

Abstract
Biochemical and genetic evidence implicate soluble oligomeric amyloid-β (Aβo) in triggering Alzheimer's disease (AD) pathophysiology. Moreover, constitutive deletion of the Aβo-binding cellular prion protein (PrPC) prevents development of memory deficits in APPswe/PS1ΔE9 mice, a model of familial AD. Here, we define the role of PrPC to rescue or halt established AD endophenotypes in a therapeutic disease-modifying time window after symptom onset. Deletion of Prnp at either 12 or 16 months of age fully reverses hippocampal synapse loss and completely rescues preexisting behavioral deficits by 17 months. In contrast, but consistent with a neuronal function for Aβo/PrPC signaling, plaque density, microgliosis, and astrocytosis are not altered. Degeneration of catecholaminergic neurons remains unchanged by PrPC reduction after disease onset. These results define the potential of targeting PrPC as a disease-modifying therapy for certain AD-related phenotypes after disease onset.
SIGNIFICANCE STATEMENT The study presented here further elucidates our understanding of the soluble oligomeric amyloid-β–Aβo-binding cellular prion protein (PrPC) signaling pathway in a familial form of Alzheimer's disease (AD) by implicating PrPC as a potential therapeutic target for AD. In particular, genetic deletion of Prnp rescued several familial AD (FAD)-associated phenotypes after disease onset in a mouse model of FAD. This study underscores the therapeutic potential of PrPC deletion given that patients already present symptoms at the time of diagnosis.
Introduction
Alzheimer's disease (AD) is the most common form of dementia worldwide, affecting >5 million Americans (Alzheimer's Association, 2012). AD is characterized by two hallmark pathologies: amyloid-β (Aβ) plaques composed of Aβ peptide and neurofibrillary tangles composed of hyperphosphorylated Tau (Braak and Braak, 1991; Selkoe, 2011). The clinical presentation of AD is characterized by progressive memory loss and early death (Mölsä et al., 1986; Mayeux, 2003). Central to AD is the inability of patients to form new memories, with synaptic dysfunction and loss being tightly correlated with symptom progression (Selkoe, 2002; Scheff et al., 2006). Therefore, understanding how synapses are lost is key to understanding AD. Genetic and biochemical evidence suggest a soluble high-molecular weight oligomeric amyloid-β peptide (Aβo) as a trigger for synaptic dysfunction in AD (Hardy and Selkoe, 2002; Sheng et al., 2012; Dohler et al., 2014; Kostylev et al., 2015). Several studies in rodent models have shown that Aβo can initiate a cascade of deleterious effects on synaptic function (Lambert et al., 1998; Walsh et al., 2002; Lesné et al., 2006; Shankar et al., 2008). These studies highlight the importance of understanding Aβo-dependent synaptotoxicity.
Recent evidence suggests Aβo-binding cellular prion protein (PrPC) as a central protein in mediating synaptotoxicity. Previous work has shown PrPC as a high-affinity binding partner of Aβo and a mediator in suppressing LTP (Laurén et al., 2009). In addition, constitutive deletion of Prnp can rescue synapse density, survival, and learning and memory deficits seen in a mouse model of familial AD (Gimbel et al., 2010). Other groups have shown PrPC to bind Aβo with high affinity (Chen et al., 2010; Dohler et al., 2014), mediate suppression of LTP (Barry et al., 2011; Freir et al., 2011; Klyubin et al., 2014), and suppress learning and memory (Chung et al., 2010; Fluharty et al., 2013). Nevertheless, conflicting reports of the role for PrPC in mediating synaptotoxicity (Balducci et al., 2010; Calella et al., 2010; Kessels et al., 2010) have prompted further studies to test the therapeutic potential of PrPC as a target.
Targeting PrPC for AD treatment holds the potential for disease-modifying therapy, as opposed to the symptomatic action of current interventions approved for AD (Yiannopoulou and Papageorgiou, 2013). Several studies have shown directly or indirectly that Aβo binding to PrPC leads to PrPC-mGluR5 coupling (Um et al., 2013; Haas et al., 2016) and subsequent activation of intracellular components including eEF2 (Um et al., 2013; Ma et al., 2014) and Fyn (Larson et al., 2012; Um et al., 2012; Rushworth et al., 2013; Kaufman et al., 2015) can lead to dendritic spine loss (Um et al., 2012; Zhang et al., 2015), suppressed synaptic plasticity (Hu et al., 2014; Haas et al., 2016), and Tau phosphorylation (Larson et al., 2012; Kaufman et al., 2015). Multiple groups have begun to develop methods to target the Aβo-PrPC interaction using small-molecule approaches (Fluharty et al., 2013; Aimi et al., 2015; Osborne et al., 2016) and immunotherapy approaches (Chung et al., 2010; Barry et al., 2011). These efforts underscore the need to understand whether the Aβo-PrPC interaction is required for AD phenotype maintenance and progression after disease onset.
To test the therapeutic potential of targeting Prnp, we decided to take advantage of a tamoxifen (TMX)-inducible Cre-lox system to partially delete Prnp early after disease onset and months after disease onset in a mouse model of familial AD. Partial deletion of Prnp was able to rescue synaptic and behavioral deficits in a mouse model of AD at 12 and 16 months. These results highlight the clinical potential of targeting the Aβo–PrPC interaction.
Materials and Methods
Animals
All mice were cared for by the Yale Animal Resource Center. Yale's institutional animal care and use committee approved all experiments. As described previously (Gimbel et al., 2010), the mouse strains used were the APPswe/PSEN1ΔE9 mice on a C57BL/6J background and ER-Cre mice (Hayashi and McMahon, 2002) on a C57BL/6J background were purchased from The Jackson Laboratory. Flox-Prnp mice on a C57BL6 background have been described previously (Tuzi et al., 2004; Bradford et al., 2009). All experiments used littermate control mice. The group receiving Prnp deletion at 12 months (hereafter referred to as the 12MD cohort) contained a 2:1 male to female sex ratio and the group receiving Prnp deletion at 16 months (hereafter referred to as the 16MD cohort) contained a 1.1:1 male to female sex ratio. The differential male to female sex ratios were not intentional, but rather were caused by random breeding and selection.
Brain tissue collection
Immunohistology.
Mice were killed and immediately perfused with ice-cold phosphate buffer saline (PBS) for 2 min, followed by a five-minute perfusion with ice-cold 4% paraformaldehyde (PFA). Brains were dissected out, cut down the midline into two hemispheres and fixed for 24 h in 4% PFA. Following fixation, brains were cut into 40 μm parasagittal sections using a Leica (Wetzlar, Germany) WT1000S Vibratome. Sections were stored in PBS at 4°C until staining.
Biochemistry.
Mice were killed as above and brains were dissected out immediately. Cortex, hippocampus, and cerebellum were dissected out separately and from each hemisphere. The left hemisphere was flash frozen in liquid nitrogen and the right hemisphere was homogenized in a volume three times the brain weight with TBS containing PhosSTOP and cOmplete-mini to extract the cytosolic fraction. Homogenate was centrifuged at 100,000 × g for 20 min at 4°C. The supernatant was collected as TBS soluble and the pellet was resuspended in equal volume as the TBS volume of RIPA buffer containing PhosSTOP and cOmplete-mini. This homogenate was centrifuged at 100,000 × g at 4°C for 20 min and the supernatant was collected as the RIPA-soluble extract. The pellet was flash frozen in liquid nitrogen and stored at −80°C. Protein concentrations of the TBS- and RIPA-soluble fractions were quantified using a BCA assay (Thermo Scientific).
Immunohistology
Free-floating 40 μm sections were washed in a 12-well plate once with 0.1% PBS–Triton X-100 for 5 min. Subsequently, sections were blocked with 5% normal horse serum (NHS) in PBS for 1 h at room temperature. Sections were then incubated in primary antibody with 5% NHS in PBS overnight at 4°C. The following primary antibodies were used: mouse anti-PrPC (8H4) (Abcam, #ab61409, 1:1000), rabbit anti-β-Amyloid (Cell Signaling Technology, #2454, 1:250), rabbit anti-PSD-95 (Invitrogen, #51-6900, 1:250), rabbit anti-SV2A (Abcam, #ab32942, 1:250), chicken anti-GFAP (Abcam, #ab4674, 1:500), and rabbit anti-Iba1 (Wako Chemicals, #019-19741, 1:250). Sections were washed five times after primary incubation with PBS for 10 min each. Sections were then incubated with fluorescent secondary antibodies (donkey anti-rabbit or donkey anti-chicken; Invitrogen, Alexa Fluor, 1:500) for 1 h at room temperature. Samples were washed three times in PBS. To quench lipofuscin autofluoresence for PrPC staining, samples were incubated in 10 mm CuSO4 in ammonium acetate for 15 min (Schnell et al., 1999) after secondary antibody. The samples were washed one with PBS and mounted onto glass slides (Superfrost Plus; Thermo Scientific) and mounted using Vectashield (Vector Laboratories).
Immunoblots and ELISA/PLISA
All samples were run on a precast 4–20% Tris-glycine gel (Bio-Rad Laboratories) and then transferred to a nitrocellulose membrane using iBlot gel transfer stacks (Novex Life Technlogies). After transferring, membranes were washed in blocking buffer (Rockland, #MB-070) for 1 h at room temperature followed by overnight incubation at 4°C with the following antibodies: mouse anti-PrPC (SAF-32; Cayman, #189720; 1:200), rabbit anti-Doppel (ab23701, 1:500), mouse anti-β-Actin (Cell Signaling Technology, #3700, 1:10,000), rabbit anti-eEF2 (Cell Signaling Technology, #2332; 1:1000), and rabbit anti-phospho-eEF2 (Cell Signaling Technology, #2331; 1:1000). The membranes were then washed 3 times in 0.1% TBS-Tween 20 and incubated with secondary antibody (Odyssey donkey anti-rabbit and donkey anti-mouse conjugated to IRDye 680 or IRDye 800 at 1:20,000) in blocking buffer for 1 h at room temperature. Finally, membranes were washed 3 times in 0.1% TBS-Tween 20 and developed using a LI-COR Infrared Imaging system. Densitometric analysis was performed using Image Studio Lite (LI-COR).
ELISA was used to measure human amyloid-β 40 (Life Technologies, #KHB3481) and human amyloid-β 42 as per the manufacturer's instructions. The TBS-soluble cortical lysate was homogenized with 1:6 (w/v) and diluted in standard diluent buffer at a 1:1 ratio. RIPA-soluble cortical lysates were homogenized as described above and diluted in standard diluent buffer at 1:5. The absorbance at 450 nm was measured using a Victor 3V plate reader (PerkinElmer Life Sciences). The absolute amount of a Aβ40 or Aβ42 was calculated using the standard curve based on serial dilutions of monomeric synthetic Aβ40 or Aβ42 and normalized to original tissue weight. A modified ELISA was used to measure PrPC-interacting Aβo as described previously (Um et al., 2012). Briefly, a plate-based assay was used for measuring PrPC-interacting Aβo to achieve higher sensitivity. Low-volume high-binding white microplates (Greiner, #784074) were coated overnight with 20 μl/well of 250 nm human PrPC AA23-111 in 30 mm Na2CO3, 80 mm NaHCO3, pH 9.6, at 4°C. After washing twice with PBST (PBS, 0.05% Tween 20), the plates were blocked with 25 μl/well protein-free T20 PBS blocking buffer (Pierce) for 4 h at room temperature. After washing 3 times with PBST, 20 μl of TBS soluble samples diluted 1:2–1:4 in PBSTB (PBS, 0.05% Tween 20, 0.5% bovine serum albumin) were applied to microplates in triplicate and incubated overnight at 4°C. Two-fold serial dilutions of synthetic Aβo in PBSTB (0–10 nm range) were included in each plate as a standard curve. Plates were then washed four times with PBST and incubated with D54D2 anti-Aβ antibody (epitope in the N terminus of Aβ peptide; Cell Signaling Technology, #8243) diluted 1:2000 in PBSTB for 2 h. After washing four times in PBST to remove the unbound primary antibody, 20 μl/well of 1:8000 dilution of Eu-N1 goat anti-rabbit IgG (PerkinElmer Life Sciences) in DELFIA assay buffer (PerkinElmer Life Sciences) was applied for 1 h. Finally, after washing five times in PBST, 20 μl of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied to each well and time-resolved europium fluorescence was measured using a Victor 3V plate reader (PerkinElmer Life Sciences).
Imaging and analysis of immunohistochemistry
An UltraVIEW VoX (PerkinElmer) spinning disc confocal microscope with 60× 1.3 numerical aperture oil-immersion lens was used to analyze PrPC, PSD-95, and SV2A staining. For PrPC staining, three images were captured from the stratum radiatum and cortex from each section with two sections per animal to calculate mean fluorescence intensity (MFI) using ImageJ software. For PSD-95 and SV2A staining, two 40 μm hippocampus containing sections were chosen at random. Three images of the mossy fibers in the dentate gyrus were collected for each slice and ImageJ was used to calculate the area occupied by immunoreactive puncta and averaged per mouse. A Zeiss AxioImager ZI fluorescent microscope (4× and 20× air-objective lens) was used to image and analyze β-amyloid, GFAP, and Iba1 staining. The hippocampus of each mouse was imaged and the immunoreactive percentage area was analyzed using ImageJ (n = averaged percentage positive area for one mouse).
Tamoxifen treatment
All mice used in the experiments were treated with tamoxifen after the first round of behavioral experiments. Tamoxifen was solubilized in sunflower oil to 10 mg/ml with heating at 37°C and sonication was repeated. All mice received a 100 mg/kg dose by oral gavage for 3 d and behavior was analyzed 21 d after treatment after the last day of treatment. Mice were treated at 12 or 16 months (see Figure 1).
Degeneration of catecholaminergic neurons
Immunohistology.
Forty-micrometer coronal sections were placed serially into 12-well plates with cryoprotectant. Tissue sections were washed with TBS and incubated with a polyclonal anti-tyrosine hydroxylase (TH; Millipore, #AB152, 1:2000) antibody overnight at 4°C. Sections were washed in TBS and incubated in a biotin-conjugated anti-rabbit (Jackson Laboratories) secondary antibody. Sections were visualized with ABC (Vector Laboratories, #PK-6100) and DAB (Sigma-Aldrich, #D4418) was used as the chromogen. Sections were then mounted, counterstained with cresyl violet, and coverslipped after a series of alcohol dehydrations.
Stereological analyses.
Stereological sampling was performed using StereoInvestigator software (MicroBrightField) as described previously (Liu et al., 2008). Cell body analyses were conducted with the optical fractionator for counting the number of neurons and the nucleator for neuronal volume analysis of neurons in the locus ceruleus (LC) (Liu et al., 2008). Sampling parameters include a 40 × 30 μm counting frame, a guard height of 2 μm, a 100 × 100 μm sampling grid, and a dissector height of 18 μm. Every fourth section was taken for neuronal analyses, yielding three to five sections for the LC region. The nucleator probes were applied to anteromedial sections of the LC. Gundersen (m = 1) coefficients of error were <0.1.
Behavioral testing
The order of behavioral testing was rotarod, novel object recognition (NOR), Morris water maze (MWM), and passive avoidance test (PAT) last. Exclusion criteria were used for each test as described below. Exclusion criteria for one test were independent of any other test.
Rotarod.
The experimenter was blind to genotype status of all animals. The rotarod used for all experimentation was done on an Economex apparatus (Columbus Instruments). Mice were brought to the testing room and allowed to adjust to the new environment for 15 min before testing began. Mice were gently placed on a stationary rod. The mice were oriented in such a way that would allow them to walk forward to maintain balance once the test began. The rod's initial speed was 2 rotations/min with an acceleration of 0.2 rotations/s. Mice were tested on the rotarod 5 times/d for a 3 d period to acclimate them to the task. On the fourth day, the mice were tested on the rotarod six times and their time until falling off the rod was recorded. Each mouse's shortest and longest time on the rod was discarded. The remaining times for each mouse were averaged together to create a single value for each mouse. If a mouse was unable to stay on the rod for longer than 10 s after 3 d of training, that mouse was not tested.
NOR.
As described in Zhang et al. (2012), mice were handled for 5 min a day for 5 d before the experiment to reduce anxiety. Mice were habituated to clean, rectangular, empty rat cages in a dimly lit behavioral testing room for 1 h. The cage was centered in front of the experimenter and oriented with the short axis perpendicular to the test administrator. During the acquisition trial, mice were removed from the behavioral cage and 2 identical objects, either a single 15 ml conical tube with orange cap or a wrapped 5 ml plastic syringe, were placed 1 inch from the edge of either end of the long axis of the cage perpendicular to the long axis. Object choice was pseudorandom; the object was recorded as the familiar object for each animal. The mouse was then replaced in cage's center not facing either object and a total timer counted up to 10 min. The mice were allowed to explore the object and the time it took to accumulate 30 s of total orofacial object exploration was recorded. Orofacial is defined as whisking or sniffing. Finally, mice were left for 10 min with the objects in the behavior cage. Then, the objects were removed and discarded and the mice were left in their cages for 1 h.
During the test trial, one of both the novel and familiar objects were placed on pseudorandom sides of the cage. Orofacial exploration of each object was timed until a combined total of 30 s was reached. After the trial and acquisition trial of each mouse, cages were cleaned to eliminate scent cues. The experimenter was blind to object novelty and genotype. Mice that did not explore both objects, had barbered whiskers, or failed to reach 30 s of exploration during either trial <6 min were eliminated.
MWM.
Animals were randomly assigned to treatment groups and the experimenter was blind to both genotype and treatment status. Each mouse was handled for 5 min for the 5 d leading up to any behavioral testing to reduce anxiety. MWM testing was completed in 3 d blocks with probe trial performed on the fourth day. Mice were repeatedly placed in an open water pool ∼1 m in diameter to find a submerged hidden platform. Clear and colorless water remained at room temperature throughout all aspects of the experiment. The location of the platform remained fixed in the center of one of the quadrants (target quadrant) of the pool throughout the entire testing period. Visual cues remained constant throughout forward and reverse swims. The mice had a total of eight attempts per day to locate the platform and training was divided into two blocks of four. The first block of four attempts was performed in the morning and the second block of four was performed in the afternoon. The order that the mice were tested in remained constant. The mice were gently placed into the pool facing the wall at one of four locations located in the opposite hemisphere from where the platform was and the latency to finding the platform was timed. The order of the four locations used to start the mice varied for each block to ensure that the mice would have to rely on spatial cues to find the platform. Once a mouse spent 1 s on the platform, the attempt was considered complete and the mouse would be removed from the pool. If a mouse did not find the platform within 60 s, it was guided to the platform and allowed to spend 10 s on the platform, after which it was removed from the pool.
Twenty-four hours after the completion of the last block, the mice were tested in a probe trial. The probe trial consisted of returning the mice to the pool to explore for a single trial of 60 s with the hidden platform removed. The start location was the point in the pool furthest from where the platform originally was placed. A reverse swim was performed with the hidden platform in a different target quadrant from the target quadrant in the forward swim. The latency to platform testing and the probe trials were recorded on a JVC Everio G-series camcorder and tracked by Panlab's Smart software.
PAT.
After the administration of NOR and MWM, mice were subject to PAT using a box with equally sized, nonelectrified light and electrified dark compartments. The guillotine door between the two collapsed with the mouse's complete movement from the light to the dark side. On day one of testing, mice habituated to the light side for 90 s before the door opened. After this, the latency in seconds to enter the dark side was measured for up to 300 s. Mice received a shock lasting 2 s with an intensity of 0.5 mA on the dark side and were left in the dark for 10 s before being returned to the home cage, as described previously (Filali and Lalonde, 2009). This was repeated after 5 min for each mouse. On day two, the shock was lowered to 0 mA and the latency to enter the dark side after habituation was measured once more as a measure of retention of negative association. Perfect retention was considered the maximum latency of 5 min.
Statistics
All data are represented as means ± SEM. Prism 6 software was used for statistical analyses. All the data were analyzed by one-way or two-way ANOVA analysis as noted, followed by post hoc Tukey's multiple-comparisons test, Holm-0Sidak's multiple-comparisons test, and Fisher's LSD multiple-comparisons test. Two-sided tests were used and all data analyzed met the assumption for the specific statistical test used. A level of p < 0.05 was deemed statistically significant.
Results
Conditional deletion reduces total PrPC levels without inducing motor deficits or altering amyloid precursor protein (APP) metabolism
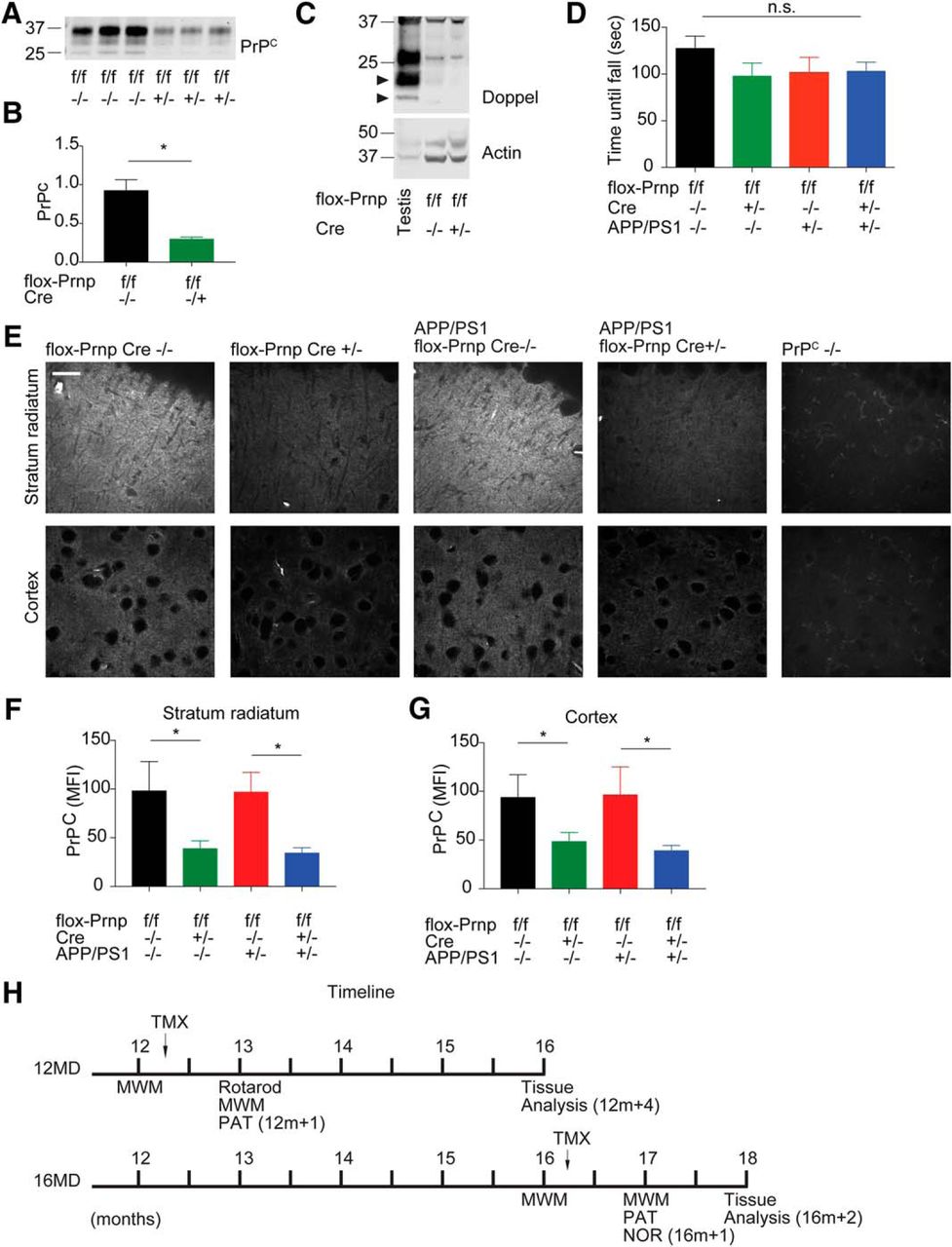
We have observed previously that constitutive deletion of PrPC rescues many of the phenotypes associated with familial AD in mice (Laurén et al., 2009; Gimbel et al., 2010). To determine the therapeutic potential of targeting PrPC after disease onset, we used a genetic approach to delete Prnp shortly after disease onset in a mouse model of familial AD (APPswe/PSEN1ΔE9; hereafter referred to as APP/PS1) and months after the disease had been established. To temporally delete Prnp, we took advantage of mice with flox-Prnp alleles (Tuzi et al., 2004) and crossed these mice to mice expressing tamoxifen-inducible Cre driven ubiquitously by an actin promoter (Hayashi and McMahon, 2002). We were able to generate flox-Prnp Cre−/−, flox-Prnp Cre+/−, APP/PS1 flox-Prnp Cre−/−, and APP/PS1 flox-Prnp Cre+/− mice on an inbred C57BL/6J background for our analysis. We established two cohorts: 12MD and 16MD (Fig. 1H). We performed behavioral experiments before a 3 d tamoxifen treatment and 21 d after the last day of treatment. Tamoxifen treatment was able to reduce the amount of immunoreactive PrPC significantly (Fig. 1A,B). Furthermore, there was no difference in the degree of PrPC reduction in males versus females (data not shown). Previous studies have shown that deletion of PrPC can have varying developmental phenotypes depending on the insertion of the targeting construct (Rossi et al., 2001; Tuzi et al., 2004). To ensure that deletion of Prnp did not induce motor deficits through Doppel (Prnd) expression, we analyzed brain lysate from the 12MD cohort 4 months after deletion (12m + 4) and observed no evidence of Doppel expression compared with a positive testis lysate control (Fig. 1C). Furthermore, we did not observe motor deficits by rotarod test after ubiquitous conditional deletion of Prnp in the 12MD cohort (Fig. 1D). To determine the spatial distribution of PrPC expression after tamoxifen treatment, we measured PrPC expression by immunohistology using an anti-PrPC antibody and quantified immunoreactive MFI for PrPC in the 12MD cohort at 12m+4 (Fig. 1E–G). flox-Prnp mice positive for Cre showed a significant decrease in immunoreactive PrPC MFI compared with Cre-negative mice independently of whether these mice expressed APP/PS1. We observed a similar level of PrPC deletion in the stratum radiatum and the cortex and found that deletion of PrPC using our system did not favor a subpopulation of neurons, but deleted PrPC in a nonspecific manner. Together, these data suggest a working system for conditional deletion of PrPC.

Tamoxifen-inducible Cre-mediated deletion of Prnp reduces PrPC expression and does not activate Doppel expression or induce motor deficits. A, Representative immunoblot of PrPC after Prnp deletion in 12MD cohort using a tamoxifen-inducible Cre system. Each lane represents cortical lysate from one mouse at 12m+4 (H). B, Densitometric analysis of immunoblot from A normalized to total protein levels. Data are graphed as mean ± SEM, n = 3 flox-Prnp Cre−/− mice, n = 3 flox-Prnp Cre−/+ mice. There was a significant decrease in PrPC levels after tamoxifen treatment (unpaired two-tailed Student's t test, *p < 0.05). C, Representative immunoblot of Doppel expression in 12-month-old mice with and without Cre activation. Arrowhead denotes Doppel band. Testis is used as a positive control. Equal amounts of protein loaded in each lane for all blots. D, Rotarod test was performed on all experimental genotypes in the 12MD cohort at 12m+1 to determine potential motor deficits. There was no significant change between genotypes by one-way ANOVA. Data are graphed as mean ± SEM, n = 4 flox-Prnp Cre−/− mice, n = 4 flox-Prnp Cre−/+ mice, n = 5 APP/PS1 flox-Prnp Cre−/− mice, n = 4 APP/PS1 flox-Prnp Cre−/+ mice. E, Representative immunofluorescent images from the stratum radiatum of the hippocampus (top) and cortex (bottom) stained with anti-PrPC (8H4) antibody from the 12MD cohort at 12m+4. Images were captured using a confocal microscope with a 60× objective. Scale bar, 10 μm. F, G, Quantification of immunoreactive PrPC MFI for the indicated genotypes in the stratum radiatum (F) and cortex (G). Two 40 μm slices per animal were analyzed. Data are graphed as mean ± SEM per animal and normalized to flox-Prnp Cre−/− mice. For F, n = 6 flox-Prnp Cre−/− mice, n = 6 flox-Prnp Cre−/+ mice, n = 5 APP/PS1 flox-Prnp Cre−/− mice, n = 5 APP/PS1 flox-Prnp Cre−/+ mice. For G, n = 6 flox-Prnp Cre−/− mice, n = 6 flox-Prnp Cre−/+ mice, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 5 APP/PS1 flox-Prnp Cre−/+ mice. flox-Prnp mice with Cre have significantly less immunoreactive PrPC MFI compared with Cre−/− controls (one-way ANOVA with uncorrected Fisher's LSD multiple-comparison test; *p < 0.05). H, Timeline representing the experimental design. Early Prnp deletion at 12 months (12MD) is represented by the top timeline and late Prnp deletion at 16 months (16MD) is represented by bottom timeline. Black arrow represents tamoxifen injection of all animals regardless of genotype.
Previous work showed that Aβo signaling through PrPC is downstream of APP processing and is independent of APP metabolism (Gimbel et al., 2010). To determine whether conditional deletion of Prnp modified APP metabolism, we measured Aβ plaque load in APP/PS1 flox-Prnp mice with or without Cre in both the 12MD cohort at 12m+4 (Fig. 2A,B) and 16MD cohort at 2 months after deletion (16m+2) (Fig. 2C,D) cohorts. We observed no change in Aβ plaque deposition. In addition to Aβ plaque load, we measured cortical RIPA-soluble Aβ40 and Aβ42 in the 12MD cohort at 12m+4 (Fig. 2E,F). We observed a significant increase in Aβ40 and Aβ42 levels in APP/PS1 flox-Prnp mice compared with WT controls, but no difference between APP/PS1 mice with or without Cre. We measured TBS-soluble Aβ40, Aβ42, and PrPC-interacting Aβo in the 12MD cohort at 12m+4 (Fig. 2G–I). Similar to the findings in RIPA extracts, there was a significant increase in Aβ40, Aβ42, and PrPC-interacting Aβo between flox-Prnp mice and APP/PS1 flox-Prnp with or without Cre. These data show that conditional deletion of Prnp does not alter APP metabolism.

APP metabolism remains unchanged with conditional deletion of Prnp. A, Representative immunofluorescent images of the hippocampus stained with anti-Aβ antibody from 12MD cohort at 12m+4. Images were captured using a fluorescent microscope with a 4× objective. Scale bar, 10 μm. B, Quantification of Aβ-immunoreactive percentage area in the hippocampus for indicated genotypes. Two slices per animal were analyzed. There is no statistical difference between genotypes by unpaired two-tailed Student's t test. Data are graphed as mean ± SEM per animal, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. C, Representative immunofluorescent images of the hippocampus stained with anti-Aβ antibody from the 16MD cohort at 16m+2. Images were captured using a confocal microscope with a 10× objective. Scale bar, 10 μm. D, Quantification of Aβ-immunoreactive percentage area in the hippocampus for indicated genotypes. Two slices per animal were analyzed. There is no statistical significance between genotypes by unpaired t test. Data are graphed as mean ± SEM per animal, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. E, F, RIPA-soluble cortical lysates from the 12MD cohort at 12m+4 were analyzed for Aβ40 (E) and Aβ42 (F) levels by ELISA. There is a significant increase between flox-Prnp Cre−/− and APP/PS1 flox-Prnp Cre−/− mice, but the presence of Cre had no effect on RIPA-soluble Aβ40 or Aβ42 levels (one-way ANOVA with uncorrected Fisher's LSD multiple-comparisons test; **p < 0.01, ****p < 0.0001). Data are graphed as mean ± SEM per animal. For E and F, n = 6 flox-Prnp Cre−/− mice, n = 6 flox-Prnp Cre−/+ mice, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. G–I, TBS-soluble cortical lysates from the 12MD cohort at 12m+4 were analyzed for Aβ40 (G) and Aβ42 (H) by ELISA and PrPC-interacting Aβo (I) levels were measured by a modified ELISA, PLISA (see Materials and Methods). There is a significant increase between flox-Prnp Cre−/− and APP/PS1 flox-Prnp Cre−/− mice, but the presence of Cre had no effect on TBS-soluble Aβ40-, Aβ42-, or PrPC-interacting Aβo levels. (one-way ANOVA with uncorrected Fisher's LSD multiple-comparisons test; ***p < 0.001, ****p < 0.0001). Data are graphed as mean ± SEM per animal. For G and H, n = 6 flox-Prnp Cre−/− mice, n = 6 flox-Prnp Cre−/+ mice, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. For I, n = 3 flox-Prnp Cre−/− mice, n = 2 flox-Prnp Cre−/+ mice, n = 4 APP/PS1 flox-Prnp Cre−/− mice, n = 3 APP/PS1 flox-Prnp Cre−/+ mice.
Conditional deletion of Prnp does not affect microgliosis and astrocytosis
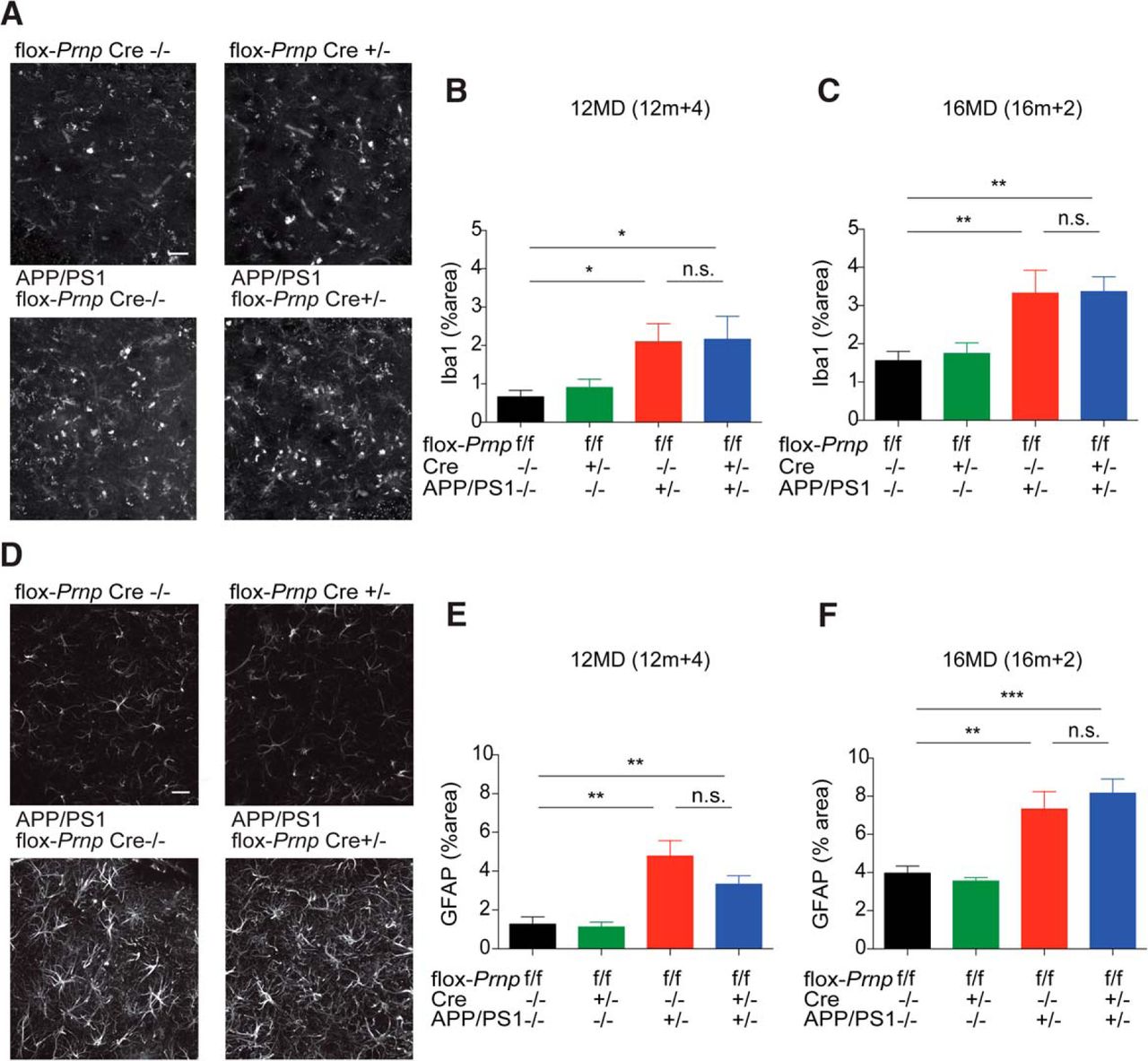
Similar to Aβ plaque load, microgliosis and astrocytosis have been shown to be separate from neuronal Aβo-PrPC signaling and are likely driven by Aβ deposition itself (Gimbel et al., 2010). We measured gliosis by immunohistology using an anti-Iba1 antibody and quantified percentage area immunoreactive for Iba1 (Fig. 3A–C). APP/PS1 flox-Prnp mice in both cohorts showed a significant increase in Iba1-immunoreactive cells compared with WT mice and this was independent of Cre status. Similarly, we measured astrocytosis by immunohistology using an anti-GFAP antibody and quantified the percent area immunoreactive for GFAP. APP/PS1 flox-Prnp mice in both cohorts showed a significant increase in GFAP immunoreactivity compared with WT controls and Cre status did not change the percentage of immunoreactivity significantly (Fig. 3D–F). These data suggest that any synaptotoxic effects due to Aβo-PrPC signaling are independent of microgliosis and astrocytosis.

Deletion of Prnp does not have an effect on microgliosis or astrocytosis regardless of time of deletion. A, Representative immunofluorescent images of the dentate gyrus stained with anti-Iba1 antibody from the 12MD cohort at 12m+4. B, Quantification of Iba1-immunoreactive percentage area in the dentate gyrus for indicated genotypes from the 12MD cohort at 12m+4. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 8 flox-Prnp Cre−/− mice, n = 7 flox-Prnp Cre−/+ mice, n = 11 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. C, Quantification of Iba1-immunoreactive percentage area in the dentate gyrus for the indicated genotypes from 16MD cohort at 16m+2. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 7 flox-Prnp Cre−/− mice, n = 7 flox-Prnp Cre−/+ mice, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. In both B and C, APP/PS1 flox-Prnp mice with or without Cre have significantly more Iba1-immunoreactive cells compared with WT controls (one-way ANOVA with uncorrected Fisher's LSD multiple-comparisons test; *p < 0.05, **p < 0.01). D, Representative immunofluorescent images of the dentate gyrus stained with anti-GFAP antibody from the 16MD cohort at 16m+2. E, Quantification of GFAP-immunoreactive percentage area in the dentate gyrus for indicated genotypes from the 12MD cohort at 12m+4. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 8 flox-Prnp Cre−/− mice, n = 8 flox-Prnp Cre−/+ mice, n = 9 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. F, Quantification of GFAP-immunoreactive percentage area in the dentate gyrus for indicated genotypes from the 16MD cohort at 16m+2. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 6 flox-Prnp Cre−/− mice, n = 8 flox-Prnp Cre−/+ mice, n = 8 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. In both cohorts, APP/PS1 flox-Prnp mice with or without Cre have significantly more GFAP-immunoreactive cells compared with WT controls (one-way ANOVA with uncorrected Fisher's LSD multiple-comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001). Data are graphed as mean ± SEM. Images were captured using a confocal microscope with a 40× objective. Scale bar, 20 μm.
Conditional deletion of Prnp reverses activated eEF2 and synapse loss in APP/PS1 mice
To further characterize temporal deletion of Prnp, we examined eEF2 activation in our 12MD cohort. Previous work has shown that phosphorylation of eEF2 at T56 suppresses translation and potentially mediates synaptic deficits seen in APP/PS1 mice (Horman et al., 2002; Ma et al., 2014). We harvested hippocampi from mice in the 12MD cohort at 12m+4 and found that p-eEF2 (T56) is significantly increased in the APP/PS1 flox-Prnp Cre−/−, as expected, and that this increase was reversed from WT levels in the APP/PS1 flox-Prnp Cre+/− (Fig. 4). These data suggest that suppression of translation through p-eEF2 (T56) is PrPC dependent and the abnormal activity can be reversed months after the disease has been established. We also observed a significant decrease by immunoblot (p < 0.01 by unpaired two-tailed t test) in Pyk2 activation from acute brain slices treated with 1 μm Aβo for 30 min from flox-Prnp Cre−/− mice (116 ± 3% of vehicle values, n = 9 slices from two mice) compared with flox-Prnp Cre+/− mice (99 ± 4% of vehicle values, n = 9 slices from two mice). Overall, conditional deletion of PrPC expression normalizes Aβo-induced signal transduction.
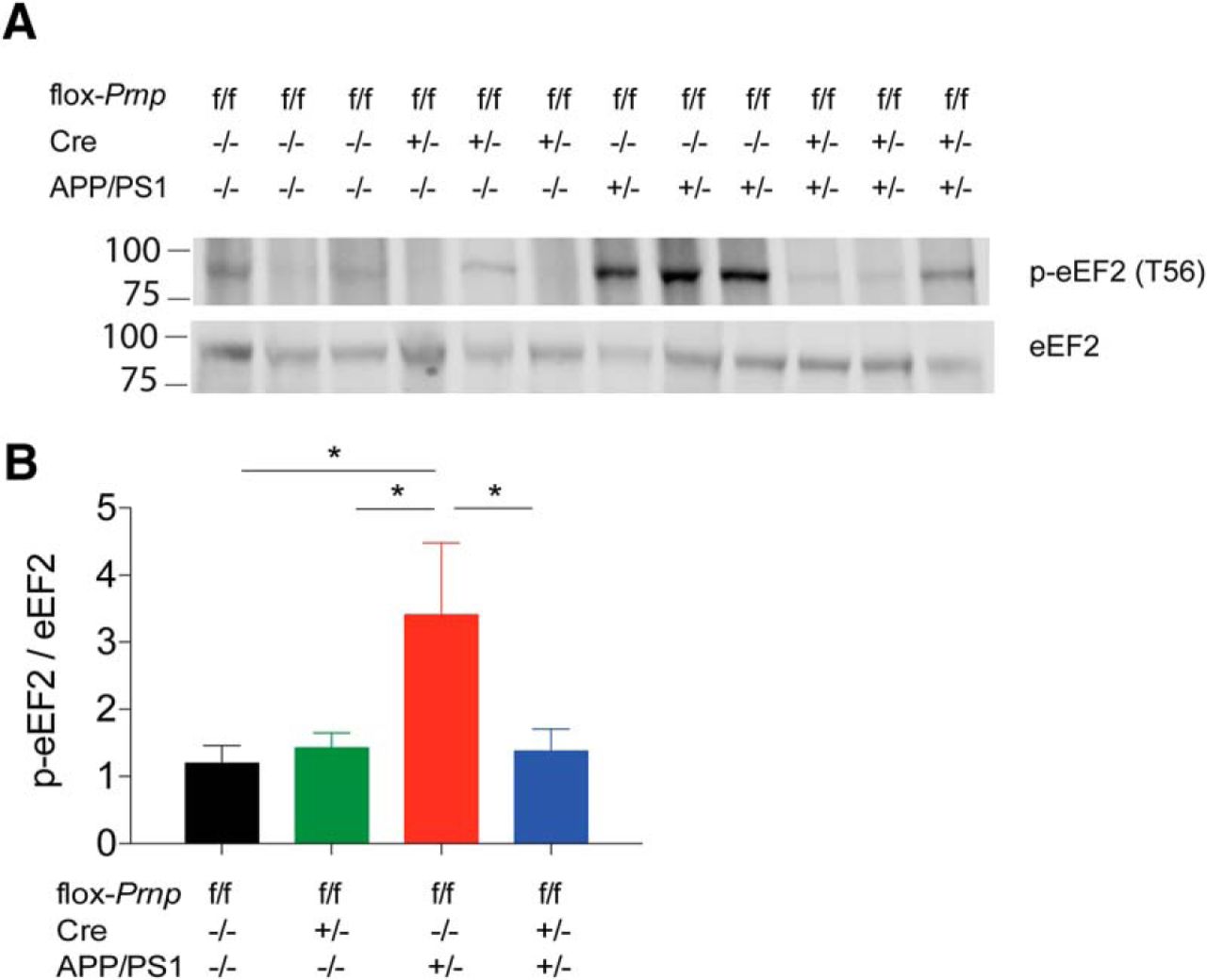
Activation of eEF2 is reversed by deletion of Prnp in the 12MD cohort. A, Representative immunoblot of p-eEF2 (T56) in mice from 12MD cohort at 12m+4. Each lane represents hippocampal lysate from one mouse of the indicated genotype. Equal amount of protein loaded in each lane. B, Densitometric analysis of immunoblot from A normalized to eEF2 levels. There was a significant increase in p-eEF2 (T56) levels in APP/PS1 flox-Prnp Cre−/− mice and this was reversed to WT levels in APP/PS1 flox-Prnp Cre+/− (one-way ANOVA with uncorrected Fisher's LSD test; *p < 0.05). Data are graphed as mean ± SEM, n = 6 flox-Prnp Cre−/− mice, n = 6 flox-Prnp Cre−/+ mice, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice.
To determine the effect of Prnp deletion on synapse density, we imaged synapses immunohistologically in both the 12MD cohort at 12m+4 and 16MD cohort at 16m+2. Although the same mice cannot be examined histologically at different ages, our previous studies (Park et al., 2006; Gimbel et al., 2010; Um et al., 2012, 2013; Haas et al., 2016; Kaufman et al., 2015; Nygaard et al., 2015) clearly showed reduced synapse density in this strain by 12 months of age. We stained brain sections using anti-PSD-95 (postsynaptic marker) or anti-SV2A (presynaptic marker) antibodies. Synapse density in the APP/PS1 flox-Prnp Cre−/− group at 12m+4 displayed a significant decrease as percentage of area immunoreactive for PSD-95 and SV2A (Fig. 5A–C). Conversely, synapse density in APP/PS1 flox-Prnp Cre+/− mice was rescued to WT levels (Fig. 5A–C). Further, the 16MD cohort at 16m+2 also displayed a significant decrease in synapse density by PSD-95 and SV2A immunoreactivity in the APP/PS1 flox-Prnp Cre−/− group (Fig. 5D–F) and this deficit was fully restored in the APP/PS1 flox-Prnp Cre+/− group to WT levels (Fig. 5D–F). Therefore, the decrease in synapse density seen in the APP/PS1 mice is PrPC dependent and conditional deletion of Prnp is able to reverse this deficit even after the disease has been fully established.

Synaptic density is reversed back to WT levels in APP/PS1 mice with early and late deletion of Prnp. A, Quantification of percentage area positive with PSD-95 immunoreactivity in the dentate gyrus of the hippocampus in 12MD cohort at 12m+4. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 10 flox-Prnp Cre−/− mice, n = 10 flox-Prnp Cre−/+ mice, n = 10 APP/PS1 flox-Prnp Cre−/− mice, n = 10 APP/PS1 flox-Prnp Cre−/+ mice. There was a significant decrease in PSD-95 immunoreactivity in APP/PS1 flox-Prnp Cre−/− mice and this decrease was reversed in APP/PS1 flox-Prnp Cre+/− mice (one-way ANOVA with Tukey's multiple-comparisons test; ***p < 0.001, **p < 0.01, *p < 0.05). B, Quantification of percentage area positive with SV2A immunoreactivity in the dentate gyrus of 12MD cohort at 12m+4. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 10 flox-Prnp Cre−/− mice, n = 10 flox-Prnp Cre−/+ mice, n = 10 APP/PS1 flox-Prnp Cre−/− mice, n = 11 APP/PS1 flox-Prnp Cre−/+ mice. There was a significant decrease in SV2A immunoreactivity in APP/PS1 flox-Prnp Cre−/− mice and this decrease was reversed in APP/PS1 flox-Prnp Cre+/− mice (one-way ANOVA with Tukey's multiple-comparisons test; *p < 0.05). C, Representative immunofluorescent images of PSD-95 immunoreactivity in the dentate gyrus of the hippocampus. Images were captured using a confocal microscope with a 63× objective. Scale bar, 10 μm. D, Quantification of percentage area positive with PSD-95 immunoreactivity in the dentate gyrus of the hippocampus in the 16MD cohort at 16m+2. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 8 flox-Prnp Cre−/− mice, n = 8 flox-Prnp Cre−/+ mice, n = 8 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. There was a significant decrease in SV2A immunoreactivity in APP/PS1 flox-Prnp Cre−/− mice and this decrease was reversed in APP/PS1 flox-Prnp Cre+/− mice (one-way ANOVA with uncorrected Fisher's LSD multiple-comparisons test; **p < 0.01, *p < 0.05). E, Quantification of percentage area positive with SV2A immunoreactivity in the dentate gyrus of 16MD cohort at 16m+2. Two slices per animal were analyzed. Data are graphed as mean ± SEM per animal, n = 6 flox-Prnp Cre−/− mice, n = 6 flox-Prnp Cre−/+ mice, n = 6 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. There was a significant decrease in SV2A immunoreactivity in APP/PS1 flox-Prnp Cre−/− mice and this decrease was reversed in APP/PS1 flox-Prnp Cre+/− mice (one-way ANOVA with uncorrected Fisher's LSD multiple-comparisons test; **p < 0.01, *p < 0.05). F, Representative immunofluorescent images of PSD-95 immunoreactivity in the dentate gyrus of the hippocampus. Images were captured using a confocal microscope with a 63× objective.
Conditional deletion of Prnp at 12 and 16 months fails to reverse monoaminergic degeneration
Although synapses can recover after conditional gene deletion, other aspects of neuronal degeneration may be either irreversible or PrPC independent. Degeneration of monoaminergic neurons has been described in the APP/PS1 mice, starting before 12 months and progressing through 18 months (Liu et al., 2008). Constitutive PrPC deletion rescued early serotonin fiber loss in the cerebral cortex (Gimbel et al., 2010). Here, we assessed TH-positive neurons in the LC for the groups with Prnp deletion at 12m+4 (12MD) and 16m+2 (16MD) months (Fig. 6). Stereological counts of TH-positive neurons were decreased by nearly 50% in the APP/PS1 mice regardless of the conditional PrPC loss. Therefore, this phenotype does not require continued expression of PrPC.

Deletion of Prnp in 12MD or 16MD cohorts did not attenuate the loss of TH-positive neurons in the LC. A, Representative immunohistological images of TH immunoreactivity in the LC. Scale bar, 50 μm. B, Quantification of TH-positive neurons in the 12MD cohort at 12m+4. Data are graphed as mean ± SEM, n = 6 flox-Prnp Cre−/− mice, n = 7 flox-Prnp Cre−/+ mice, n = 8 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. Both APP/PS1 groups with or without Cre displayed a significant decrease in TH-positive neurons. C, Quantification of TH-positive neurons in the 16MD cohort at 16m+2. Data are graphed as mean ± SEM, n = 8 flox-Prnp Cre−/− mice, n = 9 flox-Prnp Cre−/+ mice, n = 8 APP/PS1 flox-Prnp Cre−/− mice, n = 4 APP/PS1 flox-Prnp Cre−/+ mice. Both APP/PS1 groups with or without Cre displayed a similar significant decrease in TH-positive neurons. Both APP/PS1 groups in both cohorts displayed ∼a 50% decrease of TH-positive LC neurons compared with non-APP/PS1 littermate controls as shown by two-way ANOVA (**p < 0.01; ***p < 0.001; ns, nonsignificant).
Conditional deletion of Prnp at 12 and 16 months is able to rescue behavioral deficits
To investigate whether deletion of Prnp of at 12 (12MD) and 16 (16MD) months could reverse familial AD phenotypes in our mouse model, we assessed both cohorts before and after deletion of Prnp (Fig. 1.D). We performed PAT only after tamoxifen treatment on the 12MD cohort at 12m+1 and the 16MD cohort at 16m+1 due to the inability to repeat this training procedure in the same mice. We did not find a difference in latency during the training portion, but observed that APP/PS1 flox-Prnp Cre−/− have a significant decrease in latency to enter the dark chamber after 24 h (Fig. 7A,B). Deletion of Prnp in APP/PS1 flox-Prnp Cre+/− mice at either 12m (Fig. 7A) or 16m (Fig. 7B) was able to prevent fully this behavioral deficit at 12m+1 and 16m+1, respectively. These data suggest that deletion of Prnp at 12 months and at 16 months, is able to rescue behavioral deficits seen in PAT.

Deletion of Prnp during early and late disease state is able to reverse behavioral deficits in PAT and NOR. A, 12MD cohort at 12m+1 was subjected to PAT after Cre activation through tamoxifen treatment. All animals were treated with tamoxifen regardless of genotype. APP/PS1 flox-Prnp Cre−/− mice showed a significant decrease in latency to enter the dark chamber, whereas the APP/PS1 flox-Prnp Cre+/− mice had a significant increase in latency similar to that of WT mice (two-way ANOVA with Holm–Sidak's multiple-comparisons test; ***p < 0.001, **p < 0.01, *p < 0.05). Data are graphed as mean ± SEM, n = 5 flox-Prnp Cre−/− mice, n = 8 flox-Prnp Cre−/+ mice, n = 5 APP/PS1 flox-Prnp Cre−/− mice, n = 9 APP/PS1 flox-Prnp Cre−/+ mice. B, 16MD cohort at 16m+1 was subjected to PAT after Cre activation through tamoxifen treatment. All animals were treated with tamoxifen regardless of genotype. APP/PS1 flox-Prnp Cre−/− mice showed a significant decrease in latency to enter the dark chamber, whereas the APP/PS1 flox-Prnp Cre+/− mice had a significant increase in latency similar to that of WT mice (two-way ANOVA with Holm–Sidak's multiple-comparisons test; ***p < 0.001, **p < 0.01). Data are graphed as mean ± SEM, n = 11 flox-Prnp Cre−/− mice, n = 12 flox-Prnp Cre−/+ mice, n = 10 APP/PS1 flox-Prnp Cre−/− mice, n = 5 APP/PS1 flox-Prnp Cre−/+ mice. C, 16MD cohort at 16m+1 was subjected to NOR. All animals were treated with tamoxifen regardless of genotype. The mice were familiarized to an object and subsequently allowed to explore both a familiar or novel object. APP/PS1 flox-Prnp Cre−/− mice did not display a preference for either object, whereas APP/PS1 flox-Prnp Cre+/− mice showed a significant preference for the novel object similar to WT mice (two-way ANOVA with Holm–Sidak's multiple-comparisons test; ***p < 0.001, *p < 0.05). Data are graphed as mean ± SEM, n = 11 flox-Prnp Cre−/− mice, n = 12 flox-Prnp Cre−/+ mice, n = 10 APP/PS1 flox-Prnp Cre−/− mice, n = 6 APP/PS1 flox-Prnp Cre−/+ mice. D, Discrimination index for C was calculated as follows: (novel − familiar)/(novel + familiar). APP/PS1 flox-Prnp Cre−/− mice displayed a discrimination index close to zero, whereas APP/PS1 flox-Prnp Cre+/− mice showed a significant increase in their discrimination index similar to WT mice (one-way ANOVA with uncorrected Fisher's LSD test; ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05).
To evaluate a distinct learning paradigm, we performed the NOR test on the 16MD cohort at 16m+1. WT mice with or without Cre preferred the familiar object, whereas APP/PS1 flox-Prnp Cre−/− mice did not have an object preference (Fig. 7C). Deletion of Prnp in the APP/PS1 flox-Prnp Cre+/− group generated a preference for the novel object similar to WT mice (Fig. 7C). The discrimination index, reflecting the difference in object exploration times, was close to zero for the APP/PS1 flox-Prnp Cre−/− mice, whereas the APP/PS1 flox-Prnp Cre+/− and WT mice with or without Cre showed a similar positive discrimination index (Fig. 7D). Together, these data show that deletion of Prnp after disease onset is able to reverse behavioral PAT deficits and reversal of behavioral deficits in NOR and PAT is achieved even when Prnp deletion occurs many months after the disease has been established.
To further examine learning and memory in our model, we performed MWM on both cohorts. We analyzed the 12MD cohort before and after deletion of Prnp. We performed the first set of 24 training swims (six blocks of four swims) to a hidden platform in the 12MD cohort at 12 months. The APP/PS1 flox-Prnp groups with or without Cre had a significantly higher latency to find the hidden platform than did the WT groups (Fig. 8A). We performed a second set of training swims to a hidden platform in a different quadrant of the pool 21 d after a 3 d tamoxifen treatment (12m+1). In contrast to the first swim set, the post-TMX behavior of the APP/PS1 flox-Prnp Cre+/− group showed significantly reduced time to the hidden platform, whereas the APP/PS1 flox-Prnp Cre−/− group did not improve (Fig. 8B). As shown in Figure 8C, for mice without Cre and thus no Cre-mediated deletion of Prnp, the second training session was as poor as the first. Conversely, the APP/PS1 flox-Prnp Cre+/− mice exhibited significantly better learning of the platform after tamoxifen treatment (Fig. 8D). A probe trial was performed 24 h after trial block six. The APP/PS1 flox-Prnp groups with or withour Cre before tamoxifen treatment spent significantly less time in the target quadrant compared with WT groups with or without Cre (Fig. 8E). After the second training set (after tamoxifen treatment), the APP/PS1 flox-Prnp Cre−/− mice spent less time in the target quadrant, similar to the first probe trial, whereas APP/PS1 flox-Prnp Cre+/− mice showed a trend to spent more time in the target quadrant (Fig. 8F). We also assessed Prnp deletion later during disease progression in the 16MD cohort at 16m and 16m+1. For this group, during the pre-tamoxifen session, the APP/PS1 flox-Prnp mice with or without Cre spent significantly less time in the target quadrant compared with WT mice with or without Cre (Fig. 8G). After tamoxifen treatment at 16 months, the APP/PS1 flox-Prnp Cre+/− spent just as much time as WT mice in the target quadrant and significantly more time compared with APP/PS1 flox-Prnp Cre−/− mice still expressing PrPC (Fig. 8H). Together, these data demonstrate that Prnp deletion can reverse synaptotoxic effects on learning and memory mediated by Aβo-PrPC signaling during early and late stages of disease progression.

Deletion of Prnp during early and late disease state is able to reverse learning and memory deficits in MWM. A, 12MD cohort at 12m+1 was subjected to MWM and latency was calculated as time to find a hidden platform. The forward swim was performed on mice before Cre activation. Both APP/PS1 mice with or without the Cre gene had a significantly higher latency compared with WT controls in the last four swims (two-way RM-ANOVA; APP/PS1 flox Prnp Cre−/− and APP/PS1 flox Prnp Cre+/−, **p < 0.01). Data are graphed as mean ± SEM, n = 21 flox-Prnp Cre−/− mice, n = 21 flox-Prnp Cre−/+ mice, n = 20 APP/PS1 flox-Prnp Cre−/− mice, n = 20 APP/PS1 flox-Prnp Cre−/+ mice. B, Cre was activated with a 3 d tamoxifen treatment. The reverse swim was performed 21 d after the last day of tamoxifen treatment. APP/PS1 flox-Prnp Cre−/− mice had significantly higher latency compared with APP/PS1 flox-Prnp Cre+/− and WT mice in the last eight swims (two-way RM-ANOVA; APP/PS1 flox Prnp Cre+/−, *p < 0.05). Data are graphed as mean ± SEM, n = 16 flox-Prnp Cre−/− mice, n = 17 flox-Prnp Cre−/+ mice, n = 17 APP/PS1 flox-Prnp Cre−/− mice, n = 15 APP/PS1 flox-Prnp Cre−/+ mice. C, Data from A with the latency from the APP/PS1 flox-Prnp Cre−/− group separated into pre-tamoxifen (black) and post-tamoxifen (red). Latency of either group was not statistically significant. D, Data from B with the latency from the APP/PS1 flox-Prnp Cre+/− group separated into pre-tamoxifen (black) and post-tamoxifen (red). APP/PS1 flox-Prnp Cre+/− mice are able to learn significantly better after Cre activation (two-way RM-ANOVA; APP/PS1 flox Prnp Cre+/− pre-tamoxifen, **p < 0.01, ***p < 0.001). E, The probe trial was performed from the cohort in A after trial 6 by removing the hidden platform. Quadrant time percentage is quantified as the amount of time spent in the target quadrant in 1 min. APP/PS1 flox Prnp mice with or without Cre spent significantly less time in the target quadrant compared with WT (one-way ANOVA with uncorrected Fisher's LSD test; *p < 0.05). F, This probe trial was performed from the same group in B after trial 6. WT groups spent significantly more time in the target quadrant compared with APP/PS1 groups (one-way ANOVA with uncorrected Fisher's LSD test; *p < 0.05). Although not significant compared with the APP/PS1 flox-Prnp Cre−/− group, the APP/PS1 flox-Prnp Cre+/− group spent a similar amount of time in the target quadrant after Cre activation as WT mice. G, Similar to the cohort in E, the probe trial was performed in the 16MD cohort at 16m+1 after trial 6 by removing the hidden platform. Before tamoxifen treatment, APP/PS1 flox-Prnp mice with or without Cre spent significantly less time in the target quadrant compared with WT controls (one-way ANOVA with Tukey's multiple-comparisons test; **p < 0.01). Data are graphed as mean ± SEM, n = 13 flox-Prnp Cre−/− mice, n = 12 flox-Prnp Cre−/+ mice, n = 9 APP/PS1 flox-Prnp Cre−/− mice, n = 7 APP/PS1 flox-Prnp Cre−/+ mice. H, After Cre activation through tamoxifen treatment, APP/PS1 flox-Prnp Cre+/− mice spent significantly more time in the target quadrant compared with APP/PS1 flox-Prnp Cre−/− mice (one-way ANOVA with Tukey's multiple-comparisons test; *p < 0.05, ****p < 0.0001, *p < 0.05). Data are graphed as mean ± SEM, n = 10 flox-Prnp Cre−/− mice, n = 5 flox-Prnp Cre−/+ mice, n = 11 APP/PS1 flox-Prnp Cre−/− mice, n = 12 APP/PS1 flox-Prnp Cre−/+ mice.
Discussion
The major finding in our study is that deletion of Prnp after disease onset reverses synaptic and behavioral deficits in a mouse model of familial AD. Specifically, temporally controlled deletion of Prnp during early onset of the disease at 12 months of age and after continued progression of the disease at 16 months of age can reverse behavioral deficits. Our system provides slightly less than complete loss of Prnp, but the widespread substantial reduction of PrPC expression is sufficient to reverse a decrease in synaptic density and behavioral deficits in a mouse model of familial AD. This work confirms and extends our previous study using a constitutive deletion model of PrPC (Gimbel et al., 2010; Haas et al., 2016). Importantly, this work underscores the clinical potential of targeting PrPC for two reasons: (1) we have shown that PrPC deletion can be efficacious after disease onset and (2) we have shown that less than total deletion of Prnp is sufficient for reversal of AD related phenotypes. Partial deletion holds greater clinical relevance because it is doubtful that any pharmacological intervention can achieve 100% blockade. Combined, these data suggest that reduction of PrPC can reverse AD-associated phenotypes in a transgenic mouse model.
As an artifact of the gene targeting, the Prnp-related gene Doppel is aberrantly expressed in certain mouse models. The CNS expression of Doppel from Prnp regulatory elements leads to Purkinje cell loss and ataxia (Rossi et al., 2001). Using our tamoxifen-inducible system, we have shown that Doppel expression remains unchanged before and after Cre activation. Therefore, these mice do not exhibit locomotor dysfunction by rotarod test or display behavioral abnormalities by several paradigms for learning and memory. We have shown that this Prnp deletion model does not display any obvious phenotype and can be used for Aβo-PrPC related studies with temporal control of PrPC expression.
Our previous work suggests that PrPC is required for synaptic and behavioral deficits seen in a mouse model of AD (Gimbel et al., 2010; Haas et al., 2016). Similarly, we have found that conditional deletion of <100% of Prnp is sufficient to reverse the same synaptic and behavioral deficits long after disease onset. In addition, conditional deletion of Prnp does not change Aβ plaque burden, RIPA- or TBS-soluble Aβ, or PrPC-interacting Aβo, suggesting that the Aβo–PrPC interaction is downstream of APP metabolism. Furthermore, we did not observe a significant effect on astrocytosis or microgliosis through Prnp deletion, suggesting that the Aβo-PrPC effects are neuron specific. Further work is necessary to fully determine whether these effects are cell autonomous within neurons. We observed that eEF2 phosphorylation was reversed, supporting previous work (Um et al., 2013; Ma et al., 2014; Haas et al., 2016; Kaufman et al., 2015) that suppressed protein translation in this familial AD mouse model is PrPC dependent. Overall, these findings confirm the hypothesis that late suppression of most PrPC expression achieves the near complete rescue observed with constitutive and complete ablation.
Multiple receptors for Aβ oligomers have been reported in addition to PrPC (for review, see Smith and Strittmatter, 2017). The data reported here indicate that PrPC is required for the continued presence of behavioral and synaptic deficits in APP/PS1 mice. Previous evidence from us and others have shown that PrPC is a major receptor for Aβo (Laurén et al., 2009; Chen et al., 2010; Dohler et al., 2014). To the extent that other receptors are required in addition to PrPC with regard to manifestation of these phenotypes, they must be co-dependent with PrPC. Work from Kostylev et al. (2015) suggests the precise nature of Aβo species vary from model to model and alternate transgenic models may be more dependent on other receptors. The current study demonstrates the genetic dependence of APP/PS1 phenotypes on the continued presence of PrPC, but they are not designed to explore whether it is acting specifically as a receptor or by an unknown indirect mechanism. Given previous work, a receptor explanation is the most parsimonious, but we cannot formally exclude the possibility that indirect mechanisms may contribute.
Synapse area is restored by PrPC deletion in aged APP/PS1 mice. This goes beyond simply arresting further damage and implies that new synapses can be formed in the adult mouse brain if pathological processes are blocked. Techniques have been developed to monitor synapse stability in vivo with transgenic markers and two photon imaging (Holtmaat and Svoboda, 2009). These methods have shown that, whereas many synapses are stable, new synapses are formed in the adult mouse brain even at advanced age (Mostany et al., 2013). We have documented adult brain synapse formation previously in aged APP/PS1 mice (Heiss et al., 2017). Therefore, if an intervention can block those mechanisms that are accelerating synapse loss in an AD model, then the existing literature provides a basis for expecting synapse restoration (Um et al., 2013; Kaufman et al., 2015).
Although synaptic loss is reversible and synapse density is fully restored by late Prnp deletion, catecholaminergic degeneration in the LC is not. This degeneration may occur by PrPC-independent mechanisms. Our previous observation of cortical serotonin axon rescue in constitutive Prnp mice at 12 months suggests that at least early degeneration for 5HT neurons is PrPC dependent(Gimbel et al., 2010). Conversely, rescue of neurodegeneration may require more complete removal of PrPC than is achieved by the conditional paradigm. Another alternative is that, once monoaminergic degeneration is under way, it cannot be reversed in the same way that the synaptic deficits are by PrPC removal. The relative role of temporal versus dose-dependent versus molecular distinctions in the resistance of monoaminergic neurons to the benefit of conditional Prnp deletion will require further investigation.
AD is primarily a disease of learning and memory and, accordingly, we were able to measure the effect of Prnp deletion on APP/PS1-dependent learning and memory phenotypes in our mouse model. Importantly, we observed that partial deletion was sufficient to reverse behavioral deficits whether gene knock-out occurred shortly after disease onset or many months later, when behavioral and anatomical deficits are robust. We observed robust learning and memory deficits by MWM in all APP/PS1 groups before tamoxifen treatment when Prnp remained intact. One month after tamoxifen treatment, this learning and memory deficit was rescued in the same mice that previously displayed this deficit. The reversal of synapse density provides an explanation for this functional recovery. We observed a similar result when we deleted Prnp at 12 or 16 months. Similarly, we observed a rescue of memory impairment in NOR test and PAT after tamoxifen treatment. The broader implications of this study suggest that targeting the Aβo–PrPC interaction after disease onset can be beneficial. Nevertheless, it remains unclear how late Prnp can be deleted and still provide therapeutic efficacy and if the moment of irreversibility is due to chronic downstream signals induced through Aβo-PrPC signaling.
In contrast to targeting the downstream effects of Aβ, numerous studies have focused on decreasing the amount of Aβ burden by using tet-off gene-regulatable systems. Researchers were able to reduce Aβ levels to WT levels by suppressing mutant APPSwe/Ind in a transgenic mouse model using doxycycline administration (Jankowsky et al., 2005). Their system successfully reduced Aβ levels, but demonstrated that early suppression of APPSwe/Ind was necessary because amyloid plaques were slow to clear once deposited. Suppression of this transgenic APP during early postnatal development rescued locomotor hyperactivity (Rodgers et al., 2012) and a separate study using 12- to 13-month-old mice with a similar tet-off gene-regulatable system found that suppressing transgenic APP expression was able to rescue impairments in both short- and long-term memory tasks (Melnikova et al., 2013). Although this study suggested that suppression of APP on the order of weeks was enough to reverse cognitive deficits seen in this mouse model of familial AD (FAD), it remained unclear whether soluble Aβ species or plaques were responsible. Fowler et al. (2014) were able to show that 2-week suppression of APP using the same tet-off gene-regulatable system was able to partially rescue short-term learning and memory deficits, whereas plaque burden remained unchanged, suggesting that a soluble form of Aβ was responsible for this rescue. Using our conditional deletion system, we also saw a reversibility of endophenotypes associated with our mouse model of FAD, suggesting that, at least in part, cognitive deficits seen in FAD mouse models is mediated by the Aβo–PrPC interaction.
Here, we demonstrate reversibility of a number of AD-related phenotypes in a transgenic mouse model. Deletion of Prnp at either 12 or 16 months was able to reverse synaptic and behavioral deficits seen in this transgenic AD model. Even though the conditional deletion of Prnp was partial, this was sufficient to reverse fully synaptic and behavioral phenotypes. This work supports clinical translation of PrPC targeting for AD treatment. Importantly, it suggests that inhibiting the Aβo-PrPC interaction during later stages of disease will be efficacious. Although PrPC targeting fully reverses synapse loss when cell loss is minimal in the APP/PS1 mouse model, its efficacy in the clinical situation might be predicted to shift from reversing early deficits dependent on synapse dysfunction to slowing progression in advanced stages when cell loss is substantial. Given the high rate of failure for AD directed therapies (Cummings et al., 2014), PrPC is a promising candidate given its cell surface location and potential for small-molecule-directed therapeutics. Nevertheless, further research is required to better understand Aβo–PrPC signaling and to develop disease-modifying therapeutics for the treatment of AD.
Footnotes
This work was supported by the National Institutes of Health, the BrightFocus Foundation, the Alzheimer's Association, and Falk Medical Research Trust (all to S.M.S.). We thank Stefano Sodi for assistance with mouse husbandry and Jillian Friedrich for technical assistance with sectioning the brains.
S.M.S. is a cofounder of Axerion Therapeutics seeking to develop PrPC-based therapeutics for Alzheimer's disease. The remaining authors declare no competing financial interests.
- Correspondence should be addressed to Stephen M. Strittmatter, CNNR Program, BCMM 436, Yale University School of Medicine, 295 Congress Avenue, New Haven, CT 06536. Stephen.Strittmatter{at}yale.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}