

Abstract
Whether synapses in appetite-regulatory brain regions undergo long-term changes in strength in response to satiety peptides is poorly understood. Here we show that following bursts of afferent activity, the neuromodulator and satiety peptide cholecystokinin (CCK) shifts the plasticity of GABA synapses in the dorsomedial nucleus of the hypothalamus of male Sprague Dawley rats from long-term depression to long-term potentiation (LTP). This LTP requires the activation of both type 2 CCK receptors and group 5 metabotropic glutamate receptors, resulting in a rise in astrocytic intracellular calcium and subsequent ATP release. ATP then acts on presynaptic P2X receptors to trigger a prolonged increase in GABA release. Our observations demonstrate a novel form of CCK-mediated plasticity that requires astrocytic ATP release, and could serve as a mechanism for appetite regulation.
SIGNIFICANCE STATEMENT Satiety peptides, like cholecystokinin, play an important role in the central regulation of appetite, but their effect on synaptic plasticity is not well understood. The current data provide novel evidence that cholecystokinin shifts the plasticity from long-term depression to long-term potentiation at GABA synapses in the rat dorsomedial nucleus of the hypothalamus. We also demonstrate that this plasticity requires the concerted action of cholecystokinin and glutamate on astrocytes, triggering the release of the gliotransmitter ATP, which subsequently increases GABA release from neighboring inhibitory terminals. This research reveals a novel neuropeptide-induced switch in the direction of synaptic plasticity that requires astrocytes, and could represent a new mechanism by which cholecystokinin regulates appetite.
Introduction
Energy intake and expenditure are regulated by a complex neuronal network that integrates incoming nutrient and hormonal signals and gates neuronal output to maintain homeostasis. The ability of synapses in this network to undergo long-term changes in strength in response to various stimuli is emerging as a potentially important contributor to appetite regulation (Zeltser et al., 2012). Such plasticity may underlie an individual's ability to match energy intake and expenditure and to adapt to changes in food availability, and has been reported in feeding circuits throughout the hypothalamus (Pinto et al., 2004; Sternson et al., 2005; Crosby et al., 2011; Liu et al., 2012; Qi and Yang, 2015).
Within these feeding circuits, the dorsomedial nucleus of the hypothalamus (DMH) is recognized as a key player in the regulation of appetite (Bellinger and Bernardis, 2002; Otgon-Uul et al., 2016; Jeong et al., 2017). Although much remains to be elucidated with respect to the identity and distribution of feeding-related neuropeptides in DMH neurons, it is well established that the satiety signal and neuromodulator cholecystokinin (CCK) is expressed in this region (Innis et al., 1979; Micevych et al., 1988; Otake, 2005; Wagner et al., 2013). Both type 1 CCK receptor (CCK1R) and type 2 CCK receptor (CCK2R) are also localized in the DMH, and intra-DMH administration of CCK triggers a long-lasting, robust suppression of food intake (Blevins et al., 2000; Chen et al., 2008). This long-lasting effect could be mediated by long-term changes in the strength of synapses in the DMH. We have previously demonstrated that CCK increases GABA release onto DMH neurons (Crosby et al., 2015), but whether CCK influences the plasticity of these synapses in response to bursts of afferent activity remains unknown.
It is well accepted that astrocytes are key players in synaptic transmission and plasticity (Araque et al., 2014). Many vital roles of astrocytes are performed in a neuronal activity-dependent manner, as follows: the clearance of synaptic glutamate (Armbruster et al., 2016); the modulation of basal synaptic transmission (Panatier et al., 2011); and the release of energy substrates for neuronal consumption (Mächler et al., 2016). Furthermore, astrocytes have been implicated in numerous complex behaviors, including sleep homeostasis (Halassa et al., 2009), the control of breathing (Gourine et al., 2010), wakefulness (Papouin et al., 2017), and the regulation of appetite (Kim et al., 2014). Because CCK2Rs are also expressed on astrocytes (Hösli et al., 1993; Müller et al., 1997) and CCK enhances astrocytic Ca2+ signaling in other brain regions (Müller et al., 1997), we hypothesized that CCK also influences the synaptic plasticity of DMH neurons through an astrocyte-dependent pathway.
Here we report that CCK shifts the plasticity of GABA synapses from long-term depression (LTD) observed under basal conditions to long-term potentiation (LTP). This LTP is presynaptic and requires the activation of CCK2Rs that act in concert with group 5 metabotropic glutamate receptors (mGluR5s) on astrocytes to trigger a rise in intracellular calcium and a release of the gliotransmitter ATP. ATP subsequently acts on purinergic receptors on GABA afferents to enhance GABA release.
Materials and Methods
Animals.
All experiments were performed using male Sprague Dawley rats [postnatal day 21 (P21) to P30] obtained from Charles River Laboratories (Rat Genome Database catalog #734476; RRID:RGD_734476) or transgenic mice that were a heterozygous cross between a cre-dependent GCaMP3 line [International Mouse Strain Resource (IMSR) catalog #JAX:014538; RRID:IMSR_JAX:014538) and a tamoxifen-inducible (ERT2) astrocyte-specific glutamate aspartate transporter (GLAST) promoter (IMSR catalog #JAX:012586; RRID:IMSR_JAX:012586). Two weeks before the imaging experiments, mice received three consecutive tamoxifen injections (100 mg/kg, 10 mg/ml corn oil stock). Animals were maintained in specified pathogen-free conditions on a 12 h light/dark cycle at 22°C with food and water available ad libitum. All experimental protocols were performed in accordance with guidelines established by the Canadian Council on Animal Care, with approval from the University of Calgary and Mount Allison University Animal Care and Use Committees.
Brain slice preparation.
Animals were anesthetized with isoflurane and decapitated, and their brains were quickly removed and placed in ice-cold slicing solution containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, and 75 sucrose saturated with 95%/5% O2/CO2. Angled horizontal slices (250 μm) containing the DMH were obtained using a vibrating slicer (Leica), and slices were incubated in 32.5°C artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, and 10 glucose saturated with 95%/5% O2/CO2 for a minimum of 60 min. Slices remained in this incubation chamber until recording commenced.
Electrophysiology.
Hypothalamic slices were submerged in a recording chamber and superfused with 32.5°C aCSF at a flow rate of 1 ml/min. DMH neurons in the compact zone were identified and visualized with an Olympus upright microscope fitted with infrared differential interference contrast optics. Whole-cell electrophysiological recordings were obtained using borosilicate glass microelectrodes (tip resistance, 4.0–6.0 MΩ) filled with an intracellular solution containing the following (in mm): 116 potassium gluconate, 8 NaCl, 2 MgCl2, 8 KCl, 1 potassium EGTA, 4 potassium ATP, 0.3 sodium GTP, and 10 HEPES, corrected to pH 7.2 with KOH. In a subset of experiments, GDPβS (1 mm) was included in the intracellular solution and sodium GTP was removed to disrupt G-protein signaling. Recordings were accepted for analysis if changes in access resistance were <15%. Electrophysiological signals were amplified using the Multiclamp700B amplifier (Molecular Devices), low-pass filtered at 1 kHz, digitized at 10 kHz using the Digidata 1322 system (Molecular Devices), and stored for off-line analysis.
Evoked IPSCs were isolated by holding the postsynaptic neuron at −70 mV and by including DNQX (10 μm) in the perfusate to block AMPA and kainate receptor-mediated glutamatergic transmission. GABAergic fibers were stimulated extracellularly with a patch pipette filled with aCSF and positioned ∼50–100 μm lateral to the recorded DMH neuron. IPSCs were evoked at a rate of 0.2 Hz, and paired-pulse responses were obtained by applying a pair of synaptic stimuli 50 ms apart.
In high-frequency stimulation (HFS) experiments, afferents were stimulated at 100 Hz for 4 s, repeated twice, 20 s apart in current-clamp mode. This protocol has been previously used to study activity-dependent plasticity in the DMH (Crosby et al., 2011). In a subset of experiments, slices were incubated in fluorocitric acid (100 μm) for 120 min following the 60 min recovery incubation in aCSF and before recording. Fluorocitric acid disrupts cellular metabolism preferentially in glial cells (Swanson and Graham, 1994), and thus is a common tool used to target and inhibit these cells. Fluorocitric acid was dissolved in aCSF containing 1.5 mm MgSO4 in place of the same concentration of MgCl2 to facilitate its dissolution. The mixture was sonicated at 30°C for 60 min and then filtered using a 0.45 μm filter before slice incubation.
Data analysis.
Evoked currents were analyzed using Clampfit 9 (pClamp, Molecular Devices; RRID:SCR_011323). The amplitude of the synaptic current was calculated from the baseline (current immediately before the evoked response) to the peak of each evoked response. Mean IPSC amplitudes (in 30 s epochs) were compared by expressing current amplitudes as a percentage of the current amplitudes obtained during the baseline recording period within each cell. The paired pulse ratio (PPR) was calculated by dividing the second peak amplitude by the first peak amplitude and is expressed in 1 min epochs as a percentage of the PPR from the baseline recording period within each cell. For clarity, the stimulus artifacts have been digitally removed from the traces depicted. IPSC traces in all figures depict IPSCs averaged from the 5 min period immediately before (baseline) and 10–20 min after HFS, with the exception of the ATPγS experiment in which traces are averaged during slice incubation in ATPγS.
Spontaneous IPSCs (sIPSCs) that were observed in the intervals between consecutive applications of synaptic stimuli were analyzed using Mini Analysis software (Mini Analysis Program, Synaptosoft; RRID:SCR_002184). The threshold for the detection of sIPSCs was five times the root mean square noise. sIPSC frequency was measured in events per minute, and amplitude was measured as the average amplitude in 1 min bins.
Calcium imaging.
Calcium imaging was performed on transgenic mice expressing GCaMP3 under the astrocyte-specific GLAST promoter (GLAST-CreERT × LSL-GCaMP3). Brain slices of the DMH were prepared as described above. Fluorescence imaging was performed on a custom two-photon laser-scanning microscope (Rosenegger et al., 2014) equipped with a Ti:Sapph laser (∼4 W average power, 670–1080 nm, Coherent; Ultra II), objectives (40×; numerical aperture, 1.0; Zeiss), a green bandpass emission filter (525–40 nm), an orange/red bandpass emission filter (605–70 nm), and associated photomultiplier tubes (GaAsP, Hamamatsu). Time-series images, to assess fluctuations in intracellular astrocyte calcium, were acquired at a single focal plane using bidirectional scanning (512 pixels2 at 1 Hz frame rate). Astrocyte calcium regions of interest (ROI) were detected in a semiautonomous manner using the GECIquant plugin for ImageJ (ImageJ; RRID:SCR_003070; Srinivasan et al., 2015). Once the ROI detection module was launched, we used an area range of 30 μm2 to infinity for the detection of astrocyte soma, and 0.2–5 μm2 for the detection of microdomain calcium. We visually verified the detected ROIs in each individual case. For ROI detection, GECIquant generated a temporal maximum intensity projection, which was then manually thresholded. Using this thresholded image, GECIquant automatically detected ROIs and added them to ImageJ ROI Manager. Intensity values for each ROI were extracted, converted to dF/F (fluorescence intensity change/basal fluorescence intensity) values, and analyzed using Mini Analysis (Mini Analysis Program; RRID:SCR_002184) to detect and measure amplitude, half-width, and frequency values for the somatic and microdomain calcium transients.
Experimental design and statistical analysis.
All grouped data consisted of neuronal or astrocytic recordings from a minimum of three rats (transgenic mice for GCamP3 experiments) from two or more different litters. For all neuronal recordings, statistics were performed on baseline measurements taken during the 5 min before HFS and 10–20 min following HFS (or during the 10 min ATPγS incubation). Results are expressed as the mean ± SEM. All data were tested for normality, and significance was determined using two-tailed paired t tests for IPSC amplitude, PPR, coefficient of variation (CV), and spontaneous frequency and amplitude data. Multiple groups were compared using a one-way ANOVA with Tukey's post-test. To assess significance for astrocyte calcium data, we performed paired t tests comparing spontaneous calcium fluctuations in individual astrocytes before and after the application of CCK. Data were considered significant at p < 0.05.
Results
CCK shifts the plasticity of GABA synapses from LTD to LTP in response to HFS
To determine whether CCK shifts the plasticity of GABA synapses in the DMH, we obtained recordings from neurons in the compact zone of the DMH in slices from male Sprague Dawley rats, as described previously (Crosby et al., 2011, 2015; Fig. 1A). We first examined the effect of HFS of GABA inputs onto DMH neurons in the absence of CCK. Consistent with our previous findings (Crosby et al., 2011), HFS (100 Hz for 4 s ×2, 0.05 Hz interval) elicited an LTD of GABA synapses, as assessed by examining the amplitude of evoked IPSCs (baseline, 87.38 ± 9.18 pA; post-HFS, 38.60 ± 11.90 pA; n = 6; t = 3.71; p = 0.014. paired t test; Fig. 1B). This depression was not accompanied by a significant change in PPR (baseline, 1.07 ± 0.10; post-HFS, 1.18 ± 0.09; n = 6; t = 1.05; p = 0.33, paired t test) or CV (baseline, 0.53 ± 0.06; post-HFS, 0.73 ± 0.16; n = 6; t = 1.14; p = 0.30, paired t test), which, again, is consistent with our previous results.

CCK shifts the plasticity of GABA synapses from LTD to LTP. A, Schematic of DMH illustrating the approximate position of recording and stimulating electrodes in the compact zone (CZ) of the DMH (f, fornix; 3V, third ventricle). B, Sample traces of averaged IPSCs before (baseline; black trace) and after (post-HFS; gray trace) HFS of afferents (top), and summary data showing a long-lasting reduction in IPSC amplitude (bottom; n = 6; 46.8 ± 13.8% of baseline). Calibration: 50 pA and 10 ms. Amplitude values were assessed during the 5 min before HFS (baseline) and 10–20 min following HFS (post-HFS). Arrow indicates the time of HFS. C, Sample traces of averaged IPSCs before (baseline; black trace) and after HFS (post-HFS; red trace; top) in a representative cell showing a long-lasting potentiation in IPSC amplitude in the presence of CCK (0.1 μm; bottom). D, Summary data showing LTPCCK following HFS in the presence of CCK (n = 6; 252.2 ± 38.2% of baseline). E, Summary PPR data (left) and CV data (right) showing a decrease following HFS in CCK-treated slices. The horizontal black bar indicates the mean. F, Sample traces of spontaneous IPSCs before (baseline; black trace) and after HFS (post-HFS; red trace) in the continuous presence of CCK and summary data showing an increase in sIPSC frequency, but no change in amplitude following HFS in CCK-treated slices. Calibration: 25 pA and 0.5 s. *p > 0.05.
To determine whether CCK can shift the direction of plasticity of these synapses, we repeated the above experiment, but with slices continuously incubated in sulfated CCK octapeptide (hereafter referred to as CCK; 0.1 μm for at least 10 min before recordings). In the presence of CCK, HFS elicited a robust and long-lasting potentiation of GABA synapses (referred to as LTPCCK; baseline, 61.34 ± 17.84 pA; post-HFS, 129.3 ± 19.50 pA; n = 6; t = 8.84; p = 0.0003, paired t test; Fig. 1C,D). We next examined the locus of this plasticity by assessing the PPR and CV of GABA synapses, as well as the frequency and amplitude of sIPSCs. LTPCCK was accompanied by a decrease in the PPR (baseline, 0.90 ± 0.05; post-HFS, 0.77 ± 0.07; n = 6; t = 2.90; p = 0.03, paired t test; Fig. 1E) and CV (baseline, 0.15 ± 0.02; post-HFS, 0.09 ± 0.01; n = 6; t = 4.11; p = 0.009, paired t test; Fig. 1E), and an increase in the frequency of sIPSCs (baseline, 3.81 ± 1.26 Hz; post-HFS, 7.42 ± 2.30 Hz; n = 6; t = 3.00; p = 0.03, paired t test; Fig. 1F), but no change in the amplitude (baseline, 13.86 ± 1.44 pA; post-HFS, 15.05 ± 1.33 pA; n = 6; t = 1.61; p = 0.17, paired t test; Fig. 1F). Together, these findings strongly suggest that CCK shifts the direction of plasticity from LTD to LTP, which is mediated by an increase in GABA release from the presynaptic terminal, rather than by altering postsynaptic receptor activity or expression.
LTPCCK requires CCK2R activation
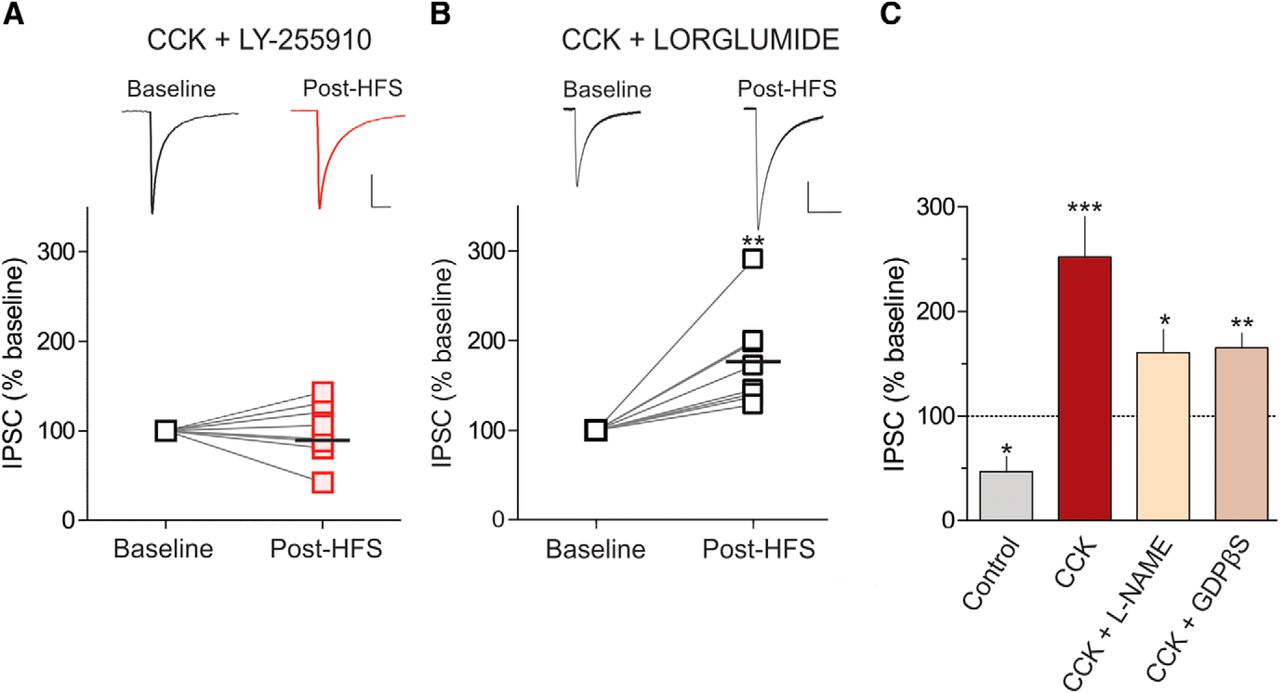
We next determined the mechanism underlying the LTP triggered by CCK. There are two CCK receptor subtypes, CCK1R and CCK2R, both of which are expressed in the DMH (Gaudreau et al., 1983; Durieux et al., 1988; Bi et al., 2004). To test whether CCK2R activation is necessary for LTPCCK, we incubated slices in the CCK2R-specific antagonist LY-225910 (1 μm) and delivered HFS in the presence of CCK. Under these conditions, HFS failed to elicit LTPCCK (baseline, 71.36 ± 15.71 pA; post-HFS, 70.49 ± 15.80 pA; n = 8; t = 0.13; p = 0.90, paired t test; Fig. 2A). As expected, no changes in PPR (baseline, 1.02 ± 0.08; post-HFS, 1.01 ± 0.05; n = 8; t = 0.28; p = 0.78, paired t test) or CV (baseline, 0.41 ± 0.06; post-HFS, 0.35 ± 0.04; n = 8; t = 0.99; p = 0.35, paired t test) were observed. To further confirm that CCK2Rs mediate LTPCCK, we blocked CCK1Rs with the selective antagonist lorglumide (1 μm) and repeated the HFS in the presence of CCK. Blockade of CCK1Rs had virtually no effect on the ability of synapses to undergo LTP in the presence of CCK (baseline, 54.02 ± 14.13 pA; post-HFS, 84.09 ± 17.99 pA; n = 8; t = 4.02; p = 0.005, paired t test; Fig. 2B). Similarly, we observed a trend toward a decrease in PPR (baseline, 1.00 ± 0.09; post-HFS, 0.86 ± 0.06; n = 8; t = 1.96; p = 0.09, paired t test), and a significant decrease in CV (baseline, 0.37 ± 0.05; post-HFS, 0.25 ± 0.03; n = 8; t = 2.63; p = 0.03, paired t test) was still observed following the blockade of CCK1Rs.

CCK2R activation is necessary for LTPCCK. A, Sample traces of averaged IPSCs before and after HFS (top), and summary data showing that LTPCCK is completely abolished with the CCK2 receptor antagonist LY-225910 (bottom; 1 μm; n = 8; 100.2 ± 11.36% of baseline). B, Sample traces of averaged IPSCs before and after HFS (top) and summary data showing that LTPCCK is not prevented with the CCK1R antagonist, lorglumide (bottom; 1 μm; n = 8; 176.4 ± 18.96% of baseline). The horizontal black bar indicates the mean. **p > 0.01. C, Summary data of the percentage change in IPSC amplitude from baseline in control slices and in the presence of CCK alone (0.1 μm; n = 6; 252.2 ± 38.2% of baseline) or with CCK and the following: l-NAME (200 μm; n = 8; 160.7 ± 20.99% of baseline) or GDPβS (1 mm; n = 6; 165.0 ± 14.42% of baseline) in the patch pipette. *p > 0.05, **p > 0.01, ***p > 0.001 compared with baseline for each group.
Our data thus far indicate that in the presence of CCK, HFS elicits LTPCCK that requires CCK2R activation and is mediated by an increase in GABA release from the presynaptic terminal. Because we have previously shown that nitric oxide (NO) can impact GABA release at these synapses (Crosby et al., 2011), we tested whether NO could be implicated in this plasticity. Surprisingly, the blockade of NO production with the NO synthase inhibitor l-NAME (200 μm) did not affect the potentiation of GABA synapses following HFS in the presence of CCK (baseline, 76.72 ± 12.14 pA; post-HFS, 123.20 ± 24.49 pA; n = 8; t = 2.55; p = 0.03, paired t test; Fig. 2C). We also observed a trend toward a decrease in PPR (baseline, 0.92 ± 0.07; post-HFS, 0.77 ± 0.06 pA; n = 8; t = 2.11; p = 0.07, paired t test) and a significant reduction in CV (baseline, 0.30 ± 0.03; post-HFS, 0.19 ± 0.03 pA; n = 8; t = 2.66; p = 0.03, paired t test). These data suggest that NO does not mediate the plasticity triggered by CCK in the DMH. To determine whether another unidentified modulator is released by CCK2Rs localized postsynaptically, we examined the impact of HFS on GABA signaling in the presence of CCK with GDPβS (1 mm) included in the patch pipette to disrupt postsynaptic G-protein signaling coupled to the CCK2R. In the presence of CCK with GDPβS in the patch pipette, the potentiation of GABA synapses was still observed following HFS of afferents (baseline, 106.9 ± 32.19 pA; post-HFS, 166.7 ± 44.02 pA; n = 6; t = 4.57; p = 0.006, paired t test; Fig. 2C), but with no significant changes in PPR (baseline, 1.25 ± 0.20; post-HFS, 1.17 ± 0.23; n = 6; t = 0.66; p = 0.54, paired t test) or CV (baseline, 0.37 ± 0.05; post-HFS, 0.32 ± 0.07; n = 6; t = 1.60; p = 0.17, paired t test). We further analyzed these data to determine whether the magnitude of the plasticity was dampened when l-NAME or GDPβS was applied in addition to CCK. A one-way ANOVA with Tukey's post-test revealed no significant difference in IPSC amplitude following HFS in CCK-treated slices compared with l-NAME plus CCK (p > 0.05) and GDPβS plus CCK (p > 0.05).
Astrocytes mediate LTPCCK
It is possible that the activation of CCK2Rs on neighboring astrocytes might trigger the release of a gliotransmitter that acts on the presynaptic terminal to stimulate GABA release. CCK2Rs are localized on astrocytes in other brain regions (Hösli et al., 1993; Müller et al., 1997), and activation of this receptor subtype on cultured astrocytes has been shown to trigger increases in intracellular Ca2+ (Müller et al., 1997) that are necessary for gliotransmitter release. To test the hypothesis that astrocytes are necessary for LTPCCK, we repeated HFS in the presence of CCK in slices that had been incubated in the gliotoxin fluorocitric acid. Fluorocitric acid is a toxin preferentially taken up by glial cells that interferes with the citric acid cycle and therefore disrupts intracellular production of ATP (Swanson and Graham, 1994). In fluorocitric acid-treated slices in the presence of CCK, HFS failed to elicit LTPCCK (baseline, 68.15 ± 20.44 pA; post-HFS, 44.07 ± 14.56 pA; n = 8; t = 1.87; p = 0.10, paired t test; Fig. 3A) or changes in PPR (baseline, 1.02 ± 0.11; post-HFS, 1.18 ± 0.13; n = 8; t = 1.44; p = 0.20, paired t test) or CV (baseline, 0.37 ± 0.04; post-HFS, 0.42 ± 0.06; n = 8; t = 0.98; p = 0.36, paired t test). We next tested whether CCK could trigger a rise in calcium from DMH astrocytes in acute brain slices, because previous data were obtained from dissociated astrocyte cultures and not from astrocytes in situ (Müller et al., 1997). Spontaneous calcium dynamics in astrocytes were measured using GLAST-CreERT LSL-GCaMP3 mice (Fig. 3B–E). We observed that the application of CCK induced an increase in both amplitude (baseline, 0.74 ± 0.02 dF/F; CCK, 0.81 ± 0.02 dF/F; n = 11; t = 2.30; p = 0.04, paired t test; Fig. 3F) and frequency (baseline, 1.41 ± 0.12 events/min; CCK, 1.71 ± 0.15 events/min; n = 11; t = 2.63; p = 0.03, paired t test; Fig. 3G) without any effect on the duration of astrocyte calcium events in microdomains (half-width; baseline, 1.58 ± 0.17 s; CCK, 1.59 ± 0.07 s; n = 11; t = 0.04; p = 0.97, paired t test; data not shown).

Astrocytes are necessary for LTPCCK. A, Sample traces of averaged IPSCs before and after HFS (top), and summary data showing the depression of GABA synapses in slices incubated in fluorocitric acid in the presence of CCK (bottom; 100 μm; n = 8; 75.4 ± 17.5% of baseline). B, Example image of GCaMP3-expressing astrocytes in the dorsomedial hypothalamus. Scale bar, 10 μm. C, D, Average intensity of field of view before (baseline; C) and after CCK application (0.1 μm; D). Scale bar, 10 μm. E, Example traces of astrocytic intracellular calcium before and after CCK application. Calibration: 0.2 dF/F and 2 min. F, CCK application increased the amplitude of individual calcium events in astrocytes (n = 11). G, CCK application increased the frequency of individual calcium events in astrocytes (n = 11). The horizontal black bar indicates the mean. *p > 0.05.
Collectively, these findings support the hypothesis that CCK2Rs, activated by bath-applied CCK, are localized on neighboring astrocytes and liberate a gliotransmitter that enhances GABA release from incoming afferents.
mGluR5 activation is required for LTPCCK
We have previously demonstrated that glutamate plays an integral role in LTP at GABA synapses in the DMH (Crosby et al., 2011). In addition, group I metabotropic glutamate receptors, particularly mGluR5s, are localized on astrocytes, and their activation triggers a significant rise in intracellular Ca2+ within these cells (van den Pol et al., 1995; Nakahara et al., 1997; Cai et al., 2000; Panatier et al., 2011; Mehina et al., 2017). We therefore hypothesized that the excess glutamate resulting from HFS activates mGluR5s secondary to CCK2R activation, ultimately elevating astrocytic Ca2+ to sufficient levels to trigger the release of a gliotransmitter. To determine whether mGluR5 activation is required for LTPCCK, we first delivered HFS to slices in the presence of CCK and the nonselective group I mGluR antagonist MCPG [(RS)-α-Methyl-4-carboxyphenylglycine; 200 μm]. This resulted in a significant reduction in IPSC amplitude (baseline, 87.13 ± 4.67 pA; post-HFS, 39.01 ± 8.67 pA; n = 6; t = 8.37; p = 0.0004, paired t test; Fig. 4A), with no change in PPR (baseline, 1.01 ± 0.09; post-HFS, 0.82 ± 0.08; n = 6; t = 1.97; p = 0.10, paired t test) or CV (baseline, 0.26 ± 0.04; post-HFS, 0.26 ± 0.03; n = 6; t = 0.04; p = 0.96, paired t test). We next examined the effect of the selective mGluR5 antagonist MTEP (3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine; 10 μm) on the ability of GABA synapses to potentiate in response to HFS in the presence of CCK. Under these conditions, HFS failed to elicit LTPCCK and instead triggered LTD (baseline, 90.79 ± 8.84 pA; post-HFS, 48.19 ± 8.80 pA; n = 6; t = 3.64; p = 0.02, paired t test; Fig. 4B), with no change in PPR (baseline, 1.17 ± 0.10; post-HFS, 1.15 ± 0.10; n = 6; t = 0.52; p = 0.65, paired t test) or CV (baseline, 0.37 ± 0.04; post-HFS, 0.25 ± 0.06; n = 6; t = 1.29; p = 0.26, paired t test). Together, these data provide evidence of the necessity of mGluR5s in the potentiation of GABA synapses in the DMH. Next, to determine whether the activation of mGluR5s is sufficient to trigger LTP, we examined the effect of HFS in the presence of the mGluR5 agonist DHPG [(RS)-3,5-Dihydroxyphenylglycine; 50 μm]. DHPG alone was not sufficient to trigger LTP in response to HFS and instead resulted in LTD, which is reminiscent of the plasticity observed under control conditions (baseline, 70.93 ± 10.63 pA; post-HFS, 29.44 ± 8.04 pA; n = 6; t = 4.04; p = 0.009, paired t test; Fig. 4C). As with the control condition, no changes in PPR (baseline, 1.43 ± 0.34; post-HFS, 1.50 ± 0.40; n = 6; t = 0.66; p = 0.54, paired t test) or CV (baseline, 0.42 ± 0.34; post-HFS, 0.65 ± 0.16; n = 6; t = 1.71; p = 0.14, paired t test) were observed.

LTPCCK requires mGluR5 activation. A, Sample traces of averaged IPSCs before and after HFS (top), and summary data showing the depression of GABA synapses in the presence of CCK and the group I mGluR antagonist MCPG (bottom; 200 μm; n = 6; 43.3 ± 8.2% of baseline). B, Sample traces of averaged IPSCs before and after HFS (top), and summary data showing the depression of GABA synapses in the presence of CCK and the mGluR5 antagonist MTEP (bottom; 10 μm; n = 6; 54.4 ± 10.9% of baseline). C, Sample traces of averaged IPSCs before and after HFS (top), and summary data showing depression of GABA synapses in the presence of the mGluR5 agonist DHPG (bottom; 10 μm; n = 6; 45.29 ± 12.74% of baseline). The horizontal black bar indicates the mean. *p > 0.05, **p > 0.01, ***p > 0.001.
ATP-induced activation of presynaptic P2X receptors is required for LTPCCK
Having determined that astrocytes are required for LTPCCK, we next asked whether the activation of CCK2Rs and mGluR5s on astrocytes triggers the release of a transmitter that acts at the GABA terminal to stimulate release. ATP is a prominent gliotransmitter that modulates synaptic transmission at numerous CNS synapses through the activation of presynaptic P2X receptors (Fu and Poo, 1991; Sperlagh and Vizi, 1991; Rogers et al., 1997). To test the hypothesis that ATP is required for LTPCCK, we first examined whether the blockade of purinergic receptors prevents the CCK-induced potentiation of GABA synapses. PPADS (pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid), a nonselective P2X purinergic receptor antagonist (30 μm) completely abolished the potentiation of IPSCs (baseline, 52.18 ± 2.45 pA; post-HFS, 26.80 ± 4.65 pA; n = 6; t = 4.80; p = 0.005, paired t test; Fig. 5A,B) and the decrease in PPR (baseline, 1.10 ± 0.15; post-HFS, 1.15 ± 0.16; n = 6; t = 0.49; p = 0.64, paired t test) and CV (baseline, 0.32 ± 0.04; post-HFS, 0.30 ± 0.07; n = 6; t = 0.45; p = 0.67, paired t test) observed with CCK. To confirm that ATP is both necessary and sufficient for LTPCCK, we applied an analog of ATP, ATPγS, to slices and examined the effect on IPSCs. ATPγS (100 μm) potentiated GABA synapses (baseline, 69.62 ± 13.54 pA; ATPγS, 126.0 ± 19.12 pA; n = 6; t = 4.37; p = 0.007, paired t test; Figs. 5C,D) and decreased PPR (baseline, 1.01 ± 0.11; ATPγS, 0.86 ± 0.07; n = 6; t = 2.86; p = 0.03, paired t test) and CV (baseline, 0.41 ± 0.06; ATPγS, 0.37 ± 0.09; n = 6; t = 0.56; p = 0.60, paired t test). Collectively, these data suggest that ATP is released from astrocytes and activates presynaptic P2X receptors on GABA terminals to facilitate release.

ATP-induced activation of presynaptic P2X receptors is required for LTPCCK. A, Sample traces of averaged IPSCs before and after HFS (top) in a representative cell (bottom) showing a long-lasting depression in IPSC amplitude in the presence of CCK and the P2X receptor antagonist PPADS (30 μm). B, Summary data showing depression in IPSCs following HFS in the presence of CCK and PPADS (n = 6; 51.9 ± 9.1% of baseline). C, Sample traces of averaged IPSCs before and during incubation with the ATP analog ATPγS (100 μm; top) in a representative cell (bottom) showing the potentiation of IPSC amplitude with ATPγS. D, Summary data showing the potentiation of GABA synapses with ATPγS (n = 6; 194.1 ± 27.6% of baseline). The shaded region represents the time and duration of ATPγS application to the slice.
Discussion
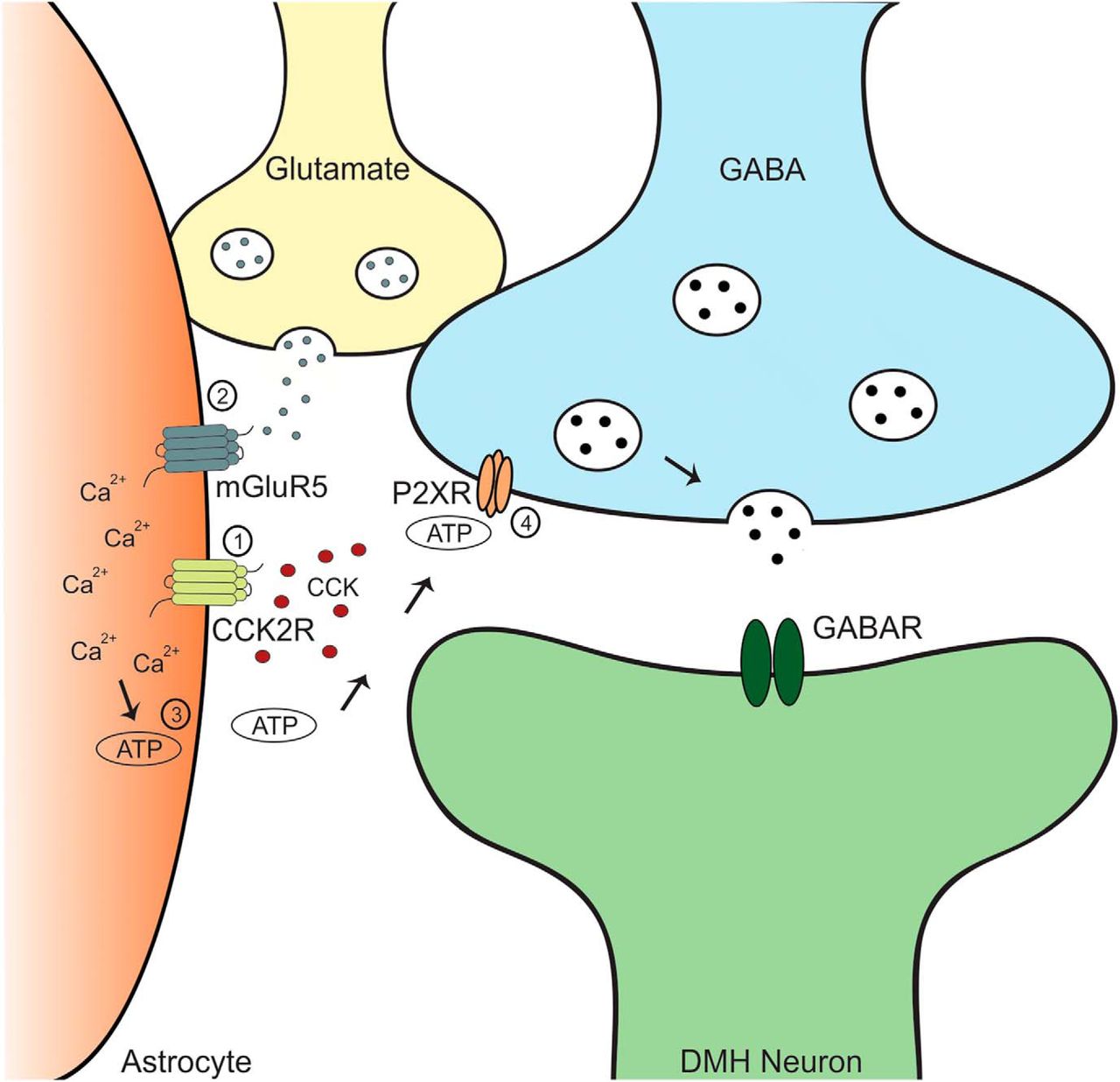
Here we report that CCK shifts the polarity of plasticity of GABA synapses in the DMH from LTD to LTP. This potentiation is mediated by an increase in the probability of GABA release from presynaptic terminals and requires the activation of CCK2Rs and mGluR5s. We propose that the activation of these receptors on astrocytes triggers a rise in intracellular Ca2+ that stimulates the release of ATP. ATP subsequently activates P2X receptors at GABA terminals to enhance release (Fig. 6). Our findings indicate a new mechanism by which CCK modulates the plasticity of GABA synapses in the DMH.

Schematic representation of the proposed mechanism, as follows: 1, In the presence of CCK, CCK2Rs are activated on astrocytes; 2, HFS triggers the release of glutamate from neighboring neurons, leading to the activation of mGluR5Rs on astrocytes; 3, the synergistic action of CCK2Rs and mGluR5s on astrocytes causes a release of calcium from intracellular stores, resulting in the release of ATP; and 4, ATP binds to P2XRs on GABA terminals, causing a prolonged increase in GABA release onto DMH neurons.
We show that CCK shifts the polarity of plasticity in response to bursts of afferent activity, such that GABA synapses exhibit LTP, as opposed to LTD. Although CCK has been previously shown to modulate GABA release in the DMH (Crosby et al., 2015) and other brain regions (Deng and Lei, 2006; Chung and Moore, 2007; Karson et al., 2008; Mitchell et al., 2011), this is the first example of a CCK-induced shift in the direction of plasticity. The physiological consequences of this shift from LTD to LTP are not yet known. CCK is a potent satiety peptide; microinjection of CCK into the DMH triggers a long-lasting inhibition of food intake (Blevins et al., 2000; Chen et al., 2008). Although the underlying mechanisms of this anorexigenic effect are not completely understood, one possible mechanism might be a CCK-induced potentiation of GABA release onto orexigenic neurons in the DMH. Indeed, there is accumulating evidence that different populations of neurons in the DMH promote food intake, including GABAergic (Otgon-Uul et al., 2016) and cholinergic (Jeong et al., 2017) neurons. In addition to appetite regulation, the DMH is also a key player in the regulation of the stress response (Crosby and Bains, 2012; Stamper et al., 2015), and compelling evidence links CCK2R activity with activation of the hypothalamic—pituitary—adrenal axis (Abelson and Young, 2003; Malendowicz et al., 2003). Thus, the plasticity reported here could also play a role in modulating the stress response.
The present data reveal a novel mechanism by which CCK enhances GABA release onto DMH neurons. CCK has previously been shown to modulate GABA release at these synapses. Specifically, we have reported that basal CCK activates postsynaptic CCK2Rs, leading to enhanced NMDAR function and subsequent NO-mediated potentiation of GABA release (Crosby et al., 2015). These distinct mechanisms by which CCK can increase GABA function under basal conditions and following HFS could potentially represent different magnitudes of inhibition of orexigenic neurons in the DMH, thus influencing feeding behavior.
Our data suggest that CCK2R activation is necessary for LTPCCK. This is consistent with our recent report illustrating that basal CCK enhances GABA release onto DMH neurons through a CCK2R-dependent mechanism (Crosby et al., 2015). Although CCK2R blockade abolished the LTPCCK, LTD was also not observed. It is possible that CCK1R activation may slightly enhance GABA release such that LTD is not observed, but our data suggest that this receptor subtype does not contribute significantly to the potentiation of GABA synapses reported in this study. Both CCK1Rs and CCK2Rs are expressed in the DMH (Gaudreau et al., 1983; Durieux et al., 1988; Bi et al., 2004); however, the physiological roles of these receptors in this region are not entirely understood. There is some evidence that DMH CCK1Rs may be important in regulating food intake. In rats lacking CCK1Rs (OLETF rats), neuropeptide Y is overexpressed in the DMH (Bi et al., 2001) and has been suggested to contribute to hyperphagia in this animal model. Replenishment of CCK1Rs specifically in the DMH in OLETF rats, however, does not significantly affect food intake, although it does normalize meal patterns and glucose homeostasis (Zhu et al., 2012). Although less is known about CCK2Rs in the DMH, it is possible that this receptor subtype is also important in appetite regulation. Indeed, CCK2R knock-out mice have been shown to develop obesity due to hyperphagia (Clerc et al., 2007). The endogenous source of the CCK available to bind to CCK2Rs is currently unknown. It is possible that the source is from DMH neurons, as we have previously reported that somatodendritic release of CCK from DMH neurons occurs following repetitive depolarization of these neurons (Crosby et al., 2015).
The present data strongly suggest that astrocytes mediate LTPCCK. Astrocytes are key players in synaptic plasticity (Verkhratsky and Nedergaard, 2018), modulating synaptic function through the release of gliotransmitters including glutamate, d-serine, and ATP (Araque et al., 2014). Release of these transmitters, which are in turn modulated by activation of a wide range of membrane-bound receptors, including mGluR5s, is largely regulated by fluctuations in intracellular Ca2+ (Panatier and Robitaille, 2016). Here, we report that CCK elevates astrocytic calcium levels and that both mGluR5s and CCK2Rs are required for LTPCCK. We propose that these receptors are localized on astrocytes, as has been reported previously (Hösli et al., 1993; Müller et al., 1997) and that their activation enhances intracellular Ca2+ concentrations and liberates ATP. Consistent with this idea, the gliotoxin fluorocitric acid completely abolished LTPCCK, and we observed a CCK-induced increase in the frequency and amplitude of calcium events in astrocytes in the DMH. Consistent with our findings, other reports have indicated that the activation of CCK2Rs mobilizes astrocytic Ca2+ from intracellular stores (Müller et al., 1997) and mGluR5 activation triggers Ca2+ events that result in the release of purines, influencing synaptic transmission (Panatier et al., 2011).
We have demonstrated that both mGluR5s and CCK2Rs are required for LTPCCK, but the link between these receptors remains elusive. Because slices are first exposed to CCK and later glutamate following HFS in this study, it is possible that the activation of CCK2Rs by CCK results in the priming of mGluR5s. Such priming would increase mGluR5 activity, and this, in combination with HFS-induced elevations in extracellular glutamate, would trigger a sufficient elevation in intracellular Ca2+ to ultimately release ATP. Both receptors are coupled to Gq proteins; thus, their activation generates diacylglycerol and inositol 1,4,5-triphosphate, which activate protein kinase C (PKC) and release Ca2+ from intracellular stores, respectively. PKC has been shown to phosphorylate mGluR5s, leading to enhanced activity (Kim et al., 2005). It is therefore possible that CCK2R activation and subsequent elevations in PKC enhance mGluR5 activity. Our observation that the mGluR5 agonist DHPG is not sufficient to trigger LTP following HFS further points to an important role for CCK and indicates that there is a potential synergistic, rather than additive, effect of CCK2Rs and mGluR5s.
We provide evidence that ATP signaling at P2X receptors is necessary for LTPCCK. There is widespread evidence that astrocytes release ATP in response to elevations in intracellular Ca2+ (Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006; Gourine et al., 2010; Covelo and Araque, 2018) and that ATP can activate presynaptic P2X receptors to increase the probability of neurotransmitter release from various synapses (Fu and Poo, 1991; Sperlagh and Vizi, 1991; Barros-Barbosa et al., 2018), including GABAergic synapses in the hypothalamus (Vavra et al., 2011; Bhattacharya et al., 2013; Haam et al., 2014). Although postsynaptic P2X receptors can also modulate synaptic transmission (Pankratov et al., 2002; Gordon et al., 2005, 2009; Baxter et al., 2011; Ferreira-Neto et al., 2015), our data are consistent with a presynaptic locus of these receptors.
Overall, the data presented here present a novel form of activity-dependent synaptic plasticity mediated by CCK in DMH neurons and provide the first demonstration of CCK-induced modulation of synaptic plasticity by astrocytes in response to bursts of afferent activity. Future studies will examine the functional implications of this plasticity, with a particular focus on the satiety effects of CCK. In addition, targeted genetic studies aimed at elucidating the functional role of astrocytic CCK2Rs will also contribute to our understanding of the actions of CCK in the brain. Finally, because these experiments were conducted in young, male rats, and age and sex can influence the physiology of eating, including CCK expression and sensitivity (Akimoto and Miyasaka, 2010; Asarian and Geary, 2013), future work can investigate whether this plasticity persists across different ages and sexes.
Footnotes
This work was supported by operating grants from the Canadian Institutes for Health Research (Q.J.P.) and the Natural Sciences and Engineering Research Council (K.M.C.), and personnel awards from Alberta Innovates Health Solutions (AIHS) to G.R.G., J.S.B., and Q.J.P. Scholarship support to K.M.C. was provided by the Natural Sciences and Engineering Research Council, AIHS, and a Hotchkiss Brain Institute Obesity Initiative Studentship. Postdoctoral fellowships from AIHS and Campus Alberta Neuroscience supported C.M.-R. We thank Mio Tsutsui and Cheryl Breiteneder for technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Quentin J. Pittman, Hotchkiss Brain Institute, University of Calgary, 3330 Hospital Drive NW, Calgary, AB T2N 4N1, Canada. pittman{at}ucalgary.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}