

Abstract
The δ opioid receptor (δR) is a promising alternate target for pain management because δR agonists show decreased abuse potential compared with current opioid analgesics that target the μ opioid receptor. A critical limitation in developing δR as an analgesic target, however, is that δR agonists show relatively low efficacy in vivo, requiring the use of high doses that often cause adverse effects, such as convulsions. Here we tested whether intracellular retention of δR in sensory neurons contributes to this low δR agonist efficacy in vivo by limiting surface δR expression. Using direct visualization of δR trafficking and localization, we define a phosphatase and tensin homolog (PTEN)-regulated checkpoint that retains δR in the Golgi and decreases surface delivery in rat and mice sensory neurons. PTEN inhibition releases δR from this checkpoint and stimulates delivery of exogenous and endogenous δR to the neuronal surface both in vitro and in vivo. PTEN inhibition in vivo increases the percentage of TG neurons expressing δR on the surface and allows efficient δR-mediated antihyperalgesia in mice. Together, we define a critical role for PTEN in regulating the surface delivery and bioavailability of the δR, explain the low efficacy of δR agonists in vivo, and provide evidence that active δR relocation is a viable strategy to increase δR antinociception.
SIGNIFICANCE STATEMENT Opioid analgesics, such as morphine, which target the μ opioid receptor (μR), have been the mainstay of pain management, but their use is highly limited by adverse effects and their variable efficacy in chronic pain. Identifying alternate analgesic targets is therefore of great significance. Although the δ opioid receptor (δR) is an attractive option, a critical limiting factor in developing δR as a target has been the low efficacy of δR agonists. Why δR agonists show low efficacy is still under debate. This study provides mechanistic and functional data that intracellular localization of δR in neurons is a key factor that contributes to low agonist efficacy, and presents a proof of mechanism that relocating δR improves efficacy.
Introduction
The δ opioid receptor (δR) is a prototypical GPCR that is an attractive target for managing neuropsychiatric disorders, including pain, anxiety, and depression. δR expressed in nociceptive neurons can reduce cAMP and inhibit Ca2+ channels to inhibit neuronal activity (Jutkiewicz et al., 2006; Cahill et al., 2007; Pradhan et al., 2011; Bardoni et al., 2014). δR agonists can cause antinociception in some conditions, and they show reduced abuse liability compared with current opioids that target the μ opioid receptor (μR) (Vanderah, 2010; Gendron et al., 2016). δR-mediated antinociception in vivo, however, requires high doses approaching concentrations that cause serious adverse effects (Danielsson et al., 2006; Jutkiewicz et al., 2006; Pradhan et al., 2011; Gendron et al., 2016). This low agonist efficacy is a critical limiting factor in developing δR as a drug target.
One explanation for this low efficacy is that there is limited surface δR in nociceptive neurons, where δR is localized mainly to intracellular structures (Svingos et al., 1995; Cheng et al., 1997; Zhang et al., 1998; Petaja-Repo et al., 2000; Cahill et al., 2001a, b; Wang et al., 2001; Bao et al., 2003; Kim and von Zastrow, 2003). Several conditions, including inflammation, δR activation, chronic opioid treatment, and pharmacological chaperones, can increase surface δR and “prime” nociceptive neurons for δR antinociception (Stein et al., 1989; Patwardhan et al., 2005, 2006; Gavériaux-Ruff et al., 2008; Rowan et al., 2009; Pettinger et al., 2013; Brackley et al., 2016). This increase in surface δR levels can be partly attributed to increased δR levels by higher transcription or translational efficiency (Zhang et al., 1998; Petaja-Repo et al., 2000; Cahill et al., 2001a, b; Wang et al., 2001; Bao et al., 2003; Gendron et al., 2015), raising the possibility that translocating δR to the surface could increase δR agonist efficacy.
Interestingly, in non-neuronal cells, such as undifferentiated PC12 cells, δR expresses well on the cell surface (Kim and von Zastrow, 2003; Gupta et al., 2014). Nerve Growth Factor (NGF), which initiates PC12 differentiation, also triggers intracellular δR retention at relatively short timescales (Kim and von Zastrow, 2003). A major pathway that affects both differentiation and trafficking is the phosphatidylinositol 3 kinase (PI3K)/phosphatase and tensin homolog (PTEN) pathway (Ross et al., 2001; Chen et al., 2012). PI3K adds a phosphate to the 3′ position on the inositol ring of phosphatidylinositides and profoundly influences trafficking and signaling by recruiting adapters and effectors that specifically bind different phosphorylated species of phosphoinositides (Song et al., 2012; De Matteis and Godi, 2004; Balla et al., 2012). In contrast to the well-studied PI3K, the role of the opposing 3′ phosphatase PTEN in neuronal differentiation and membrane trafficking is less known (Christie et al., 2010; Liu et al., 2010; Song et al., 2012). An increase in PTEN expression, seen at around P7 in mice, is required for the differentiation of neurons in vivo and for NGF-induced differentiation of PC12 cells in culture (Lachyankar et al., 2000; Ross et al., 2001). Interestingly, although acute NGF activates PI3K, persistent NGF downregulates PI3K, facilitating neuronal differentiation (Chen et al., 2012). Paralleling this timescale, long-term NGF increases δR-induced inhibition of postsynaptic currents in brainstem slices (Bie et al., 2010), whereas short-term NGF causes intracellular δR retention (Kim and von Zastrow, 2003). Therefore, we hypothesized that the PI3K-PTEN balance might coordinate δR surface levels, with 3′ phosphoinositides increasing surface delivery and PTEN causing intracellular retention.
In this study, we tested the key prediction of this hypothesis: that inhibiting PTEN will drive δR to the neuronal surface and increase δR agonist efficacy. We show that, in peripheral sensory neurons, PTEN inhibition reduces intracellular δR and increases the functional pool of δR on the neuronal surface both in culture and in vivo. PTEN inhibition increases SNC-80 efficacy in a mouse model of pain without detectable side effects. Together, our results provide an explanation for why δR agonists are largely ineffective in vivo, and a strategy to improve δR antinociception by stimulating surface δR delivery.
Materials and Methods
Cell lines and DNA constructs.
The primary cell line used in this investigation was the pheochromocytoma-12 (PC12) cell line isolated from rat adrenal medulla tissue. This cell line was purchase and verified by ATCC (catalog #CRL-1721, RRID:CVCL_0481). These cells were cultured in F12K medium (Invitrogen) supplemented with 10% horse serum and 5% FBS. Stable and transient PC12 cell lines expressing the full-length δR with an N-terminal signal sequence Flag epitope (SSF-δR) have been created to study the intracellular retention of δR upon NGF (100 ng/ml) treatment in a fixed cell assay. The full-length μR with an N-terminal signal sequence Flag epitope (SSF-μR) was used to control for receptor specific effects. For transfection and maintenance of a stable cell lines, the lipofection reagent Lipofectamine 2000 (Invitrogen) and the selection antibiotic Geneticin were used.
Pharmacological experimental details.
For all fixed-cell pharmacological experiments, the treatments were conducted for 1 h followed by fixation and immunofluorescence microscopy that is detailed in additional methods. The concentrations and usage specifications are listed in Table 1.
Concentrations and usage specifications
Primary trigeminal ganglion cell isolation.
Glass coverslips (Corning) were placed into 6-well plates and precoated with poly D-lysine and laminin to enhance neuron adhesion and promote cell survival before plating. Adult mice (p9–15, C57BL/6J, RRID:IMSR_JAX:000664) were obtained from the animal facility and killed via CO2 asphyxiation. Using large scissors, the head is removed. With small scissors, the skin covering the skull is removed. Cutting away the top of the skull then exposes the brain. The brain is removed by lifting toward the posterior and by cutting cranial nerve V. With the brain removed, the trigeminal ganglia (TG) is visible as longitudinal bundles of tissue. These bundles are cut into small pieces and placed into ice-cold HBSS. A sterile filtered papain solution is applied and incubated for 12 min at 37°C. The papain solution is removed, and a dispase/collagenase is applied for 15–20 min at 37°C. Upon completion of digestion, the TG tissue is triturated with a fire polished Pasteur pipet 3–5 times, or until the neurons appear visual dissociated. The TG cells are then plated onto the poly D-lysine-coated coverslip within a cloning ring to create a more densely populated cell area. After 5 h, the cloning ring can be removed, and fresh neural basal media (Invitrogen) can be added to each well. The animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care-approved facilities, and studies used protocols that have received institutional review and approval from the Institutional Animal Care and Use Committee at the respective institutions.
Transfection of primary neurons.
Lipofectamine 2000 lipofection reagent (Invitrogen) was used per manufacturer's instructions for DNA transfections. Briefly, 2 μg of DNA plasmid and 5–7 μl of Lipofectamine reagent were incubated in 250 μl of Opti-MEM (Invitrogen) for 5 min at room temperature. These mixtures were then combined and left to complex for 25 min. The neuronal growth media was removed but saved, and the DNA-Lipofectamine solution was added dropwise to the cells containing 1 ml of additional Opti-MEM and left to incubate at 37°C for 4–5 h. Upon completion of the transfection, the reagent-containing medium was removed and replaced with saved neuronal growth medium. Identical conditions and protocol were performed for PC12 cell transfections; however, after transfection, F12K + 10% horse serum and 5% FBS-containing medium was used.
Immunofluorescence and fluorescence microscopy.
Cells were plated on poly-D-lysine-coated glass coverslips (Corning) and grown for 24–48 h. For PC12 SSF-δR intracellular retention experiments, cells were treated with NGF (100 ng/ ml) for 1 h followed by fixation in 4% PFA, pH 7.4. The cells were blocked in calcium magnesium containing PBS with 5% FBS, 5% 1 m glycine, and 0.75% Triton X-100. The SSF-δR was labeled with anti-FLAG M1 antibody (1:1000, Sigma-Aldrich catalog #F3040, RRID:AB_439712) conjugated with Alexa-647 (Invitrogen catalog #A-21244 also A21244, RRID:AB_141663), and a trans-Golgi marker, anti-trans-Golgi network (TGN)-38 (1:2000, Sigma-Aldrich catalog #T9826, RRID:AB_796176) rabbit polyclonal for 1 h in the blocking buffer. The coverslips were washed 3 times in calcium magnesium PBS followed by addition of Alexa-568 goat anti-rabbit secondary (1:1000, Thermo Fisher Scientific catalog #A-11011, RRID:AB_143157) antibody in blocking buffer for 1 h. The coverslips were again washed 3 times in calcium magnesium PBS and mounted onto coverslips in calcium magnesium containing anti-fade mounting medium. Following mounting, confocal imaging was performed at 60× magnification using a Nikon TE-2000 inverted microscope with live-cell imaging capabilities and Andor IQ 2 software (Andor iQ, RRID:SCR_014461). Images of representative fields were taken. For all fixed-cell imaging experiments, a minimum of the average fluorescence ratios of 100 cells was quantified to ensure a representative population. Additionally, a minimum of three biological replicates were performed to confirm all results.
Cycloheximide assay.
PC12 SSF-δR cells were plated onto poly-D-lysine-coated coverslips in a 24-well plate and incubated overnight at 37°C. NGF (100 ng/ml) was added to all appropriate samples for 2 h to achieve complete intracellular retention of δR. The media was changed to Opti-MEM + Glutamax (Invitrogen) with cycloheximide (3 μg/ml) for 1 additional hour ± NGF. The cells were then fixed in 4% PFA and blocked in the immunofluorescence-blocking buffer. Immunofluorescence of SSF-δR and intracellular compartmental markers were performed as described previously.
Live-cell imaging of NGF-induced retention and PTEN IH release of δR.
To visualize δR in live cells, eGFP was N-terminally tagged to δR in place of the FLAG-tag previously described. PC12 cells or TG neurons were transfected with eGFP-δR using Lipofectamine 2000 (Invitrogen) as described previously. Before imaging, cells were transferred to poly-D-lysine-coated coverslips. Cells were imaged at 60× magnification using a Nikon TE-2000 inverted microscope with a 37 degree heated enclosure and 5% CO2 environment. Images were acquired every minute for 1.5–2 h. Following 5 min of baseline imaging, pharmacological treatment was added to the coverslip. The images were analyzed and quantified using ImageJ analysis software. In live-cell analysis, due to enhanced consistency from internal controls within a cell, a minimum of 10 cells were quantified from three or more biological replicates to provide sufficient statistical power.
Live-cell imaging of cAMP activity.
We evaluated activation of δR by its specific agonist [d-Ala2, d-Leu5]-enkephalin (DADLE) with a live-cell Förster resonance energy transfer (FRET) reporter of cAMP, exchange protein directly activated by cAMP (EPAC) (de Rooji et al., 1998; Ponsioen et al., 2004). The EPAC sensor contains a cyan fluorescent protein (CFP) (405 nm excitation, 470 nm emission) and yellow fluorescence protein (515 nm excitation, 530 nm emission) (YFP) linked by a cAMP binding domain. When cAMP binds to the sensor, a decrease in the FRET signal (CFP excitation at 405 nm and yellow fluorescence protein emission at 530 nm) is observed. Experiments followed a validated paradigm for cAMP measurements (Sneddon et al., 2000; Bin et al., 2013). Baseline cAMP was stimulated with forskolin (5 μm), and subsequent inhibition following δR agonist addition was calculated via the EPAC CFP/FRET ratio. Additionally, blocking the pool of surface receptors with the irreversible δR antagonist, chlornaltrexamine (CNA) (1 μm), can prevent the basal surface pool of δR from signaling to isolate the response induced following forced surface trafficking of δR.
Ca2+ imaging and electrophysiology.
TG neurons from adult female Sprague Dawley rats (Harlan) (RRID:RGD_5508397) were prepared as previously described (Vaughn and Gold, 2010). Two hours after plating, neurons were transferred to an L15-based media (Vaughn and Gold, 2010; Harriott et al., 2012), where they were stored at room temperature for no more than 8 h. For Ca2+ imaging, neurons were loaded with fura-2 AM in pluronic F127 as previously described (Lu et al., 2006). They were then transferred to bath solution containing vehicle or bpV(Phen) (10 μm) where they were incubated at 35°C for 30 min before study. Depolarization-induced Ca2+ transients were evoked with high K+ (30 mm, 800 ms) every 2 min. SNC80 (1 μm) was then applied 1 min before the application of SNC80 with high K+. The variance around the mean baseline response of all neurons studied in each group was used to identify neurons responsive to SNC80, where a decrease >2 SDs from the mean change observed at baseline was considered a response to SNC80. At the end of each experiment, neurons were challenged with capsaicin (500 nm for 500 ms). Neurons used for voltage-clamp experiments were processed in the same way as those used for Ca2+ imaging, except they were not loaded with fura-2. Inclusion/exclusion criteria were preestablished and were based on the resting Ca2+ levels, which in healthy neurons is <150 nm, the stability of the magnitude of the evoked Ca2+ transient, which, in healthy neurons, is neither consistently increasing nor consistently decreasing over at least 3 applications of high K+ at an interstimulus interval of 3 min, and the rate at which in intracellular Ca2+ level returns to baseline after each depolarizing evoked transient, where the recovery is complete within 4 min in healthy neurons. Voltage-gated Ca2+ currents were recorded with bath and electrode solutions composed to minimize voltage-gated Na+ and K+ currents, as previously described (Vaughn and Gold, 2010). Currents were evoked from a holding potential of −60 mV, to a test potential of −5 mV every 5 s. When currents stabilized after obtaining whole-cell access, SNC80 (1 μm) was applied via a gravity perfusion system. A decrease >2 times the SD from the mean baseline current was considered a response to SNC80.
Fluorescent deltorphin II-Cy3.29.
A cysteine-extended Deltorphin II was custom ordered from www.Lifetein.com for conjugation to Cy3.29 maleimide. Cysteine-extended Deltorphin II (7 mg) was dissolved in 2 ml of dry DMF. Cy3.29 maleimide (10.5 mg = 1.5 equivalents) was dissolved in 2 ml dry Dimethylformamide (DMF). The Deltorphin II and Cy3.29 maleimide solutions were combined with the addition of diisopropylethylamine (60 μl) to keep the reaction from acidifying. The reaction mixture was kept at 37°C for 2 h and then left it at room temperature overnight. The reaction mixture was added drop wise to 50 ml of ethyl acetate to precipitate the product and remove the DMF. The organic phase was decanted. The residue containing the Cy3-labeled Deltorphin II was purified by HPLC on a μ-Bondapak C18 7.8 × 300 mm column; flow 3 ml/min; acetonitrile/water 0.1% TFA gradient. The product peak was further analyzed by UPLC on an Acquity BEH C18 column 1.7μ 2.1 × 50 mm. Following product fraction collection, the pH was adjusted to 8.4 with sodium bicarbonate and concentrated to a volume of 2 ml. The product was desalted by loading the solution on a Waters Sep-Pak C18 column, washing with 5 ml of water and eluting the product with 4 ml of water/20% ethanol. The final product solution was lyophilized to give 2 mg of high purity Cy3 maleimide-labeled Deltorphin II. The product was further characterized my mass spectrometry on a Thermo Fisher LTQ-XL ESI/APCI Linear Ion Trap instrument. The final molecular weight of the cysteine-extended Deltorphin II Cy3.29 was 1637.9.
In vivo PTEN IH and surface receptor quantification.
To establish a time point in mice (C57BL/6J, RRID:IMSR_JAX:000664) for PTEN IH-induced surface trafficking of δR and visualize newly trafficked receptors, we subcutaneously injected a saline control, or the PTEN IH bpV(Phen) at 3 mg/kg, and returned the mice to their home cage. At 2 h intervals (2, 4, 6 h), TG neurons were dissociated following the aforementioned protocol. Instead of plating onto coverslips for in vitro culturing, we isolated the dissociated neurons and incubated them with Deltorphin II Cy3 (100 nm) and Hoechst stain for 10 min on ice. The neurons were then centrifuged at 5× gravity and washed with PBS. The labeled neuron suspension was then transferred to a coverslip for live-cell confocal imaging performed at 37°C and 20× magnification using a Nikon TE-2000 inverted microscope. The percentage of Hoechst-positive Deltorphin II Cy3-expressing cells was determined at each time point.
In vivo analgesia assay.
Subjects were male C57BL/6J mice (RRID:IMSR_JAX:000664), between 9 and 12 weeks old. Mechanical hyperalgesia was tested using the manual von Frey test following standard protocols established by the Pradhan laboratory (Pradhan et al., 2013). Baseline mechanical responses were determined, and inflammatory pain was induced by injecting 13 μl of CFA into the plantar surface of the left hindpaw. On the test day, mice were randomly allocated to different test groups and injected with vehicle or bpV (Phen) (3 mg/kg s.c.), and returned to their home cages. Four hours after injection, mice were injected with vehicle or SNC80 (2 mg/kg i.p.) and mechanical hyperalgesia was assessed 45 min later. To confirm specificity to δR, we treated mice with naltrindole (10 mg/kg, s.c.), a δR-selective antagonist, 1 h before SNC80. The investigator was partially blinded and was not aware whether animals received bpV (Phen) or vehicle. All experiments were done in accordance with Institutional Animal Care and Use Committee-approved protocols at the respective institutions. Animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care-approved facilities with unrestricted access to food and water.
Statistics and data analysis.
Statistical analyses were performed using Prism 5 software (GraphPad, RRID:SCR_002798), and appropriate statistical tests were chosen based on sample size and distribution. For statistical analysis of the quantified imaging data, two-tailed unpaired t tests were performed between the different experimental conditions and controls. For calcium imaging in neurons, sample size was based on a preliminary analysis of control neurons in which we determined the proportion of neurons in three fields of view from each of 2 animals, that were responsive to SNC80, where a neuron was considered responsive to SNC80 if the depolarization evoked Ca2+ transient in the presence of SNC80 was reduced by >2 SDs from the mean of at least 3 transients evoked before the application of SNC80. These preliminary results were used in a power analysis in which we estimated the number of neurons needed to detect a significant increase in the proportion of SNC80-responsive neurons. To compare the population changes in δR-positive neurons, a two-tailed Fisher's Exact test was performed. For the animal data, a power analysis was performed based upon our initial experiment, and a sample size of >5 was estimated to be adequate. We choose to have our minimum sample size be 10 to exceed concerns due to limited sample size. To analyze the significance of the in vivo animal data, a one-way nonparametric ANOVA was performed followed by a Kruskal–Wallis post-test comparing all columns. A p value of <0.05 was considered statistically significant. All images were quantified using macros contained within the ImageJ software package (ImageJ, RRID:SCR_003070). Figures were constructed with ImageJ and Adobe Photoshop CS6 (Adobe Photoshop, RRID:SCR_014199).
Results
PTEN inhibition drives δR relocation to the surface in TG neurons
We initially used neuroendocrine PC12 cells expressing N-terminally FLAG-tagged δR as an experimental system to start addressing regulated δR trafficking. This expression system has been highly useful to study many GPCRs, including δR, and we and others have confirmed that tagged receptors are functional (Guan et al., 1992; Kim and von Zastrow, 2003; Arttamangkul et al., 2008; Puthenveedu et al., 2010; Soohoo and Puthenveedu, 2013; Vistein and Puthenveedu, 2013; Bowman et al., 2015). Consistent with previous observations, FLAG-δR expressed in PC12 cells showed predominantly surface expression (Fig. 1A). NGF treatment for 1 h caused δR accumulation in an intracellular compartment broadly overlapping with the TGN marker TGN-38 (Fig. 1A). Quantitation of the fraction of cells with intracellular δR showed a significant increase after NGF treatment (Fig. 1B). This intracellular accumulation was not a general effect, as NGF did not induce accumulation of the related μ opioid receptor (FLAG-μR) under identical conditions (Fig. 1C,D). To establish that this was a transport block of newly synthesized receptors, we first accumulated δR in the TGN by treating cells with NGF for 1 h to induce Golgi retention, and then chased this accumulated pool by blocking the synthesis of new δR with cycloheximide. This chase was performed either in the absence or presence of continued NGF. The intracellular pool was rapidly chased out in the absence of NGF, but, in the presence of NGF, it persisted even when the synthesis of new δR was blocked, indicating that NGF signaling blocks export of δR from these intracellular compartments that overlap with the Golgi (Fig. 1E).
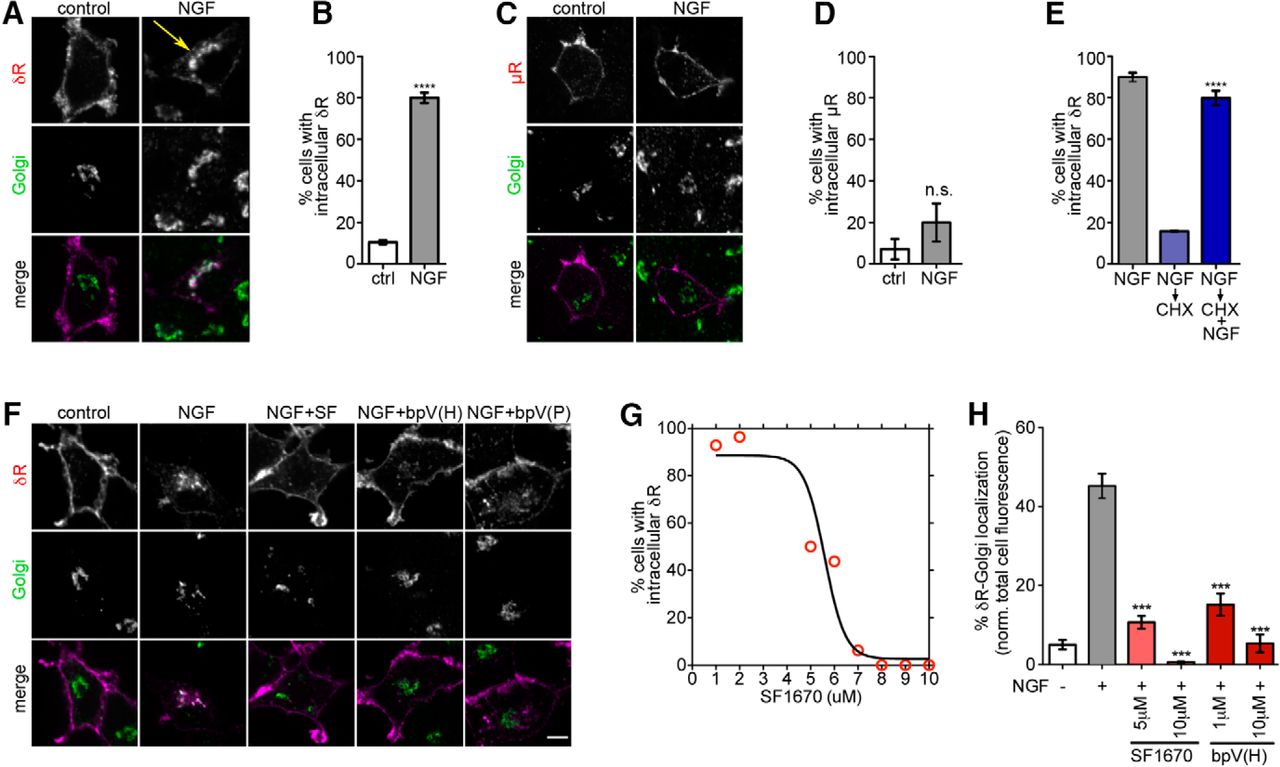
PTEN inhibition releases Golgi-retained δR and promotes surface delivery in PC12 cells. A, Example images of PC12 cells showing surface δR expression (red in merge) and intracellular accumulation of FLAG-δR (arrow) after NGF treatment (60 min at 100 ng/ml), colocalizing with TGN-38 (Golgi, green in merge). B, Quantitation showing an increase in the percentage of cells with δR Golgi localization after NGF (n >100 cells each). Data are mean ± SEM. ****p < 0.0001. C, Example image of a PC12 cell showing surface FLAG-μR as a control. NGF treatment (60 min at 100 ng/ml) causes no internal μR accumulation. D, Quantitation showing that NGF does not increase the percentage of cells showing μR retention (n >50 cells each). Data are mean ± SEM. E, NGF-treated PC12 cells, chased with cycloheximide (CHX) for 1 h in the presence and absence of NGF, to isolate the effect of NGF independent of δR biosynthesis and transport. The percentage of cells with Golgi-localized δR is shown (n >100 cells in each condition). Data are mean ± SEM. ****p < 0.0001, CHX treatments with NGF versus without NGF. F, Example images for control and NGF-treated PC12 cells with inhibition of PTEN by 10 μm of SF1670 (SF), bpV HOptic (bpV(H)), or bpV (Phen) (bpV(P)). PTEN inhibition abolishes Golgi retention of δR. G, A dose-dependent effect of SF1670-mediated loss of Golgi-localized δR in NGF-treated PC12 cells. H, Image analysis and quantification show a significant reduction in the percentage of total δR fluorescence that overlaps with the Golgi upon PTEN inhibition (n >100 cells each). Data are mean ± SEM. ***p < 0.001.
We next asked whether PTEN inhibition would block NGF-mediated δR retention in this experimental system. We tested three different PTEN inhibitors, SF1670, bpV(HOptic), and bpV(Phen), all of which abolished NGF-induced δR retention and induced surface δR delivery (Fig. 1F). Quantitation of the fraction of cells with intracellular δR showed a dose-dependent decrease with SF1670. As a more objective index of intracellular retention of δR, we quantitated the fraction of total cellular δR that overlapped with TGN-38, using TGN-38 fluorescence as a mask. The fraction of δR that was retained in response to NGF was significantly attenuated by PTEN inhibition (Fig. 1H), indicating that PTEN inhibition can counter NGF-induced retention of δR.
Importantly, we next tested whether PTEN inhibition could drive the intracellular pool of δR to the cell surface in physiologically relevant TG neurons. We cultured TG neurons isolated from adult mice and transfected them with FLAG-δR. Because previous reports indicated that δR was predominantly expressed intracellularly in small-diameter neurons, we limited our experiments to neurons with a diameter between 10 and 20 μm (Wang et al., 2010; Pettinger et al., 2013). Consistent with these reports, FLAG-δR was predominantly seen in intracellular compartments that overlapped with the Golgi in these neurons (Fig. 2A). NGF treatment for 1 h did not significantly modify the localization of FLAG-δR or of PTEN, which showed a diffuse cytoplasmic distribution. Treatment with the PTEN inhibitor SF1670 for 1 h caused a loss of intracellular FLAG-δR localization and the appearance of δR on the surface of neurons, regardless of whether NGF was present or not (Fig. 2A). This loss corresponded with a significantly higher fraction of neurons that showed surface FLAG-δR fluorescence (Control = 28.57 ± 12.53%; PTEN IH = 92.86 ± 7.14%; p < 0.001). Quantitation of the fraction of δR that overlapped with the Golgi showed a significant decrease after PTEN inhibition (Fig. 2B). This loss of intracellular δR was also detected as a loss of colocalization between the Golgi marker and FLAG-δR, as measured by Pearson's coefficients across cells (Fig. 2C). As a control, FLAG-μR expressed in TG neurons, and analyzed in an identical manner, did not show intracellular receptor pools, regardless of whether NGF was added or not (Fig. 2D). This suggests that the intracellular localization is an active process that is specific for δR. We next directly visualized the redistribution of δR in neurons following PTEN inhibition, by live-cell confocal microscopy of mouse TG neurons expressing GFP-δR. In TG neurons, GFP-δR was primarily localized to intracellular pools, with minimal surface localization. PTEN inhibition by bpV(Phen) induced loss of the intracellular δR pool and increased the membrane localization (Fig. 2E). A radial profile analysis for each time point showed, quantitatively, that the fluorescence intensity for the intracellular pool (smaller radii) decreases and the plasma membrane (larger radii) increased (Fig. 2F,G). Quantitation of intracellular δR fluorescence in multiple neurons showed a significant decrease in the intracellular δR pool after PTEN inhibition (Fig. 2H). These data demonstrate that PTEN inhibition reduces intracellular δR retention and stimulates δR surface trafficking in mouse TG neurons.

PTEN inhibition releases Golgi-retained δR and promotes surface delivery in TG neurons. A, Example immunofluorescence images demonstrating the localization of expressed FLAG-δR in mouse TG neurons with or without NGF (100 ng/ml for 1 h), with and without the PTEN inhibitor (SF1670, 10 μm). PTEN inhibition drives δR to the surface. B, The fraction of Golgi-localized δR in primary TG neurons decreased significantly upon PTEN inhibition (SF1670, 10 μm) (n >10 neurons each). Data are mean ± SEM. **p < 0.01. ***p < 0.001. C, Pearson's correlation coefficients show decreased colocalization between δR and the Golgi upon SF1670 addition (n >10 neurons each). Data are mean ± SEM. *p < 0.05. D, Example immunofluorescence images of expressed FLAG-μR in mouse TG neurons with or without NGF (100 ng/ml for 1 h). μR was localized to the surface with minimal intracellular fluorescence. E, Example frames from a live-cell movie of mouse TG neurons expressing GFP-δR before and after addition of PTEN inhibitor bpV(Phen) (10 μm). F, Radial profile analysis of the images (schematic) revealed a decrease in the fluorescence intensity of the center and an increase in the periphery over time following PTEN inhibition (added at t = 1 min). Time is depicted as a color gradient from red to blue. G, PTEN inhibition causes an increase in GFP-δR fluorescence on the surface and a decrease in fluorescence in Golgi regions. H, Quantitation across multiple neurons shows a decrease in the percentage of intracellular δR normalized to the total cell fluorescence (n = 10). Data are mean ± SEM. **p < 0.01. Scale bars, 5 μm.
δR delivered to the surface by PTEN inhibition is competent for signaling
To test whether PTEN inhibition increased functional δR on the surface, we measured cAMP inhibition by δR as a primary readout of Gi coupling. Dynamic changes in cAMP levels were detected using the EPAC FRET sensor (DiPilato et al., 2004), simultaneously with activation and endocytosis of δR (Fig. 3A). δR activation was measured as attenuation of forskolin-stimulated cAMP. In NGF-treated PC12 cells expressing the FLAG-δR, PTEN inhibition caused a stronger inhibition of cAMP in response to DADLE, which was ∼48% lower than the NGF only response, consistent with increased availability of δR on the surface (Fig. 3B).

The δR delivered to the surface by PTEN inhibition is competent to inhibit cAMP. A, Schematic of ratiometric analysis for cAMP measurement using the Epac sensor. The CFP, FRET, and FLAG-δR were imaged and followed live in the same PC12 cells. FRET ratio image is mapped to blue for low cAMP and red for high cAMP. B, Inhibition of Fsk-mediated increases in cAMP by DADLE was measured as an index of the activity of surface δR. In NGF-treated PC12 cells, DADLE-mediated inhibition was significantly enhanced by PTEN inhibition (SF1670, 10 μm), as seen by a lower Fsk response (n >15 each). Data are mean ± SEM. **p < 0.01. C, Schematic of experiments to isolate the functional effect of δR delivery by PTEN inhibition after surface δR is blocked by CNA (1 μm). D, The percentage of inhibition of cAMP response under the four conditions shows recovery of DADLE-mediated inhibition by PTEN inhibition after surface δR is blocked by CNA, and that the inhibition requires δR. Data are mean ± SEM. ****p < 0.0001. E–J, Example images (E, G, I) from a time lapse movie showing FRET changes and δR localization after DADLE in the three experimental conditions. The average FRET change (F, H, J) over time from multiple cells for each experimental condition is shown. Red lines indicate curve fits for the Fsk response, DADLE response, and desensitization, over time. Error bars indicate SEM. Scale bars, 5 μm.
To directly test whether the increase in activity was due to release of NGF-retained δR by PTEN inhibition, we blocked preexisting surface δR with the irreversible antagonist CNA. This allowed us to isolate signaling from newly delivered receptors in a defined period of time (schematic in Fig. 3C). Following NGF-induced δR retention, pretreatment with CNA significantly reduced DADLE-mediated δR endocytosis and cAMP inhibition, from ∼80% to 20%, in cells expressing δR (Fig. 3D). Following the NGF-induced δR retention and CNA blockade of surface δR, PTEN inhibition was sufficient to recover agonist-induced δR signaling back to ∼80% (Fig. 3D, time series in Fig. 3E–J). In PC12 cells not exogenously expressing the FLAG-δR construct, DADLE did not inhibit the forskolin-stimulated cAMP, instead causing a 12 ± 11% increase (Fig. 3D), confirming that, in FLAG-δR cells, cAMP inhibition in response to DADLE is specific to δR. Together, our data indicate that the δR delivered to the surface from intracellular pools by PTEN inhibition was competent to bind agonists and induce signaling.
To test whether PTEN inhibition induced an increase in the levels of endogenous functional δR in neurons, we evaluated the ability of PTEN inhibition to increase the δR-mediated inhibition of evoked calcium channel transients in adult rat TG neurons. Following pretreatment of isolated TG neurons with the PTEN inhibitor bpV(Phen) for 30 min, agonist-induced (SNC-80) activation of δR caused a robust inhibition of Ca2+ transients (Fig. 4A,B). To better quantitate the effect of PTEN inhibition, we used the calcium indicator dye fura-2 AM to measure high K+ evoked responses in neurons before and after δR activation. KCl-induced Ca2+ transients were observed as a brief increase in fluorescence intensity followed by decay over time (Fig. 4C). Neurons were then characterized as either nonresponsive (Fig. 4D) or responsive (Fig. 4E) based on the decrease in evoked KCl-induced Ca2+ transients following SNC80 (calculated as more than twice the SD of the initial responses). The percentage of neurons that responded to the δR agonist SNC80 increased significantly following pretreatment with the PTEN inhibitor bpV(Phen), compared with control neurons (∼52% responders following PTEN inhibition compared with ∼13% in control) (Fig. 4F). The magnitude of inhibition across TG neurons did not change significantly. As a control, PTEN inhibition did not increase the fraction of TG neurons that responded to capsaicin in this same pool. A similar trend of increased responses to SNC80 was also observed in adult mouse DRG neurons. Interestingly, in DRG neurons, the magnitude of SNC80-mediated inhibition increased from 29 ± 3.4% in control neurons to 38 ± 4.3% after PTEN inhibition, indicating that the resting levels of δR might vary between neuronal cell types or species. As a control, PTEN inhibition did not change the fraction of responding neurons or the magnitude of inhibition by DAMGO in DRG neurons (50.1 ± 3.2 in control neurons and 47.0 ± 2.9% after PTEN inhibition). Together, our results demonstrate that, by driving endogenous δR to the surface through PTEN inhibition, we can increase functional responses of peripheral neurons to δR agonists.

PTEN inhibition increases the functional pool of endogenous δR in TG neurons. A, Example calcium current traces from a voltage-clamped rat TG neuron after PTEN inhibition, before and after SNC80, showing robust decrease in current after SNC80. B, Time course of decrease in current after SNC80 bath application to a TG neuron after PTEN inhibition. C, Live imaging of calcium transients of rat TG neurons in response to high K, detected by fura-2 AM loading. D, E, Example traces of fura-2 AM responses showing a neuron that is nonresponsive to SNC80 (D) and one that is responsive (E). F, Quantification of the fraction of neurons that respond to SNC80 with and without pretreatment with bpV(Phen). The number of cells in each condition is noted. PTEN inhibition induced a significant increase in the fraction of neurons that respond to SNC80. ****p < 0.0001.
PTEN inhibition stimulates surface delivery of endogenous δR in TG neurons in vivo
We next tested whether PTEN inhibition drives endogenous δR to the surface of TG neurons in vivo. Because the lack of specific antibodies made the detection of endogenous δR challenging, we conjugated the fluorophore Cy-3 to the C terminus of the δR-specific agonist Deltorphin II (delt-Cy3) (Fig. 5A). This approach has been used to detect endogenous GPCRs previously (Arttamangkul et al., 2000), and our conjugation strategy preserved the binding interface of Deltorphin II to δR. To test whether PTEN inhibition released the intracellular retention of endogenous δR in TG neurons, TG neurons were incubated with delt-Cy3 for 10 min following 60 min of PTEN inhibition or control. The cells were then imaged live via confocal microscopy to quantify the amount of fluorescent delt-Cy3 labeling, as an index of the amount of δR present on the cell surface. Neurons positive for delt-Cy3 showed endosome staining due to receptor internalization (Fig. 5B). For neurons that were treated with the PTEN inhibitor bpV(Phen), the integrated density of delt-Cy3 fluorescence per cell was significantly higher compared with control treated neurons (Fig. 5C). Additionally, the number of endosomes observed per neuron also increased significantly following PTEN inhibition, consistent with increased surface binding and internalization of delt-Cy3. Together, this suggests that PTEN inhibition increases the surface levels of endogenous δR in TG neurons.

PTEN inhibition increases the functional surface pool of endogenous δR in TG neurons in vivo. A, Structure of the δR-specific agonist Deltorphin II C-terminally conjugated to Cy3. The final UPLC purification spectrum is shown below and demonstrates a single pure product peak. B, Example images (of n = 3 independent experiments) are shown of cultured mouse TG neurons that were preincubated with delt-Cy3 for 10 min following 1 h control and PTEN inhibitor (bpv(Phen) 10 μm) treatment conditions. Scale bars, 5 μm. C, Quantitation across multiple cells shows that the delt-Cy3 fluorescence integrated density per cell increases following PTEN inhibition (Control, n = 10 neurons; bpV(H), n = 29 neurons). Data are mean ± SEM. *p < 0.05 (two-sided t test vs Control). D, The number of delt-Cy3-positive endosomes per neuron increased following PTEN inhibition (Control, n = 10 neurons; bpV(H), n = 29 neurons). Data are mean ± SEM. ***p < 0.001 (two-sided t test vs Control). E, Mice were injected subcutaneously with saline as a control or the PTEN inhibitor bpV(Phen) (3 mg/kg). TG neurons were isolated 2, 4, and 6 h after injection. Representative images are shown for cells labeled live with Hoechst DNA stain (blue) and Deltorphin II-Cy3 (red). Scale bars, 15 μm. F, Quantitation shows a significant increase in the fraction of delt-Cy3-positive cells 4 and 6 h after PTEN inhibition (contingency plot is shown; saline, n = 234 neurons; bpV(H) 2 h, n = 94 neurons; bpV(H) 4 h, n = 132 neurons; bpV(H) 6 h, n = 147 neurons). ****p < 0.0001. n.s., Not significant (p > 0.05, by Fisher Exact test vs saline).
To determine whether PTEN inhibition would increase the pool of endogenous δR in vivo, we injected adult mice subcutaneously with the PTEN inhibitor bpV(Phen), or saline as a control, isolated TG neurons 2, 4, or 6 h after injection, and labeled them live with delt-Cy3 to detect surface δR by confocal imaging. The cells were counterstained with Hoechst DNA stain to assess the fraction of TG neurons labeled by delt-Cy3. In saline-injected mice, only a small fraction (∼14%) of neurons were labeled with delt-Cy3 (Fig. 5E,F). The fraction (∼21%) was approximately comparable (not significant by Fisher's Exact test) after 2 h of PTEN inhibition. In contrast, after 4 h of PTEN inhibition, ∼80% of neurons were labeled with delt-Cy3 (p < 0.0001). This increase persisted after 6 h, although the fraction (∼48%) was lower than that observed at 4 h (Fig. 5E,F). These data show that PTEN inhibition drives a functional pool of δR to the surface of TG neurons and that the maximal effect was observed 4 h after PTEN inhibition in vivo.
Stimulated δR delivery unmasks antihyperalgesic efficacy of SNC80 in mice
Because δR is an attractive target for pain management, we next tested whether stimulated δR delivery increases receptor availability enough to increase the efficacy of δR agonists. We chose the complete Freund's adjuvant (CFA) model of chronic inflammatory pain in mice because it is a well-accepted and commonly used paradigm for chronic pain and one in which δR agonists have shown partial effects at high doses. In addition, previous studies using conditional knock-out mice have shown that δR-expressing neurons in peripheral ganglia are critical for the pain-relieving effects of the δR agonist SNC80 within the CFA model (Gavériaux-Ruff and Kieffer, 2011). To test antihyperalgesia, mice were injected with CFA in the left hindpaw, and the experiments were performed 72 h later. Mechanical threshold analysis was performed using von Frey filaments 45 min after the final treatment condition, as described previously (Fig. 6A) (Pradhan et al., 2013). We first tested multiple doses of the PTEN inhibitor bpV(Phen) pretreatment to evaluate whether PTEN inhibition on its own altered the CFA-induced mechanical hyperalgesia. A PTEN inhibitor concentration of 3 mg/kg (s.c.) did not alter the CFA-induced mechanical hyperalgesia, and was chosen as an appropriate dose for further studies. In CFA-injected mice, neither bpv(Phen) alone nor the dose of SNC80 tested (2 mg/kg i.p.) relieved CFA-induced mechanical hyperalgesia (Fig. 6B). Our previous experiments in naive mice suggested that bpv(Phen) would be most effective at potentiating SNC80 4 h after administration. Preadministration of bpv(Phen) 4 h before testing significantly increased the efficacy of this dose of SNC80 (Fig. 6B). Importantly, in these conditions, robust antihyperalgesia was observed without noticeable adverse effects, such as convulsions.

PTEN inhibition unmasks the antihyperalgesic efficacy of the δR agonist SNC80. A, The threshold for mechanical hyperalgesia was determined using a manual von Frey hair, in mice after intraplantar injection of CFA. A schematic shows the timeline and doses used in our experiments. B, Mechanical thresholds in mice show that pretreatment with PTEN inhibitor unmasked the antihyperalgesic effects of SNC80 (mean ± SEM). **p < 0.01. SNC80 or bpv (Phen) on their own did not show antihyperalgesia compared with control vehicle-injected mice. The mechanical threshold before CFA treatment was used as baseline (green dashed line). C, A schematic showing the timeline and doses used in testing whether naltrindole blocks the bpV(Phen)-mediated increase in SNC80 efficacy. D, Naltrindole abolishes the increase in SNC80-mediated antihyperalgesia caused by PTEN inhibition (mean ± SEM). **p < 0.01. E, Proposed model for how PTEN/PI3K balance provides a Golgi checkpoint to regulate δR export and surface availability. Following neuronal signals, such as NGF, δR is retained within the TGN due to disruption of the 3′ phosphoinositide balance by either inhibiting PI3K or increasing PTEN activity. By blocking PTEN activity via pharmacologic inhibition, 3′ phosphorylation of phosphoinositides is maintained and δR surface trafficking is stimulated. Increased δR surface trafficking via PTEN inhibition results in greater receptor surface localization, increased agonist-induced δR function, and unmasks the antihyperalgesic effects of SNC80.
To confirm that the antihyperalgesic effect induced by PTEN inhibition was caused by increased activation of δR, we tested whether naltrindole, a δR-selective antagonist, could block the effect of SNC80 in these conditions. We followed a similar paradigm as in Figure 6A, with an additional injection of vehicle or naltrindole (10 mg/kg) 1 h before SNC80 (Fig. 6C). bpV(Phen)-treated animals injected with vehicle before SNC80 showed a significantly increased antihyperalgesic response, comparable with what was seen in Figure 6B. Strikingly, the SNC80-induced antihyperalgesia caused by bpV(Phen) was completely abolished by naltrindole, indicating that this antihyperalgesia was likely δR-mediated (Fig. 6D). Animals treated with naltrindole, without bpV(Phen), also did not show antihyperalgesia with SNC80. Together, our data support the physiological relevance of our model that PTEN inhibition bypasses a critical Golgi export checkpoint to drive the intracellular pool of δR to the neuronal surface and increase the efficacy of δR agonists (Fig. 6E).
Discussion
It is well established that controlling the number of surface receptors, by removal or delivery, is a critical method to control the potency and efficacy of signals. Our understanding of this process has largely been limited to studies of receptor endocytosis and recycling, where many mechanisms and pathways of regulation have been described (Sorkin and Zastrow, 2002; Marchese and Trejo, 2013). The current results, by identifying PTEN as an important factor that controls the delivery of newly synthesized δR, show that regulated delivery from the biosynthetic pathway can control δR physiology.
Intracellular retention of δR has been reported in a variety of neurons, but the details vary depending on the systems tested (Zhang et al., 1998; Cahill et al., 2001b; Bao et al., 2003; Kim and von Zastrow, 2003; Walwyn et al., 2005; Cahill et al., 2007; Mittal et al., 2013; Gendron et al., 2015). In DRG neurons and induced chromaffin cells, δR is localized to large dense-core vesicles (Guan et al., 2005; Wang et al., 2010). In PC12 cells and hippocampal neurons, δR is localized to the Golgi (Kim and von Zastrow, 2003), similar to what we report in this study. It is possible that these differences reflect cell-type-dependent differences in receptor behavior. The intracellular localization is reduced upon tagging the receptors on the C-terminal tail with GFP (Scherrer et al., 2009; Wang et al., 2010) but is retained in HA and Myc-tagged δRs in small-diameter DRG neurons (Wang et al., 2008, 2010; Zhang et al., 2015). This differential subcellular localization seen with different tags suggests that intracellular retention of δR is an actively regulated process, dependent on sequences along the cytoplasmic surface of the receptor, which is shared across multiple neuronal types.
Nevertheless, the regulation of Golgi export of δR might be diverse and neuron-specific. In mature neurons, δR is localized primarily to intracellular structures even without extraneous NGF signaling (Fig. 1A) (Zhang et al., 1998; Cahill et al., 2001b; Bao et al., 2003; Kim and von Zastrow, 2003; Walwyn et al., 2005; Cahill et al., 2007; Gendron et al., 2015). This suggests that neuronal differentiation causes persistent changes in cellular signaling that maintain the regulated receptor trafficking through this Golgi checkpoint. In brainstem slices, however, chronic NGF treatment of >4 h increased the deltorphin-dependent suppression of EPSCs (Bie et al., 2010). It is possible that NGF elicits different functional responses in central neurons compared with peripheral neurons, as the increased response was prevented by a PI3K inhibitor (Bie et al., 2010). At these timescales, however, NGF might also increase expression of many genes, including δR, via transcriptional and epigenetic changes (Huang and Reichardt, 2003; Chen et al., 2008), and it is conceivable that the total response could increase, even though the majority of receptors are retained in intracellular pools. Consistent with this, NGF and neuronal differentiation, via a complex of proteins associated with ARMS/Kidins220, can increase the surface pools of glutamate receptors and secreted cargo, such as the human growth hormone (Arévalo et al., 2010; López-Benito et al., 2016). Conditions, such as chronic inflammation, or pharmacological chaperones, which increase total δR mRNA and protein expression, are also associated with a proportional increase in surface δR, which could enhance the potency of δR agonists (Bao et al., 2003; Cahill et al., 2003; Kim and von Zastrow, 2003; Patwardhan et al., 2005; Cahill et al., 2007; Pettinger et al., 2013). Inflammation-induced priming of neurons to δR also involves a GRK2-RIPK-mediated modulation of δR function, presumably independent of trafficking (Brackley et al., 2016). It is possible that δR function is modulated by both regulated trafficking to the cell surface as well as a separate priming step at the cell surface, using overlapping mechanisms.
The physiological necessity of intracellular δR retention in neurons, however, is not well understood. A key difference between δR and μR is that δR is internalized and largely degraded after activation by many ligands, whereas μR is largely recycled and reused. Neurons rely on the delivery of newly synthesized δR for recovery of signaling. The intracellular δR could therefore be a “reserve” pool that can be readily delivered to the surface, on demand, much faster than if the receptor had to be synthesized de novo after internalization and degradation of existing surface δR. Consistent with this, δR activation itself might serve as a cue for increasing δR responses, through ROCK-LIMK-mediated changes in the actin cytoskeleton, which regulates agonist-dependent coupling of δR to voltage-dependent calcium channels (Mittal et al., 2013). In our hands, the ROCK pathway was not involved in NGF- or PTEN-regulated δR transport in the absence of agonists, suggesting that the PI3K/PTEN-mediated release and agonist-induced release play distinct roles in δR function on the cell surface.
In addition to serving as a reserve pool, δR in the Golgi could serve an as-yet undiscovered signaling function. Recent data suggest that signals from the related catecholamine receptor, the β2-adrenergic receptor, are spatially encoded, with the same downstream second messengers at different cellular locations activating distinct subsets of genes (Tsvetanova and Zastrow, 2014; Bowman et al., 2016). It is conceivable that the Golgi pool of δR serves a similar function by spatially separating signals originating from the cell surface and the Golgi. Attempts to activate intracellular δR using agonists that are membrane permeable, however, have been largely unsuccessful. Therefore, while the intracellular δR might be competent for signaling, it is not sufficient to cause analgesia, and the analgesic response requires surface localization of δR. In this context, the signaling-regulated checkpoint for TGN export of δR export at the TGN, which we identify here, provides a target to independently manipulate surface delivery of δR and increase antihyperalgesic responses to δR agonists.
Our results provide an important proof of concept: that independently manipulating trafficking of δR is a viable strategy to improve the bioavailability of δR on the surface and to increase the efficacy of δR agonists. Previous attempts at targeting δR to manage pain have mainly focused on developing better δR agonists (Scherrer et al., 2009; Gavériaux-Ruff and Kieffer, 2011; Bardoni et al., 2014). However, these efforts have largely been unsuccessful, as available agonists show poor in vivo efficacy, necessitating the use of high doses that cause adverse effects. Our study provides a critical example on which to base future strategies to improve the bioavailability of δR and increase the responsiveness of neurons to the many δR agonists already developed. Engineered relocation of receptors may therefore provide a general blueprint to precisely manipulate the physiology of many GPCRs that are localized to intracellular pools.
Footnotes
A.A.P. was supported by National Institutes of Health DA031243. M.A.P. was supported by National Institutes of Health Grants DA024698 and GM117425, and National Science Foundation Grant 117776. We thank Claudia Almaguer, Zara Weinberg, and Drs. Rachel Vistein, Amanda Soohoo, Shanna Bowman, and Cary Shiwarski for technical help and comments; Dr. Alison Barth, Joanne Steinmiller, Jen Dry-Henich, Dr. Rebecca Seal, and Adam Goldring for providing help with isolation of TG neurons; Drs. Nathan Urban, Adam Linstedt, Tina Lee, Peter Friedman, Guillermo Romero, Jean-Pierre Vilardaga, and Alessandro Bisello for reagents, comments, and suggestions; and Drs. Mark von Zastrow, John Williams, and Seksiri Arttamangkul for helpful discussions and reagents.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Manojkumar A. Puthenveedu, Department of Biological Sciences, Carnegie Mellon University, Pittsburgh, PA 15213. map3{at}andrew.cmu.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}