
Abstract
Intraneuronal calcium stimulates the calpain-dependent conversion of p35 to p25, a CDK5 activator. It is widely believed that amyloid β peptide (Aβ) induces this conversion that, in turn, has an essential role in Alzheimer's disease pathogenesis. However, in vivo studies on p25 generation used transgenic mice overexpressing mutant amyloid precursor protein (APP) and presenilin (PS). Here, using single App knock-in mice, we show that p25 generation is an artifact caused by membrane protein overexpression. We show that massive Aβ42 accumulation without overexpression of APP or presenilin does not produce p25, whereas p25 generation occurred with APP/PS overexpression and in postmortem mouse brain. We further support this finding using mice deficient for calpastatin, the sole calpain-specific inhibitor protein. Thus, the intracerebral environment of the APP/PS mouse brain and postmortem brain is an unphysiological state.
SIGNIFICANCE STATEMENT We recently estimated using single App knock-in mice that accumulate amyloid β peptide without transgene overexpression that 60% of the phenotypes observed in Alzheimer's model mice overexpressing mutant amyloid precursor protein (APP) or APP and presenilin are artifacts (Saito et al., 2014). The current study further supports this estimate by invalidating key results from papers that were published in Cell. These findings suggest that more than 3000 publications based on APP and APP/PS overexpression must be reevaluated.
- amyloid
- calpain
- Nav1.1
- p25
Introduction
Since 1995 (Games et al., 1995), experimental AD research in cell and animal models has suffered from artifactual findings caused by protein overexpression paradigms (Nilsson et al., 2014; Saito et al., 2014). Membrane protein overexpression results in chronic ER stress that elevates cytoplasmic calcium concentrations (Chaudhari et al., 2014; Borkham-Kamphorst et al., 2016). For instance, Seo et al. (2014) used transgenic mice that overexpressed APP and presenilin carrying 5 familial AD-associated mutations (5XFAD mice; Oakley et al., 2006). Although the two transgenes inserted impair at least two gene loci of the host animals, these and other transgenic mice have never been sequenced, to our knowledge. In this case, the asserted disease mechanisms were evidently studied without sufficient mouse genetic controls.
Materials and Methods
Mutant mice.
AppNL-F/NL-F and calpastatin (Cast) KO mice were maintained as described previously (Saito et al., 2014). All the mice used in the experiments were male and established on a C57BL/6J background. All animal experiments were performed according to the RIKEN Brain Science Institute's guidelines for animal experimentation.
Western blot analysis.
We prepared brain extracts from postmortem mouse brain excised at 1 or 2 h after demise as a positive control for calpain activation (Taniguchi et al., 2001). AD model mouse brains were prepared as described previously (Higuchi et al., 2005). We subjected the samples to Western blot analysis using anti-p35/25 antibody (C64B10, Cell Signaling Technology) and anti-β-tubulin antibody (SAP.4G5, Abcam).
We also analyzed the expression levels of Nav1.1. Brain extracts prepared as described previously (Ogiwara et al., 2013) were subjected to immunoblotting using rabbit anti-C-terminal Nav1.1 (IO1; Ogiwara et al., 2007) and anti-β-tubulin antibody. Each set of experiments was repeated at least three times. The band intensity was determined with a densitometer (LAS4000, Fujifilm).
Statistical analysis.
All data are shown as means ± SEM. For comparisons among four groups, one-way ANOVA followed by post hoc test (Scheffe's F test) was used, using Statcel 3 (add-in software on Microsoft Excel).
Results
We crossed APP-overexpressing mice with Cast KO mice and observed peculiar phenotypes (Higuchi et al., 2012), including early lethality, where half of the mice died in 10 weeks. This result contradicts the chronic, progressive nature of AD. Conversely, when we crossed mutant humanized App knock-in mice, overproducing Aβ42 without overexpressing APP, with Cast KO mice, the double mutant mice lived as long as wild-type mice, or >2 years (Saito et al., 2014), indicating that the phenotype of early lethality was an overexpression artifact. More than 3000 papers have been published using these old generation APP mouse models, and we conclude that the results described in these papers require reevaluation using new generation models. As a first step, we examined Aβ-induced calpain-dependent p25 generation (Oakley et al., 2006; Seo et al., 2014), based on studies of the calpain–calpastatin system (Saido et al., 1994).
We confirmed conversion of p35 to p25 by calpain in postmortem mouse brain as a positive control (Taniguchi et al., 2001; Fig. 1, first through fourth lanes). This conversion is caused by intraneuronal ATP depletion resulting in an elevation of cellular calcium concentration (Lipton, 1999). We then examined aged 24-month-old AppNL-F/L-F mice (Saito et al., 2014; Fig. 1, fifth and sixth lanes) and observed no conversion of p35 to p25 despite massive Aβ deposition (Saito et al., 2014). This finding disagrees with the results of Oakley et al. (2006) and Seo et al. (2014).

Generation of p25 in App knock-in and APP-overexpressing mice. We performed a Western blot analysis of wild-type postmortem brain, wild-type control brain, 24 month old AppNL-F/NL-F brain, AppNL-F/NL-F X Cast KO brain, and APP-overexpressing (APP23) brain using antibodies to p35/p25 and to β-tubulin. The band intensities were quantified as shown in the graph (n = 4). *p < 0.05; **p < 0.01 (one-way ANOVA, Scheffe's F test). Data represent mean ± SEM.
We crossed AppNL-F/NL-F mice with Cast KO mice, in which calpain is hyperactivatable (Higuchi et al., 2005; Takano et al., 2005). Even in the absence of calpastatin, we saw no conversion of p35 to p25 (Fig. 1, seventh and eighth lanes) despite the observation that calpastatin deficiency increases Aβ amyloidosis (Saito et al., 2014). Thus, we conclude that p25 generation in 5XFAD mice is an artifact. Consistently, we observed p25 generation in APP23 mice that overexpress APP (Fig. 1, ninth and tenth lanes, bottom).
Protein overexpression paradigms are risky in biological science particularly with membrane proteins. In this regard, APP and PS possess one and nine transmembrane domains, respectively (Chen, 2015). In contrast, overexpression of mutant tau, a cytosolic protein, exhibited no effect on p25 levels (data not shown). Membrane protein overexpression results in chronic ER stress that elevates cytoplasmic calcium concentration (Chaudhari et al., 2014; Borkham-Kamphorst et al., 2016). This nonspecific calcium rise probably activates calpain, resulting in p25 generation in 5XFAD mice (Oakley et al., 2006; Seo et al., 2014). Likewise, 5XFAD mice exhibit intraneuronal Aβ that is supposed to cause neurodegeneration (Oakley et al., 2006). Indeed, when we crossed APP-overexpressing mice with Atg-7 KO mice lacking autophagy, we observed intraneuronal Aβ and enhanced neurodegeneration (Nilsson et al., 2013). However, autophagy deficiency, which induced intraneuronal Aβ accumulation in single App knock-in mice, caused no neurodegeneration (unpublished data).
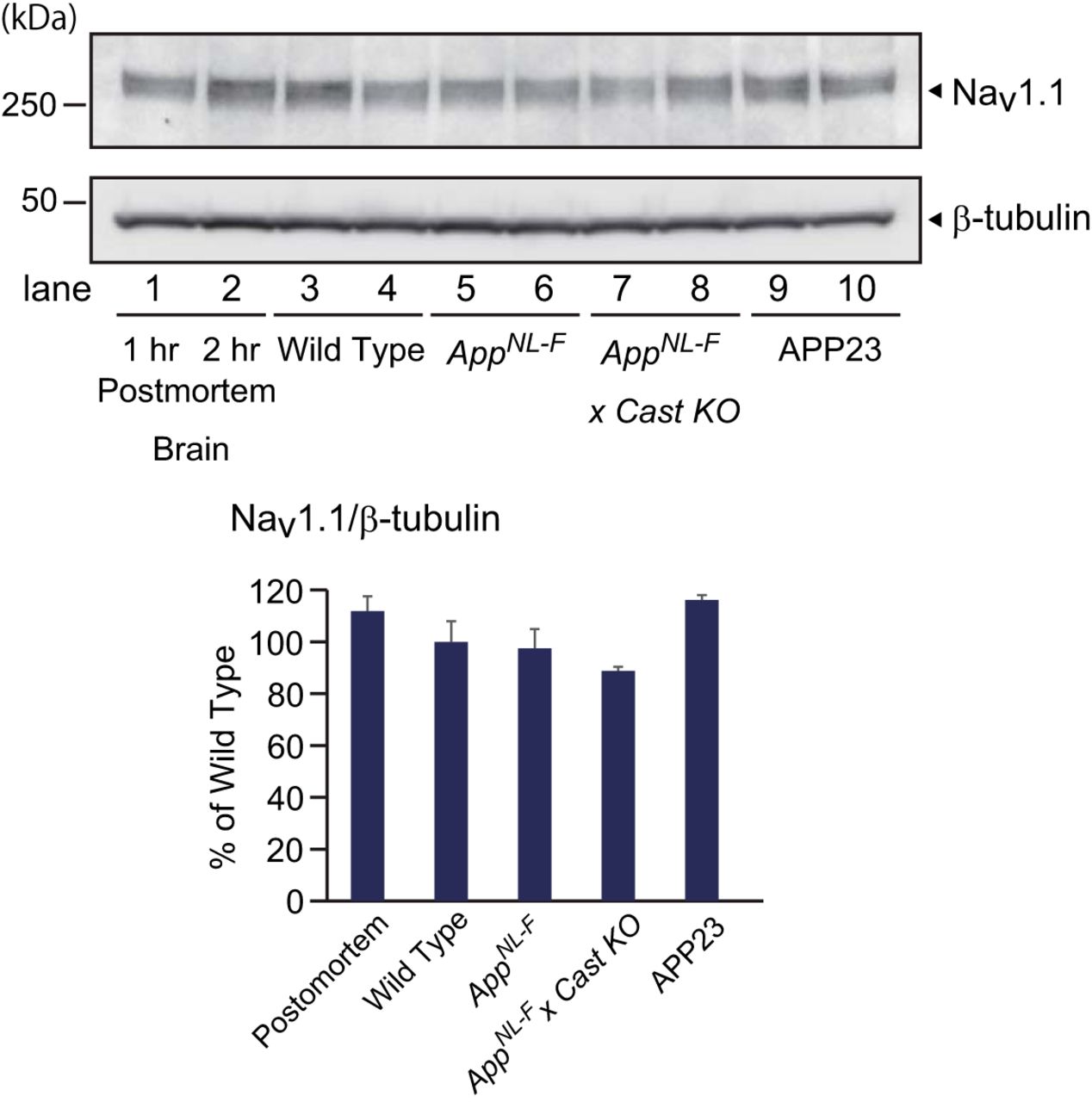
The overexpression paradigm may also explain the epileptic phenotypes of APP-overexpressing mice, which show a downregulation of Nav1.1, a sodium channel expressed in PV-positive interneurons (Verret et al., 2012), because Nav1.1 may also be another substrate of calpain (Ebensperger et al., 2005). Conversely, we observed no changes in Nav1.1 levels in App knock-in, APP-overexpressing, or postmortem brains (Fig. 2), suggesting that Nav1.1 downregulation is a phenomenon unique to the J-20 model mice.
{kind=link}
{kind=link}
Expression levels of Nav1.1 in App knock-in and APP-overexpressing mice. We performed a Western blot analysis of wild-type control brain, 24 month old AppNL-F/NL-F brain, AppNL-F/NL-F X Cast KO brain, and APP-overexpressing (APP23) brain using antibodies to Nav1.1 and to β-tubulin. The band intensities were quantified as shown in the graph (n = 4). Data represent mean ± SEM.
Discussion
Of the five phenotypes that we observed in APP-overexpressing mice crossbred with Cast KO mice (Higuchi et al., 2012), only two were reproduced using single App knock-in mice crossbred with Cast KO mice (Saito et al., 2014). This result allows an estimation that ∼60% of the phenotypes observed using APP-overexpressing mice may be artifacts. We emphasize that both basic and clinical research communities must accept and remit this reality for the reasons outlined below.
All APP-overexpressing mice overproduce an APP fragment generated by β-secretase [C-terminal fragment β (CTF-β); Saito et al., 2014, their Supplemental Fig. 4a], which does not accumulate in the AD brain (Nilsson et al., 2014). CTF-β is more neurotoxic than Aβ (Mitani et al., 2012). To our knowledge, almost all therapeutic anti-Aβ antibodies bind to CTF-β because Aβ and CTF-β share common epitopes near the amino terminus or midregion of the Aβ sequence (Lannfelt et al., 2014). Therefore, the experimental passive immunization of APP-overexpressing mice may have improved cognitive function by removing CTF-β rather than Aβ. If this proves true, the immunotherapeutic prevention trials conducted on thousands of human volunteers may not provide clear information about its beneficial effect. Alternatively, therapeutic antibodies may have bound to vascular amyloid in humans because immunotherapy marginally reduced Aβ burdens only in apolipoprotein E4 genotype carriers (Salloway et al., 2014), where apolipoprotein E4 genotype is an independent risk factor for cerebral amyloid angiopathy (Shinohara et al., 2016).
In addition, essentially all candidate medications that failed in clinical trials (Mangialasche et al., 2010) have depended on old generation models for preclinical studies. Therefore, we suggest that a careful revalidation of all the results of scientific and clinical importance obtained using APP-overexpressing mice are urgently required. Depending on expression levels, not all AD model mice may produce artificial phenotypes. In this context, p25 generation can be one criterion in the reevaluation.
Footnotes
- Received June 14, 2016.
- Revision received July 8, 2016.
- Accepted August 5, 2016.
This work was supported by a Grant-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (T.S.); the Japan Science and Technology Agency Precursory Research for Embryonic Science and Technology (T.S.); and the Brain Mapping by Integrated Neurotechnologies for Disease Studies project of the Japan Agency for Medical Research and Development (T.C.S.). We thank Kazuhiro Yamakawa for providing antibodies to Nav1.1 and Charles Yokoyama for editing this manuscript.
T.S., Y.M., and T.C.S. serve as advisor, director, and CEO, respectively, for RIKEN BIO Co. Ltd., a RIKEN venture based at the Brain Science Institute, Central Building, Wako, Japan.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
- Correspondence should be addressed to Takaomi C. Saido at the above address. saido{at}brain.riken.jp
- Copyright © 2016 Saito et al.
This article is freely available online through the J Neurosci Author Open Choice option.