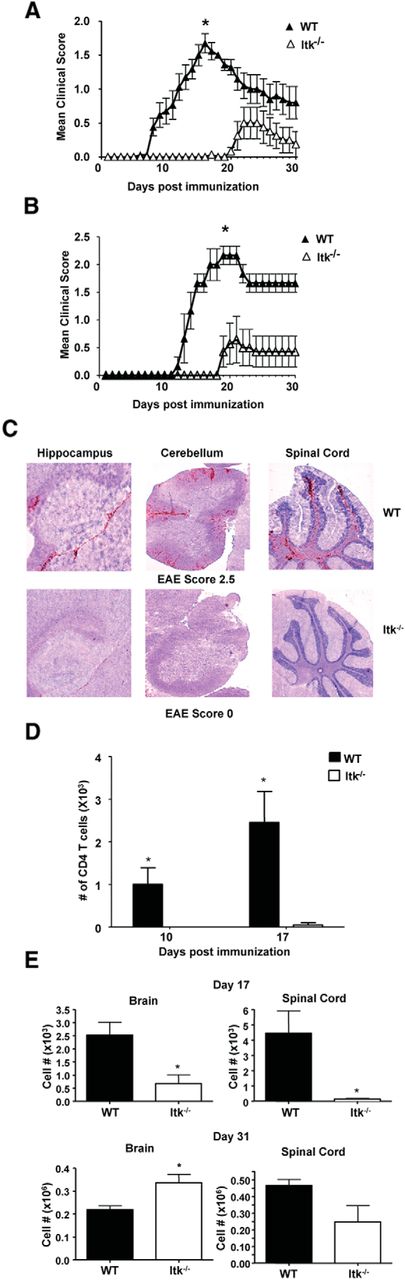
We investigated the role of Itk in the MOG-induced model of EAE. We observed that disease in Itk−/− mice was significantly attenuated compared with their WT counterparts (Fig. 1A). Moreover, onset of symptoms of EAE in Itk−/− mice was delayed by 10–20 d, and incidence of EAE was significantly less compared with WT mice (Fig. 1A; Table 1). The environment in which mice are housed has a significant effect on immune-mediated diseases. Our experiments shown in Figure 1A were performed in our “clean” animal facility, resulting in lower EAE scores in the WT mice. Experiments performed in an animal room that has a higher level of accepted pathogens (i.e., a dirtier environment) resulted in WT mice developing a more severe EAE profile, but Itk−/− mice had similar disease profile as that shown in Figure 1A (Fig. 1B). Clinical signs of EAE become evident after inflammatory immune cells invade the CNS and cause destruction of myelin tissue. Consistent with the absence of clinical signs of disease and delayed onset, Itk−/− mice had very few cells in their brain and spinal cord on day 17 after EAE induction compared with WT mice as determined by immunohistochemistry (Fig. 1C,D) and fluorescence-activated cell sorter (FACS) analysis (Fig. 1E). Analysis of the small proportion of Itk−/− mice that had developed disease by day 31 revealed that they had CD4+ T cells in the brain and spinal cord in numbers similar to WT mice (Fig. 1E). The appearance of CD4+ T cells in the CNS of Itk−/− mice at day 31 after EAE induction is consistent with the first appearance of clinical signs of disease, which occurs between days 20 and 30 in those mice. Together, these findings suggest that Itk signaling plays an important role in the promotion of autoimmunity and neuroinflammation.

Itk promotes autoimmunity and lymphocyte migration into the CNS. A, B, WT (n = 17) and Itk−/− (n = 14) mice were immunized to develop EAE and scored daily for clinical signs of EAE based on a five-point scale assessing ascending paralysis. Values are means ± SEMs, *p < 0.05 by two-way ANOVA. A, “Cleaner” animal facility. B, “Dirtier” animal facility. C, CNS sections (hippocampus, cerebellum, and spinal cord) from day 17 post-EAE induction WT (top row) and Itk−/− (bottom row) mice were stained with α-CD45 to detect immune cell infiltration in the CNS after disease induction (red) versus a nuclear background (blue/gray). D, CNS sections from day 10 and 17 post-EAE induction WT and Itk−/− mice were stained with α-CD4 and quantified using light microscope from different regions of brain and spinal cord and quantified (n = 3, each time point). Values are means ± SEMs, *p < 0.05 by unpaired Student's t test. E, Cells were isolated from the brain and spinal cord of WT and Itk−/− mice at day 17 and 31 after EAE induction and quantified for the number of CD4+ T cells by FACS analysis. Values are means ± SEMs, *p < 0.05 by unpaired Student's t test.
Itk signaling promotes EAE
We next determined whether protection conferred by Itk deficiency was solely attributable to the fact that Itk−/− mice have fewer CD4+ T cells in the periphery (Liao and Littman, 1995). We transferred equal numbers of WT or Itk−/− CD4+ T cells into mice that lack endogenous T cells (TCRα−/− mice), followed by induction of EAE. TCRα−/− recipients of WT CD4+ T cells had a higher EAE incidence and developed significantly more severe disease compared with recipients of Itk−/− CD4+ T cells (Fig. 2A; Table 2). Consistent with this, TCRα−/− recipients of Itk−/− CD4+ T cells also had fewer immune cell infiltrates in the CNS (Fig. 2B). These results indicate that Itk signaling promotes the development of autoimmune pathologies during EAE, and this is attributable to a CD4+ T-cell intrinsic requirement for Itk.

WT but not Itk−/− CD4+ T cells confer disease to TCRα−/− recipients. A, Three to 4 × 106 WT or Itk−/− CD4+ T cells were transferred into TCRα−/− recipients that were subsequently immunized to develop EAE and scored daily for clinical signs of EAE. Values are means ± SEMs, *p < 0.05 by two-way ANOVA, n = 5. B, CNS sections from the indicated mice at day 31 after EAE induction were stained with α-CD4 (red), and the cerebellar parenchyma (left column) and hippocampal area (right column) of the CNS were assessed for the presence of CD4+ T-cell infiltrates from representative mice that received WT or Itk−/− donor cells. Arrows indicate areas with pronounced CD4+ staining.
Itk signaling plays a cell intrinsic role in CD4+ T cells in promoting EAE
Reduced Th1 and Th17 effector cells in the CNS of Itk−/− mice
Signaling through Itk is critical for the production of IL-17A by Th17 cells (Gomez-Rodriguez et al., 2009), and murine and human CD4+ T cells that lack Itk rapidly upregulate IFNγ (Kannan et al., 2013). Both IFNγ and IL-17A play important roles in EAE and MS pathogenesis (Korn et al., 2009). To determine whether Itk regulates the production of these cytokines in EAE, we isolated CD4+ T cells from brain and spinal cord of WT and Itk−/− mice on days 17 and 31 after EAE induction to assess their cytokine profile by intracellular cytokine staining using the gating strategy shown in Figure 3A. We found fewer effector cells infiltrating the CNS of Itk−/− mice, with lower numbers of IFNγ+ cells in the brain and spinal cord at day 17 and lower numbers of IFNγ+ and IL-17A+ T cells at day 31 after induction of EAE (Fig. 3B,C). These results suggest that Itk signaling is important for the generation and elaboration of effector responses of autoreactive CD4+ T cells and suggests that diminution in effector Th1 and Th17 cells may in part be responsible for the attenuated disease in Itk−/− mice.

Th1 and Th17 effector cells are decreased in the CNS of Itk−/− mice. A, Gating strategy to identify cytokine-producing cells and Foxp3+ Treg cells. B, C, Cells isolated from the brain and spinal cord of the indicated day 17 (B) and day 31 (C) immunized mice were stimulated with PMA/ionomycin in the presence of Brefeldin A for 5 h, and CD4+ T cells were analyzed for the expression of IFNγ and IL-17A by FACS (top rows) and quantified for number of cytokine-producing cells (bottom rows). Values are means ± SEMs, n = 3–5, *p < 0.05 by unpaired Student's t test.
Itk signaling is critical for regulating the differentiation and function of CD4+ T cells during EAE
Prompted by the paucity of pathogenic CD4+ T cells (Th1 and Th17) in the CNS of Itk−/− mice, we next determined whether there was a defect in Th1 and/or Th17 CD4+ T-cell generation in the peripheral lymphoid organs of Itk−/− mice. We found that, consistent with the attenuated disease and lack of infiltrating pathogenic Th1 or Th17 cells in the CNS, there were significantly higher numbers of effector Th1 cells when WT splenocytes were stimulated with MOG peptide compared with those from immunized Itk−/− mice 10 d after EAE induction. However, there was little evidence of effector Th17 cells generated ex vivo at this time point (Fig. 4A–D). This is especially interesting considering the fact that Itk−/− naive CD4+ T cells are primed for IFNγ production, with higher basal levels of IFNγ (Hu and August, 2008; Kannan et al., 2013). Furthermore, by day 17 after EAE induction, WT CD4+ T cells had undergone a switch from Th1 to Th17, producing more IL-17A and less IFNγ in response to MOG peptide restimulation in vitro. However, we found little evidence for such a switch in Itk−/− CD4+ T cells. Similarly, fewer Itk−/− CD4+ T cells expressed IL-17A ex vivo on day 31 after EAE induction (Fig. 4B,C). Similar results were obtained when cells were restimulated with PMA and ionomycin (Fig. 4D). In vitro, WT splenocytes from MOG peptide-immunized mice also exhibited higher proliferation, incorporating more H3-thymidine compared with Itk−/− splenocytes in response to MOG peptide. Additionally, WT CD4+ T cells underwent more divisions (as measured by dilution of CFSE) than Itk−/− CD4+ T cells in response to MOG peptide (Fig. 4E). Collectively, these data suggest that WT CD4+ T cells have better recall responses during stimulation with MOG peptide and that Itk signaling regulates the generation of MOG-specific effector Th1 and Th17 cells.

Itk signaling is critical for regulating the differentiation and effector function of CD4+ T cells during EAE. A, B, Splenocytes isolated from day 10, day 17, and day 31 immunized WT and Itk−/− mice were stimulated with 5 μg/ml MOG peptide for 72 h. The cells were then restimulated with 10 μg/ml MOG peptide in the presence of Brefeldin A for 5 h, and CD4+ T cells were analyzed for the expression of IFNγ and IL-17A by FACS. Representative flow plots from day 10 is shown in A. B, Cytokine-producing cells quantified for IFNγ (left) or IL-17A (right) stimulation index (fold change in percentage of IFNγ or IL-17A-producing cells in response to MOG peptide compared with media controls). C, Cell culture supernatants of cells treated as in A were analyzed for the levels of IFNγ and IL-17A protein by ELISA. D, Splenocytes from A were stimulated with PMA/ionomycin (50 ng/1 μg/ml; Sigma) in the presence of Brefeldin A for 5 h and quantified for IFNγ (left) or IL-17A (right) stimulation index (fold change in percentage of IFNγ or IL-17A-producing cells in response to MOG peptide compared with media controls). E, Splenocytes obtained from day 17 immunized WT and Itk−/− mice were loaded with CFSE and stimulated with 5 μg/ml MOG peptide for 72 h. Alternatively H3-thymidine was added during the last 18 h of culture. CFSE-labeled cells were analyzed for proliferation of CD4+ T cells by FACS (top row), and data are represented as percentage CFSE diluted or fold increase in thymidine incorporation over media control (bottom row). Values are means ± SEMs, *p < 0.05 by unpaired Student's t test.
Itk−/− CD4+ T cells exhibit altered migration velocity and are ineffective in crossing the BBB in vitro
In EAE and MS, pathogenic immune cells, in particular CD4+ T cells, must traverse the brain endothelium and enter the CNS parenchyma and participate in immune responses, resulting in tissue damage (Kuchroo et al., 2002; Mills et al., 2008, 2012). Given that Itk−/− CD4+ T cells exhibit defects in CNS infiltration, which is important for inflammation and resultant CNS pathology, we next determined whether Itk signaling in CD4+ T cells is important for their traversal across the BBB. We performed migration studies using an in vitro BBB model to evaluate the migratory capacity of WT and Itk−/− CD4+ T cells. Itk−/− CD4+ T cells showed defective migration across the in vitro brain endothelial barrier compared with WT CD4 T cells (Fig. 5A). This was not attributable to reduced mobility of Itk−/− CD4+ T cells, because these cells traveled at a rate of speed approximately three times faster than WT CD4+ T cells on a planar surface (Fig. 5B). Thus, reduced ability to migrate across the BBB could partially explain the lack of CD4+ T cells in the CNS of Itk−/− mice and suggest that Itk signaling may also play an important role in T-cell migration across CNS barriers.

Itk−/− mice show inefficient migration across the BBB during EAE and in vitro. A, CD4+ T cells (2.5 × 105) isolated from MOG peptide-immunized WT and Itk−/− mice were added to a layer of mouse brain endothelial cells (bEnd3) cultured as a model of the brain endothelial barrier, and the number of cells migrating across the layer was determined at 1 and 24 h after addition of T cells. *p < 0.05, means ± SEMs, by unpaired Student's t test. B, WT or Itk−/− CD4+ T cells (1 × 105) were plated on coverglass, and the movement of cells was recorded by time-lapse video microscopy. The time (t) to travel 100 μm (d) was measured and the velocity calculated based on the equation v = d/t. C–F, CD4+ T cells isolated from MOG–TCR transgenic mice (2D2-TCR-Tg) mice were stimulated with 5 μg/ml MOG peptide for 72 h into Th0, Th1, Th2, and Th17 cells. Cells (5 × 105) were loaded onto a layer of mouse brain endothelial cells (bEnd3) cultured as a model of the brain endothelial barrier, and the number of Th1 (C), Th17 (D), Th0 (E), and Th2 (F) cells migrating across the layer was determined at 1 and 24 h after the addition of T cells. *p < 0.05, means ± SEMs, by unpaired Student's t test (n = 3).
Inhibition of Itk signaling decreases MOG-specific Th1 and Th17 cell migration across the BBB
We next determined whether inhibition of Itk signaling alters specifically the migration of MOG-specific Itk-sufficient effector T cells. We isolated T cells from MOG–TCR transgenic mice (2D2-TCR-Tg) and induced their differentiation to Th0, Th1, Th2, or Th17 in vitro. These Th cells were treated with an Itk inhibitor and evaluated for the ability to migrate across our in vitro BBB. We found that significantly lower numbers of both Th1 and Th17 cells treated with the Itk inhibitor were recovered from the bottom of transwells at both early and later time points compared with vehicle controls (Fig. 5C,D). In contrast, migration of Th0 and Th2 cells were not affected (Fig. 5E,F). This confirms that inhibition of Itk signaling alters the migration of effector CD4 T cells across brain endothelial barrier cells. These results are consistent with the initial observation of reduced numbers of immune cells in the CNS and attenuated disease in Itk−/− mice and in recipients of Itk−/− CD4 T cells.
Displacement of F-actin in Itk−/− CD4 T cells occurs specifically under conditions of peptide/major histocompatibility complex (MHC) Class II–TCR interactions
Cytoskeletal reorganization is necessary for cell movement, cell function, and communication with other cells (Lafouresse et al., 2013). Our data indicate that Itk plays a role in both CD4+ T-cell response to myelin antigen (MOG) stimulation (Fig. 4), as well as in their ability to migrate across the BBB (Fig. 5). Previous studies have shown that T cells lacking Itk are defective in their ability to polymerize actin, to become polarized, and to reorganize their cytoskeleton in response to TCR engagement, contributing to defective T-cell activation (Labno et al., 2003; Gomez-Rodriguez et al., 2007; Singleton et al., 2011). To further investigate the role of Itk in actin cytoskeletal reorganization in CD4+ T cells during myelin antigen restimulation, we isolated CD4+ T cells from WT and Itk−/− mice immunized previously with MOG peptide, stimulated them in vitro with MOG peptide (in the presence of antigen presenting cells), and stained for F-actin and the CD4 coreceptor. We observed that MOG peptide-stimulated Itk−/− CD4+ T cells exhibit distinct displacement of actin away from the CD4 coreceptor. This is in stark contrast to MOG peptide-stimulated WT CD4+ T cells, which showed complete colocalization between actin and the CD4 coreceptor (Fig. 6A,C). Unstimulated WT or Itk−/− CD4+ T cells, stimulation with varying concentrations of anti-CD3, or stimulation with anti-CD3 in the presence of APCs (data not shown) did not result in displacement of F-actin from the CD4 coreceptor at the time points examined (Fig. 6B). These findings strongly suggest that Itk signaling induces actin colocalization with CD4 coreceptor specifically under conditions of antigen/MHC Class II–TCR interactions. Put another way, actin displacement in Itk−/− CD4+ T cells occurs maximally under conditions of peptide/MHC Class II–TCR interaction.

Antigen-specific defect in F-actin/CD4 colocalization in the absence of Itk. CD4+ T cells isolated from WT or Itk−/− mice immunized with MOG peptide were stimulated with 5 μg/ml MOG peptide for 24 h (left) or 72 h (right) (A) or plate-bound α-CD3 (coated at the indicated concentrations α-CD3) for 24 h (left) or 72 h (right) (B). Cells were fixed and stained with Alexa Fluor 488 α-CD4 and Alexa Fluor 568 phalloidin to visualize F-actin. Representative images taken from each group. C, Images from each treatment group were analyzed to quantify the colocalization or displacement of F-actin from CD4 coreceptor (n = 100 per each group).
LatB partially rescues migration of Itk−/− CD4+ T cells across the BBB
Cell migration strongly depends on the organization and degree of actin filament polymerization (Lafouresse et al., 2013). LatB is a marine toxin that dose dependently inhibits actin polymerization by sequestering globular actin (G-actin) and thus prevents F-actin assembly (Wakatsuki et al., 2001). For cells to migrate efficiently, the actin cytoskeleton needs to be dynamic, (i.e., polymerizing into F-actin and depolymerizing to G-actin; Abu Shah and Keren, 2013). At high concentrations, LatB completely prevents F-actin assembly and disrupts actin polymerization, thus blocking cell migration. However, at lower concentrations, LatB allows more dynamic changes in F-actin polymerization and depolymerization (Wakatsuki et al., 2001). Thus, low concentrations of LatB can enhance the actin dynamics. We wanted to determine whether mild disruption of actin polymerization using low concentrations of LatB in Itk−/− cells would in part rescue signaling downstream of Itk and promote more efficient migration across brain endothelial cell monolayers. We treated MOG peptide-activated Itk−/− CD4+ T cells with a low concentration (1 μm) of LatB before their migration. We observed a significant increase in LatB-treated Itk−/− T cells recovered at the bottom of the transwell after their transmigration across the in vitro BBB (Fig. 7A). This is compared with lower numbers of vehicle-treated cells. These results further confirm that the lack of Itk signaling, at least in large part, attenuated disease in Itk−/− mice by hampering the migration of pathogenic T cells into the CNS, thereby reducing the collateral tissue damage in the CNS.

LatB partially rescues the transmigration capacity of CD4+ T cells by actin–cytoskeletal reorganization. A, CD4+ T cells isolated from Itk−/− mice immunized with MOG peptide were stimulated with 5 μg/ml MOG peptide for 72 h and pretreated with DMSO or LatB (1 μm) for 1 h. Cells were loaded onto mouse brain endothelial barrier, and the number of cells migrating across the layer was determined at 1 and 24 h after the addition of T cells. *p < 0.05, means ± SEMs, by unpaired Student's t test (n = 4). B, MOG peptide-stimulated Itk−/− CD4+ T cells were treated with DMSO or LatB (1 μm) for 1 h, and cells were fixed and stained with Alexa Fluor 488 α-CD4 and Alexa Fluor 568 phalloidin to visualize F-actin. MOG peptide-stimulated WT CD4+ T cells were also used as control.
We next determined whether LatB induces discernible changes in F-actin morphology. We observed that Itk−/− CD4+ T cells activated with MOG peptide and treated with LatB (as above) display a distinct change in expression and colocalization of F-actin in relation to the CD4 coreceptor compared with LatB untreated Itk−/− CD4+ T cells (Fig. 7B). There was less detectable F-actin in LatB-treated Itk−/− CD4+ T cells (Fig. 7B). Moreover, it appears that LatB sequestered F-actin in a dense area in the cell that is positioned in close proximity to the CD4 coreceptor (Fig. 7B). Thus, we hypothesize that Itk-regulated actin positioning in the proximity of CD4 is critical for the function of CD4+ T cells.
Itk signaling regulates Treg/Th17 axis to exacerbate EAE
Foxp3+ Treg cells can suppress inflammation and ameliorate pathogenesis during EAE (Fletcher et al., 2009). We wanted to determine whether Treg cells played a role in the disease phenotype observed in Itk−/− mice. The number of Treg cells in brain and spinal cord of WT and Itk−/− mice at day 30 were not statistically significant (Fig. 8A). However, although there were no differences in the number of Treg cells, we found that the ratio of conventional T (Tconv)/Treg cells was perturbed in the periphery of naive Itk−/− mice, which was maintained during the progression of EAE (Fig. 8B). The absence of Itk signaling also resulted in better expansion of Treg cells ex vivo in response to MOG peptide, with a significantly higher proportion of Treg cells in cultures of Itk−/− splenocytes isolated at either day 10 or day 17 after EAE induction (Fig. 8C).

Itk signaling regulates the Treg/Th17 axis to exacerbate EAE. A, Cells were isolated from the brain and spinal cord of day 31 immunized mice and analyzed for CD4+Foxp3+ Treg cells by FACS analysis, three mice per group. B, Splenocytes from the indicated WT and Itk−/− mice were analyzed for CD4+ T cells (Tconv) and CD4+ Foxp3+ T cells (Treg), and the number and relative proportion of Tconv to Treg cells was calculated. C, Splenocytes of WT and Itk−/− mice isolated from day 10 (left) and day 17 (right) immunized mice were stimulated in vitro with 5 μg/ml MOG peptide for 72 h and analyzed for the ratio of Tconv/Treg cells as in B. n = 3, values are means ± SEMs, by unpaired Student's t test. D, WT or Itk−/− mice were immunized to develop EAE and scored daily for clinical signs of EAE. Itk−/− mice were injected with either vehicle (PBS) or 200 μg α-CD25 (PC61 mAb) antibody every 4 d. Note that four of five mice injected with α-CD25 developed disease. n = 5, values are means ± SEMs, *p < 0.05 by two-way ANOVA.
To determine whether this Tconv/Treg cell ratio contributed to the reduced susceptibility of Itk−/− mice to developing EAE, we depleted Treg cells in vivo by administration of α-CD25 and then induced EAE in Itk−/− mice. Interestingly, ablation of Treg cells resulted in increased disease severity in Itk−/− mice, although the onset of EAE was still considerably delayed (Fig. 8D), suggesting that Itk regulates autoimmune pathologies during EAE in part by controlling the Treg/T effector balance.
Discussion
In this study, we investigated the role of Itk in the CD4+ T cell-mediated neuroinflammatory disease, EAE, a murine model of MS. Both EAE and MS are characterized by invasion of inflammatory Th1 and Th17 autoreactive cells into the CNS that cause damage to myelin tissue, resulting in paralysis and neuronal damage and loss (Keegan and Noseworthy, 2002; Kohm et al., 2002; El-behi et al., 2010). Furthermore, defects in Treg cell numbers or function are also pathogenic for both EAE and MS (Kohm et al., 2002; Viglietta et al., 2004; Haas et al., 2005; Fletcher et al., 2009). We found that the absence of Itk signaling is protective in EAE, suggesting that Itk plays an important role in the generation of autoreactive CD4+ T cells that are pathogenic in EAE. This work supports and extends previous work by Gomez-Rodriguez et al. (2009) showing that Itk is essential for the production of IL-17A by Th17 cells via activation and nuclear translocation of nuclear factor of activated T-cell 1 (NFATc1). In addition to potentiating Th17 responses in vivo, we also show that Itk is essential for the elaboration of effector function of autoreactive Th1 cells and that Itk signaling regulates the balance between Treg and T effector cells to exacerbate autoimmune pathologies during EAE. Furthermore, we show that Itk is important for the ability of effector CD4+ T cells to migrate into the CNS and critical for proper activation of CD4+ T cells in part by regulating colocalization between CD4 and F-actin.
The progression of EAE follows an initial Th1 response, which transitions to an IL-17-driven response (Jäger et al., 2009). Our results suggest that Itk may play a more critical role in the acute phase of EAE, because the clinical signs and number of CD4+ T cells in the CNS of the Itk−/− mice eventually catch up with the WT animals at later stages of development of the disease. In the Itk−/− mouse model, this may be attributable to the presence of other populations of T cells that can partially compensate for the reduced Th differentiation, because our control experiments transferring purified Itk−/− CD4+ T cells into T cell-deficient mice revealed an even more profound defect in the ability of these cells to cause disease, supporting the conclusion that Itk regulates the ability of CD4+ T cells to cause pathology in this model. Indeed, although we found that Itk−/− T cells have the capacity to become producers of IFNγ, they are less likely to migrate to the brain and contribute to the pathology of the disease. We and others have suggested that Itk−/− CD4+ T cells retain Th1 responses (Fowell et al., 1999; Miller et al., 2004; Sahu et al., 2008; Kannan et al., 2013), and so we were also surprised that Itk was also required for Th1 responses and IFNγ secretion in this disease, because this has not been identified previously as a defect in these cells. This could be secondary to Itk regulation of Ca2+ responses and activation of nuclear factors such as NFAT (Schwartzberg et al., 2005), both of which are critical for the rapid production of effector cytokines IFNγ and IL-17A by differentiated Th cells during TCR stimulation (Murphy and Reiner, 2002). It is also possible that the role of Itk in the initial stimulation and differentiation of these cells to the Th1 lineage is different compared with its role in already differentiated Th1 cells, a point we are investigating further. Nevertheless, our results suggest that Itk−/− T cells, in either Itk−/− mice or the T-cell transfer model, exhibit delayed induction of pathogenesis related to reduced IFNγ production, and the transition to IL-17A production does not occur.
EAE development is dependent on the ability of inflammatory CD4+ T cells (and other immune cells) to migrate to the CNS in which they mount inflammatory responses against myelin tissue, resulting in tissue damage (Mills et al., 2008, 2012; Kurkowska-Jastrzębska et al., 2013; Lin et al., 2014). We and others have shown previously that Itk signaling can promote migration of T cells via the chemokine stromal cell-derived factor 1α by driving actin rearrangements downstream of CXC chemokine receptor 4 (Fischer et al., 2004; Takesono et al., 2004). Consistent with this, we found reduced numbers of Itk−/− CD4+ effector T cells in the CNS and spinal cord. Moreover, we demonstrated that Itk−/− CD4+ T cells isolated from MOG-immunized mice were less efficient in migrating across brain endothelial cells in an in vitro BBB model. We also observed that treatment of WT MOG-specific Th1 and Th17 cells with an Itk inhibitor led to a decrease in migration across brain endothelial cell monolayers. Furthermore, treatment of Itk−/− CD4+ T cells with low concentrations of LatB, which enhances the turnover of actin and facilitates cell migration, led to enhanced migration, suggesting that regulation of the actin cytoskeleton by Itk in part regulates these events. This further confirms that Itk signaling potentiates and/or promotes CD4+ T-cell migration into the CNS and resultant neuroinflammation/neurodegeneration. We also found that activation of effector CD4+ T cells in the absence of Itk is affected by alterations in the actin cytoskeleton. Although the defects in actin cytoskeletal rearrangement downstream of the TCR is well established in the absence of Itk (Grasis and Tsoukas, 2011), what is not known is how these defects affect CD4 coreceptor localization. We observed disrupted interaction between CD4 and F-actin in the absence of Itk, and this is only observed in MOG-activated CD4+ T cells but not under conditions of α-CD3-induced T-cell activation. This antigen-specific defect may contribute to the defect in activation of CD4+ effector T cells and could result in the reduced pathogenicity of these cells. This strongly suggests that Itk signaling can be a therapeutic target for pharmacological modulation to regulate inflammatory T-cell entry into the CNS, such as in MS.
In this work, we also observed that the ratio of Tconv/Treg cells is perturbed in naive Itk−/− mice compared with WT controls. Itk−/− mice have fewer Tconv cells per Treg cell at the basal level and maintain this altered ratio throughout the progression of EAE. Gomez-Rodriguez et al. (2014) have shown recently that Itk regulates the balance between Treg cells and Th17 cells, and we have also shown that the absence of Itk signaling results in enhanced development of Treg cells (Huang et al., 2014a). Our data suggest that this regulation of Th17 versus Treg cells may control, in part, the ability to develop EAE. Thus, in addition to dampening detrimental effector responses, absence of Itk signaling may also favor the expansion and/or survival of Treg cells to the detriment of Th17 cells. Foxp3+ Treg cells have been shown to be critical for suppression and regulation of inflammation during EAE (Kohm et al., 2002). Consistent with this, we find that depletion of Treg cells during and after EAE immunization resulted in more severe disease pathophysiology in Itk−/− mice, albeit delayed. The fact that Treg-depleted Itk−/− mice still exhibit delayed disease onset would suggest that the protection we see is primarily attributable to a defect in effector T-cell function and that this is further enhanced by the presence of a lower ratio of Tconv/Treg cells. These results suggest that inhibiting Itk may tip the balance in favor of anti-inflammatory responses, which would be highly favorable in conditions such as inflammatory, autoimmune diseases, and prevention of transplant rejection.
These results are interesting when taken in the context of published work on the regulation of Th cell fate by mammalian target of rapamycin (mTOR) signaling. Recent work has suggested that mTOR signaling can promote the differentiation of effector CD4+ T cells, whereas absence of mTOR signaling results in preferential expansion of Treg cells (Chi, 2012; Powell et al., 2012). mTORC1 and mTORC2 promotes differentiation of Th1 and Th17 cells, whereas mTORC2 potentiates Th2 responses (Lee et al., 2010; Delgoffe et al., 2011; Chi, 2012). mTORC1 and mTORC2 have been shown to inhibit induction of Foxp3 and subsequent differentiation of Treg cells (Lee et al., 2010; Delgoffe et al., 2011). Our work is also of interest given the findings of Gomez-Rodriguez et al. (2014) that Itk signals repress the expression of phosphatase and tensin homolog, which can control mTOR signaling downstream of the TCR to affect the balance between effector Th17 CD4+ T-cell and Treg responses. Our recent work also supports this conclusion because we have found that inhibition of the kinase activity of Itk enhances the development of Treg cells (Huang et al., 2014a). However, although the function of inducible Treg cells is not affected by Itk (Gomez-Rodriguez et al., 2014), we have shown that the function of natural or thymic derived Treg cells is dependent on Itk (Huang et al., 2014a). Note that it is not clear whether Treg cells involved in regulating the development of EAE are inducible or thymic/natural Treg cells, which may determine how effective the balance between suppressive and pathogenic T-cell responses are in the Itk−/− mice developing EAE. Nevertheless, it is clear that the absence of Itk signaling not only dampens effector CD4+ T-cell responses but also tips the balance in favor of anti-inflammatory responses by promoting the expansion of regulatory T cells.
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-deficient mice spontaneously develop autoreactive T cells that infiltrate various organs, and Jain et al. (2013) reported recently that the absence of Itk results in the accumulation of autoreactive CTLA-4-deficient T cells in secondary lymph nodes, alleviating the autoimmune destruction of pancreas in models of type I diabetes (Jain et al., 2013). These data suggest that Itk may control the ability of activated T cells to leave the lymph nodes and access sites of auto-antigen for pathogenic destruction of tissue. Our findings support these conclusions but also indicate that Itk regulates other aspects of the T-cell immune response and biology. We propose that the absence of Itk signaling protects against neuroinflammation during EAE through a number of mechanisms. First, signaling through Itk is essential for the generation of autoreactive effector Th1 and Th17 cells that are central to neuroinflammation. Second, Itk signaling regulates the balance between pathogenic Th1/Th17 and tolerogenic Treg cells to exacerbate EAE. Finally, Itk signaling may also play a role in how antigen-specific cells migrate across the BBB. Therefore, this work has implications for understanding Itk as a potential therapeutic target. In light of this, inhibitors of Itk could be attractive options for the treatment of Th1/Th17-mediated autoimmune pathologies such as MS and in treatment regimens in which it would be beneficial to expand regulatory CD4+ T cells.
Footnotes
This work was funded by National Institutes of Health Grants R01 NS063011 (M.S.B.) and AI51626 (A.A.).
- Correspondence should be addressed to either Margaret S. Bynoe or Avery August, Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853. msb76{at}cornell.edu, averyaugust{at}cornell.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}