

Abstract
Mice lacking the serine protease tissue plasminogen activator (tPA) are resistant to excitotoxin-mediated hippocampal neuronal degeneration. We have used genetic and cellular analyses to study the role of tPA in neuronal cell death. Mice deficient for the zymogen plasminogen, a known substrate for tPA, are also resistant to excitotoxins, implicating an extracellular proteolytic cascade in degeneration. The two known components of this cascade, tPA and plasminogen, are both synthesized in the mouse hippocampus. tPA mRNA and protein are present in neurons and microglia, whereas plasminogen mRNA and protein are found exclusively in neurons. tPA-deficient mice exhibit attenuated microglial activation as a reaction to neuronal injury. In contrast, the microglial response of plasminogen-deficient mice was comparable to that of wild-type mice, suggesting a tPA-mediated, plasminogen-independent pathway for activation of microglia. Infusion of inhibitors of the extracellular tPA/plasmin proteolytic cascade into the hippocampus protects neurons against excitotoxic injury, suggesting a novel strategy for intervening in neuronal degeneration.
Neuronal cell death can lead to devastating human pathologies that are associated with severe cognitive and motor deficits. One mechanism by which neuronal death is induced is overstimulation with glutamate, the primary excitatory neurotransmitter in the CNS. Recent evidence suggests that various mediators contribute to excitotoxin action, including production of reactive oxygen intermediates, nitric oxide, p53, and cytokines (Coyle and Puttfarcken, 1993; Lipton and Rosenberg, 1994; Ankarcrona et al., 1995; Morrison et al., 1996). An excitotoxic pathway is implicated in ischemic neuronal death (Oyzurt et al., 1987; Meldrum, 1990) and may also be involved in other neurodegenerative diseases (Lipton and Rosenberg, 1994).
A region of the mammalian brain that is especially sensitive to neuronal degeneration is the hippocampus. The serine protease tissue plasminogen activator (tPA) has been correlated with hippocampal function, because the levels of tPA mRNA are rapidly increased in the rat and mouse hippocampus on stimulation of neuronal activity (Qian et al., 1993; Carroll et al., 1994). Excessive neuronal activity can lead to neuronal death, suggesting a possible connection between tPA and the degenerative process. Consistent with this idea, mice deficient for tPA are resistant to neuronal destruction induced by excitotoxins acting via all three subtypes of glutamate receptors (kainate, AMPA, and NMDA) (Tsirka et al., 1995). This finding indicates that tPA participates in the pathway of excitotoxin-mediated neuronal death.
In this report, we show that plasminogen-deficient mice are also resistant to kainate-induced degeneration, implicating this zymogen substrate for tPA as an effector of this pathway. Plasminogen mRNA, which is primarily synthesized in the liver, is also produced in the hippocampus (Sappino et al., 1993). Plasminogen mRNA is found exclusively in neurons, whereas tPA mRNA is expressed in both neurons and microglia. tPA and plasminogen proteins are also found in the hippocampus, suggesting that their local synthesis is playing a role in mediating degeneration. Mice lacking tPA show attenuated microglial activation, but plasminogen-deficient mice exhibit normal microglial activation. This result indicates that the influence of tPA on microglial cells is independent of the activation of plasminogen. Finally, infusion of α2-antiplasmin, an inhibitor of the protease generated by tPA, into the hippocampus of wild-type mice confers neuroprotection to excitotoxic damage.
Our findings indicate that one element of excitotoxic damage is initiation of an extracellular proteolytic cascade whose product is plasmin. Promotion of plasmin formation or action facilitates neuronal degeneration, whereas inhibition retards this process. This model has implications for new therapeutic approaches to excitotoxin-mediated neuronal injury.
MATERIALS AND METHODS
Intrahippocampal injections. Adult male mice, weighing ∼25 gm, were injected intraperitoneally with atropine (0.6 mg/kg of body weight) and then were anesthetized deeply with 2.5% avertin (0.02 ml/gm of body weight). They were placed in a stereotaxic apparatus and injected unilaterally with 1.5 nmol of kainic acid in 0.3 μl of PBS into the hippocampus (Andersson et al.,1991; Tsirka et al., 1995). The coordinates of the injection were bregma −2.5 mm, medial-lateral 1.7 mm, and dorsoventral 1.6 mm. The excitotoxin was delivered over 30 sec. After kainic acid was delivered, the injection needle remained at the above coordinates for another 2 min to prevent reflux of fluid. After variable lengths of time (indicated in each experiment), the animals were anesthetized and perfused through the heart with PBS followed by 4% paraformaldehyde. The brains were removed and left in 30% sucrose in fixative overnight at 4°C. Coronal tissue sections (30 μm) were prepared and mounted onto slides and dehydrated through increasing ethanol gradients. They were then stained with cresyl violet, which stains rough endoplasmic reticulum in neuronal cell bodies.
Plasmin (150 ng) was delivered (0.3 μl) unilaterally into wild-type mice by intrahippocampal injection, as described for the delivery of kainate.
Intrahippocampal delivery of α2-antiplasmin.Adult wild-type male mice were anesthetized as above and placed in a stereotaxic apparatus, and a micro-osmotic pump containing artificial cerebrospinal fluid (aCSF) (for control animals) or 100 μl of α2-antiplasmin in aCSF (1 mg/ml; Sigma, St. Louis, MO) was placed subcutaneously in the back of the animals. A brain infusion cannula connected to the pump was positioned at coordinates bregma −2.5 mm, medial-lateral −0.5 mm, and dorsoventral 1.6 mm to deliver the compound near the midline. The infusion rate was 0.5 μl/hr. The pump was allowed to infuse the designated solution for 2 d, and then kainate was injected as described above. Five days after the kainate injection, the mice were killed, and their brains were examined for neuronal survival, microglial activation, and tPA activity.
Immunohistochemistry. Brain sections of the mice, manipulated as described above, were incubated with (1) antiserum against mouse plasminogen raised in sheep (a gift from E. Reich), at 1:20 dilution, and normal sheep serum (detection of plasminogen protein), (2) antibody to the mature macrophage/microglia-specific antigen F4/80 (Harlan Bioproducts, Indianapolis, IN), at the dilutions recommended by the suppliers (1:10), or (3) antibody to rat tPA (Chemicon, Temecula, CA), at the dilutions recommended by the suppliers (1:100). Biotinylated secondary antibodies were used (Vector Laboratories, Burlingame, CA), and the avidin–biotin–peroxidase complex (ABC reaction) was visualized with diaminobenzidine and hydrogen peroxide (Vector Laboratories), as described previously (Tsirka et al., 1995).
In situ mRNA hybridization. Plasminogen cDNA was a generous gift of S. Degen. tPA and plasminogen antisense mRNA probes were prepared from linearized plasmids and labeled with digoxygenin-11-UTP. Hybridization and stringency washes were performed as described (Hébert et al., 1994), except that an anti-digoxygenin Fab fragment (0.3 mg/ml) was used, followed by biotinylated secondary antibody and the ABC reaction as above. To verify probe specificity, antisense mRNAs to bacterial lacZ,mouse REST (Chong et al., 1995), PLD1 (Hammond et al., 1995), or PLD2 (W. Colley, T. Sung, S. Hammond, Y. Altshuller, D. Bar-Sagi, A. Morris, M. Frohman, unpublished observations) were used as negative controls.
3H-kainate binding. Determination of3H-kainate binding was performed as described (Miller et al., 1990): 12 μm cryostat coronal brain sections (n= 9) were preincubated at 4°C with 50 mm Tris-citrate buffer, pH 7.0, for 1 hr. They were then incubated with 50 nm3H-kainate (specific radioactivity 60 Ci/mmol) in buffer for 30 min at 4°C. The slides were rinsed four times with buffer, dried rapidly, and exposed to3H-sensitive film in x-ray cassettes for a minimum of 20 d. To verify specific binding, adjacent sections were incubated with a mix of the labeled ligand and 50 μm unlabeled kainate and were treated in a fashion identical to that of the experimental sections. The autoradiograms were scanned using a BioRad imaging densitometer, and the intensity of signal over the areas of specific hybridization was quantified.
Quantification of neuronal loss over CA1–CA3 hippocampal subfields. Wild-type (n = 15), tPA−/− (n = 15), plasminogen−/− (plg−/−) (n= 4), uPA−/− (n = 2), and wild-type infused with α2-antiplasmin (n = 2) mice were injected, and the tissue was processed as above. Serial sections of 30 μm were prepared and stained with cresyl violet. Five sections from the dorsal hippocampus of each genotype and treatment of mice were matched, and the linear distances of intact (completely spared), partially lost (few intact neurons present), and lost (completely eliminated) pyramidal cell layers were determined on each section. Distances were digitized from camera lucida drawings of the hippocampus. The numbers for each category over each hippocampal region were averaged across subjects in a group.
In situ labeling of DNA fragmentation. The terminal transferase-mediated biotinylated-UTP nick end-labeling (TUNEL) technique was used to assess cell death in the hippocampus of wild-type, plg+/−, and plg−/− mice injected unilaterally with kainate. At 48 hr after injection, the mice were killed, and their brains were frozen quickly. Cryostat coronal brain sections (12 μm) through the hippocampus were fixed in 4% paraformaldehyde and processed using the in situ cell death detection kit, POD, as recommended by the manufacturer (Boehringer Mannheim, Indianapolis, IN).
RESULTS
Plasminogen-deficient mice are resistant to excitotoxin-mediated neuronal degeneration
In addition to its proteolytic activity, tPA can bind via its N-terminal noncatalytic region to a number of molecules, including annexin (Hajjar et al., 1994), low density lipoprotein receptor-related protein (Bu et al., 1992; Orth et al., 1992), and heparin (Andrade-Gordon and Strickland, 1986), as well as a protein component on the surface of the mouse oocyte (Carroll et al., 1993). It was possible therefore that the role of tPA in neuronal degeneration was mediated not by its proteolytic activity but by non-enzymatic interaction with other hippocampal proteins.
If tPA was functioning as a protease in the hippocampus, it is likely that its action would be via activation of plasminogen, the known physiological substrate for tPA (Astrup and Permin, 1947). In this case, mice deficient for plasminogen, which are viable and develop normally (Bugge et al., 1995; Ploplis et al., 1995), should display the same excitotoxin-resistant phenotype as the tPA−/− mice. Therefore, plg−/− mice were tested for their response to excitotoxin by injecting the glutamate analog kainate into the hippocampus and analyzing neuronal survival 5 d later by cresyl violet staining. As shown in Figure 1a (low magnification), control mice heterozygous for the plasminogen mutation were sensitive to neuronal degeneration (comparable to wild-type mice, not shown). Their homozygous plg−/− litter mates, however, were resistant to excitotoxic injury (Fig. 1b). The extent of resistance of the plg−/− mice was comparable to that of tPA−/− mice (Table 1).

Plasminogen-deficient mice are resistant to kainate-induced neuronal degeneration. Cresyl violet-stained coronal sections through the hippocampus reveal the neuronal degeneration generated by kainate. a, Hippocampus from heterozygous plg+/− mouse (wt) 5 d after the injection, showing substantial degeneration on the injected side (ipsilateral), whereas the uninjected (contralateral) side remains unaffected (number of mice injected = 15). CA1, CA2, andCA3 denote the hippocampal subfields; DG, dentate gyrus. b, Hippocampus fromplg−/− mouse 5 d after the injection, showing minimal degeneration on the injected side (n = 4). Arrows show the site of injection. c, High-magnification photomicrograph of part of the ipsilateral CA1 subfield of hippocampus from heterozygous plg+/− mouse (wt) 12 hr after the injection (n = 3). The pyknotic and refractile neuronal cells in the wild-type mouse have disappeared completely by 24 hr, showing that they are not glial cells. d, High-magnification photomicrograph of part of the ipsilateral CA1 subfield of hippocampus from heterozygous plg−/− mouse (wt) 12 hr after the injection (n = 3). e, TUNEL labeling, indicating DNA fragmentation in dying cells (arrows), is observed in the CA1 pyramidal subfield in wt mice (n = 2) and is absent in plg−/− mice (f) 2 d after kainate injection (n = 2).
Genotype and exogenous administration of α2-antiplasmin modulate sensitivity to neuronal degeneration
Examination of the injected hippocampus at high magnification showed that 12 hr after kainate injection, the dying neurons in the control mice were pyknotic and refractile (Fig. 1c). In contrast, neurons in the plg−/− mice had a normal appearance (Fig.1d), similar to the equivalent cells on the uninjected side, which are largely unaffected by kainate (Andersson et al., 1991). This normal appearance persists for at least 30 d (data not shown). In addition to their normal morphology, the neurons exhibit normal staining with cresyl violet, which visualizes ribosomal RNA.
The TUNEL labeling procedure, indicating the presence of fragmented DNA and therefore dying/dead neurons, revealed degenerating neuronal cells in the CA1 pyramidal subfield of the hippocampus of kainate-injected wild-type mice (Fig. 1e, arrows). The contralateral side did not show TUNEL staining, nor did control mice injected with PBS (data not shown). The staining specific in CA1 is in agreement with the region in which apoptotic death has been detected by other investigators after subcutaneous injection of kainate using the same detection method (Morrison et al., 1996). When plg−/− mice were subjected to TUNEL labeling, no staining was observed over the hippocampal neurons, indicating absence of neuronal death in these mice. These results demonstrate that the neurons that persist in the tPA−/− and plg−/− mice are viable and healthy.
The similarity in phenotype between tPA−/− and plg−/− mice, coupled with the documented interaction of these two proteins, argues that a proteolytic cascade involving tPA and plasminogen participates in mediating neuronal cell death.
Mice resistant to neuronal degeneration have normal levels of kainate receptors
One explanation for resistance to kainate in tPA−/−and plg−/− mice could be attenuated expression of kainate receptors. In this scenario, decreased numbers of receptors would result in less binding of excitotoxin, less depolarization of neurons, and ultimately less death. To evaluate this possibility, brain sections from tPA−/− mice were incubated with3H-kainate. Strong 3H-kainate binding was observed on the hippocampal formation, cortex, and caudate-putamen (Miller et al., 1990) in both wild-type and tPA−/− brain sections. The higher affinity sites of hippocampal binding along the CA3 region and dentate gyrus were quantitated by scanning these regions of the autoradiographic slides. As shown in Table 2, the binding capacity of kainate in the wild-type hippocampus was indistinguishable from that in tPA−/− hippocampus. This experiment shows that mice resistant to the degenerative actions of kainate bind the excitotoxin at levels equivalent to those in wild-type animals, ruling out a gross deficit of the receptors as an explanation of resistance. Moreover, immunohistochemical experiments using antibodies against GluR6 (a gift from Dr. J. Prives, State University of New York, Stony Brook) showed no difference in the levels of this glutamate receptor subtype between wild-type and tPA−/−hippocampi (data not shown).
3H-kainate binding to the hippocampus in wild-type and tPA−/−mice
Mice deficient in uPA or fibrinogen, other components of the fibrinolytic system, are not resistant to excitotoxin-mediated neuronal degeneration
Mice express two forms of plasminogen activator, tPA and urokinase plasminogen activator (uPA). Examination of whole mouse brain (Rickles and Strickland, 1988) and specifically the hippocampus (Sappino et al., 1993) has not revealed any uPA mRNA or activity. However, injury in the brain, such as the injection of excitotoxin, leads to the immigration of macrophages that can express both tPA and uPA (Vassalli et al., 1984; Hart et al., 1989). This infiltration of macrophages into the brain may result in both plasminogen activators being necessary to mediate neuronal degeneration. If so, mice deficient for uPA should exhibit resistance to excitotoxicity similar to that of tPA−/− mice.
To examine this possibility, mice deficient for uPA (Carmeliet et al., 1994) were tested using the unilateral, intrahippocampal delivery of kainate (data not shown). The hippocampal neurons of these mice displayed wild-type sensitivity to kainate (Table 1). We also tested mice deficient for fibrinogen (fibrinogen−/− mice) (Suh et al., 1995). Fibrinogen, a coagulation factor, is the precursor of fibrin, which binds to tPA and can enhance its activity under certain conditions. The fibrinogen−/− mice were also susceptible to kainate injection (data not shown) at a level comparable to that of wild-type mice (no spared neurons were observed over the CA1, CA2, or CA3 hippocampal subfields). These findings indicate that deficiency in these members of the fibrinolytic system does not confer excitotoxin resistance to hippocampal neurons.
Plasminogen mRNA is produced in hippocampal neurons, whereas tPA mRNA is produced in both neurons and microglia
Two aspects of the above results prompted a closer examination of the site of synthesis of tPA and plasminogen. (1) Plasminogen is an abundant serum protein synthesized primarily in the liver (Raum et al., 1980) and is too large to traverse the blood–brain barrier (BBB) without a specialized transport system. Therefore, plasminogen in the hippocampus could be derived from the blood-borne protein via leakage or transport, or from local synthesis. The latter was suggested by Sappino et al. (1993), who detected low levels of plasminogen mRNA in the hippocampus by RNase-protection assay. In light of the resistance of the plg−/− mice to excitotoxins, determining whether plasminogen is synthesized in the brain would help advance the understanding of the pathway of degeneration. (2) Previous work has indicated that cells that synthesize both a plasminogen activator and plasminogen suffer deleterious consequences, possibly attributable to the generation of intracellular proteolysis (Sandgren et al., 1991). It was therefore of interest to determine whether the production of tPA and plasminogen was segregated by cell type.
To determine the sites of synthesis of tPA and plasminogen, antisense mouse tPA and plasminogen digoxygenin-labeled RNA probes were hybridized to control or kainate-injected brain sections (Fig.2). tPA mRNA was detected along the neuronal cell layers in the hippocampus (Fig. 2c), as described previously (Qian et al., 1993; Sappino et al., 1993). At higher magnification, the pattern of tPA mRNA along the neuronal cell layer had a complex appearance (Fig. 2e), suggesting that synthesis might be occurring in more than one cell type. In addition, after destruction of neurons by kainate injection, the staining in the larger cells (pyramidal cells) disappeared but remained in the darker and smaller cells (Fig. 2g).

tPA mRNA is present in neurons and microglia, whereas plasminogen mRNA is found only in neurons. Coronal sections through the hippocampus displaying the site of synthesis of tPA (c, e, g) and plasminogen (d, f, h). Low magnification of coronal sections (a–d) and higher magnification of similar sections (e, f). Other mice were unilaterally injected with kainate to cause neuronal degeneration on one side, and subjected to mRNA ISH 5 d later (g, h). Cresyl violet staining of kainate-injected consecutive sections confirmed the complete elimination of the neurons in CA1–CA3 (data not shown). Ina, the probe was control plasmid (pBluescript KS II, Stratagene, La Jolla, CA), and in b the probe was digoxygenin-labeled sense tPA cDNA. The arrows inf indicate dendritic localization of plasminogen mRNA.
Several lines of evidence indicate that these smaller cells are microglia. (1) Transgenic mice that express the bacteriallacZ gene under the control of mouse tPA 5′ regulatory sequences (tPA/lacZ; Carroll et al., 1994) express β-gal along the neuronal pyramidal cell layer in cells ∼10 μm in diameter, smaller than pyramidal neurons, but of average size for satellite microglial cells. (2) In the tPA/lacZtransgenic mice, adjacent coronal brain sections were doubly stained for β-gal and with antibodies to detect markers specific for the three non-neuronal cell types in the hippocampus (data not shown). β-gal-expressing cells colocalized with perineuronal microglia (F4/80), but did not colocalize with oligodendrocytes (anti-myelin basic protein antibody) or astrocytes (anti-glial fibrillary acidic protein).
These results demonstrate that tPA mRNA is produced both in neurons (the fraction of the staining that disappears after injection of kainate) and in the satellite microglia that overlie the neuronal cell layer (the fraction that persists after kainate injection). The distribution of tPA mRNA in both cell types is in agreement with the studies on the localization of tPA in the rat cerebellum and brain stem (Ware et al., 1995).
Plasminogen mRNA was also detected in the hippocampus along the neuronal cell layers, indicating local transcription of this gene (Fig.2d). Plasminogen mRNA, however, was detected only in the larger pyramidal neurons (Fig. 2f), and the mRNA staining was completely abolished by kainate destruction of neurons (Fig. 2h).
An intriguing finding was that plasminogen mRNA was detected in dendrites emanating from the neuronal cell bodies (arrows in Fig. 2f and data not shown). This cellular distribution of plasminogen mRNA suggests transport of the message into the dendrites, which could direct local protein production on neuronal activity (Miyashiro et al., 1994; Link et al., 1995; Steward, 1995).
Taken together, these data demonstrate that plasminogen mRNA is synthesized in the brain exclusively by neurons, whereas tPA mRNA is produced in both neurons and perineuronal microglia.
Plasminogen protein is expressed in a subset of hippocampal neurons
Because post-transcriptional control can often regulate protein expression, it was of interest to determine whether tPA and plasminogen proteins were also present in the hippocampus. The presence of tPA protein has been examined previously by Sappino et al. (1993), who determined tPA activity by zymography on tissue sections. Even though tPA mRNA is observed in the CA1–CA3 regions of the hippocampus and in the dentate gyrus (Sappino et al., 1993) (Fig. 2c), tPA enzyme activity is confined to CA2–CA3 and the dentate and is not detected in the CA1 region. This distribution of tPA activity was corroborated by subsequent, independent experiments (Tsirka et al., 1995; Gualandris et al., 1996). Sappino et al. (1993) also showed that extracts of the CA1 region contained tPA activity; because preparation of the extracts would dissociate enzyme-inhibitor complexes, they concluded that inhibitors in the CA1 were masking the tPA activity in that region. These elegant studies suggest that tPA activity in the hippocampus is under regulation by inhibitors. Antibodies against tPA revealed the presence of tPA protein over the CA2–CA3 pyramidal subfields and dentate gyrus in neuronal as well as microglial cells (Fig. 3c,e). Injection of kainate resulted in stronger tPA microglial expression at the ipsilateral side (Fig.3e), thus confirming that tPA activity is upregulated in activated microglia (Tsirka et al., 1995).

tPA and plasminogen proteins are synthesized locally in the mouse hippocampus. Immunodetection of tPA and plasminogen proteins in coronal sections through the hippocampus 12 hr after kainate injection of a wild-type mouse unilaterally into the hippocampus. High-magnification photomicrographs of the CA1 field reacting with (a) IgG, (b) normal sheep serum, (c) anti-tPA antibody at the contralateral side, (d) anti-plasminogen antibody at the contralateral side, (e) anti-tPA antibody at the ipsilateral side, and (f) anti-plasminogen antibody at the ipsilateral side. Scale bar, 50 μm.
We have investigated the presence of plasminogen protein by immunohistochemistry. Plasminogen was detected in neurons in the pyramidal cell layer (Fig. 3). Although plasminogen mRNA is present in most pyramidal neurons, only a small subset of neurons stained for plasminogen protein (Fig. 3d). Soon after injection of kainate, the levels of plasminogen protein expression appear to be increased (Fig. 3f), suggesting that post-transcriptional regulation controls its expression. Such regulation may represent a mechanism for enhanced expression associated with neuronal activity, in agreement with proposed functions of tPA/plasmin in plasticity and restructuring in the brain (Krystosek and Seeds, 1981; Carroll et al., 1994; Frey et al., 1996).
Mice deficient for plasminogen exhibit normal microglial activation
The injection of kainate into the hippocampus results in local activation of microglial cells (Andersson et al., 1991), made manifest by changes in morphology and increased expression of the surface antigen F4/80 (Lawson et al., 1990) (Fig.4a–d). Mice deficient for tPA displayed activation of microglial cells after kainate administration, but the response was attenuated compared to that of wild-type animals, both in F4/80 staining intensity and in morphological changes (Tsirka et al., 1995). These results suggested that tPA participates in the activation pathway of microglial cells, which in turn could be affecting the degeneration process.
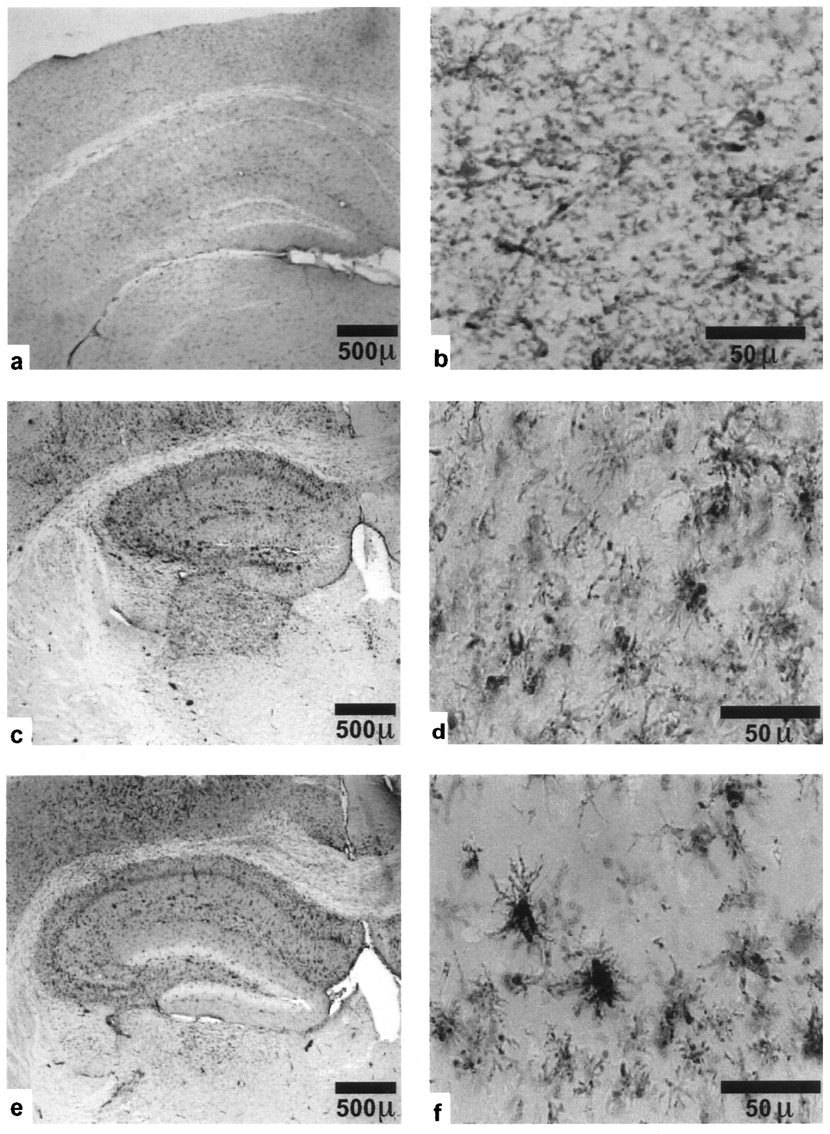
Normal activation of microglia in kainate-injected plasminogen-deficient mice. Left panels, Low-magnification F4/80 immunostaining of coronal sections through the hippocampus 5 d after injection of a wild-type mouse with PBS (a, b) or with kainate (c, d), and of a plg−/− mouse (e, f) with kainate. Extensive activation is observed on the ipsilateral side (shown) and around the injection site. Microglia on the contralateral side are also activated (not shown), but to a much lower level. Right panels, High-magnification photomicrographs of representative activated microglia in the CA1 field of stratum radiatum on the ipsilateral side of a wild-type (d) and a plg−/− mouse (f).
To examine the effect of plasminogen on microglial activation, F4/80 immunostaining was performed on brain sections from kainate-injected plg−/− mice. Both the staining intensity and morphology of microglial cells from plg−/− mice were comparable to those of wild-type cells (Fig. 4c–f). These results show that resistance to neuronal degeneration can occur in spite of normal microglial activation, and that a critical proteolytic event is downstream of activation. They also suggest that the role of tPA in microglial activation is independent of its activation of plasminogen.
Inhibition of plasmin confers resistance to excitotoxin-mediated neuronal degeneration in wild-type mice
The above results identify a genetic and cellular pathway, involving tPA and plasminogen, that can result in neuronal cell death after excitotoxic injury. To evaluate directly whether plasmin, the product generated by the action of tPA on plasminogen, is mediating neuronal death, an inhibitor of the activity of plasmin (α2-antiplasmin) was delivered to the brain. Because tPA is required acutely after excitotoxic insult to mediate neuronal degeneration (Tsirka et al., 1996), inhibition of tPA/plasmin proteolytic activity might retard neuronal cell death. Wild-type mice were infused with α2-antiplasmin, injected 2 d later with kainate, and then evaluated for their neuronal status in the hippocampus. Control mice infused with buffer were sensitive to neuronal degeneration in the hippocampus (Fig. 5,top). In contrast, α2-antiplasmin retarded neuronal degeneration induced by kainate (Fig. 5, bottom). The resistance of the protease inhibitor-infused mice was comparable to that observed with untreated tPA−/− mice (Table 1). Therefore, plasmin is a product of the proteolytic cascade that promotes hippocampal excitotoxic neuronal death, and inhibition of plasmin activity in the adult animal can protect against degeneration.

α2-antiplasmin can prevent kainate-induced neuronal degeneration. Cresyl violet-stained coronal sections through the hippocampus of wild-type mice. The mice were infused with buffer (aCSF) or α2-antiplasmin for 2 d, kainate was injected, and then the infusion continued for 5 d more, and the mice were analyzed. Top, Section from a wild-type mouse infused with aCSF showing extensive kainate-induced degeneration. Bottom, Section from a wild-type mouse infused with α2-antiplasmin, showing resistance to kainate-induced degeneration. Arrowheads point to the site of injection; arrows indicate the site of infusion.
DISCUSSION
Extracellular proteases and neuronal degeneration
The results presented here demonstrate that extracellular proteolysis plays a central role in excitotoxin-mediated hippocampal neuronal degeneration. A critical observation in defining this pathway is the protection against degeneration in mice that lack plasminogen. The comparable resistant phenotype conferred by both tPA and plasminogen deficiency is consistent with the function of these enzymes in other contexts: namely, they operate sequentially within a cascade whose final proteolytic product is plasmin. This view is strengthened by the results of local delivery of α2-antiplasmin, because this inhibitor confers resistance to wild-type mice.
Previous work has implicated proteases in neuronal destruction. (1) The degeneration of ganglion neuronal cells after transection of the optic nerve can be retarded by the injection of protease inhibitors into the vitreous body (Thanos, 1991; Thanos et al., 1993). (2) Protease nexin-1, an inhibitor of plasminogen activators and plasmin, can protect cultured hippocampal neurons against hypoglycemic damage, suggesting that proteases can modulate vulnerability to neurotoxicity (Smith-Swintosky et al., 1995). (3) In the cerebellum of the mouse mutant weaver, increased neuronal cell death is evident, coinciding with 10-fold higher than normal tPA activity. This cell death is also observed in cultured cerebellar weaver neurons and can be prevented by inclusion of aprotinin, a serine protease inhibitor, in the culture medium (Murtomäki et al., 1995). The weaver phenotype results from a mutation in the potassium channel gene Girk2 (Patil et al., 1995), which alters the specificity of the channel (Slesinger et al., 1996) and may result in depolarization of granule neurons (Goldowitz and Smeyne, 1995). Such a depolarization could lead to increased tPA levels, because we have found that chemically induced depolarization of PC12 cells leads to an enhanced rate of tPA secretion (Gualandris et al., 1996).
Neurons and microglia in neuronal degeneration
Both neurons and microglia synthesize tPA, but it is not known whether the function of the enzyme from these two sources is the same or distinct. There are previous reports that microglia participate directly in neuronal degeneration (Thanos, 1991; Lang and Bishop, 1993). Activated microglia may contribute to neuronal death by secretion of glutamate (Streit et al., 1992; Patrizio and Levi, 1994), reactive oxygen intermediates (Piani et al., 1992), or cytokines (Prehn and Krieglstein, 1994). A requirement for microglia in degeneration might limit damage to those areas in which both neurons and microglia have received the appropriate stimulus. It is possible, however, that microglial tPA does not act directly in the degeneration pathway and that neuronal tPA is the effector that mediates neuronal death.
The synthesis of plasminogen by neuronal cells and of tPA by both neurons and microglia cells suggests that both cell types may be necessary to effect neuronal degeneration in the hippocampus. Such a neuronal–microglial cross-talk may limit degeneration to those areas in which both cell types receive the appropriate stimulus. On neuronal injury, macrophages cross the BBB and accumulate in the brain. If these cells were responsible solely for neurotoxicity, they might cause extensive neuronal degeneration along their migration path. Instead, the participation of the injured neuronal cells in their own demise (by presenting the plasminogen component of the cascade) could provide a means to localize destructive/phagocytic microglia to those areas in which neurons have been injured. To support this hypothesis, infusion of tPA alone does not cause neuronal degeneration in the side contralateral to kainate injection (Tsirka et al., 1996), presumably because increased plasminogen is also a necessary element in the pathway.
Another reason for the participation of adjacent cell types is that production of both tPA and plasminogen in the same cell could lead to intracellular proteolysis and cell death (Sandgren et al., 1991). We do not believe, however, that intracellular generation of plasmin plays a significant role in this system, because administration of α2-antiplasmin retards kainate-induced degeneration, and this inhibitor can inhibit proteases only in the extracellular space. Moreover, although tPA and plasminogen mRNAs are expressed by neurons, it is possible that the two proteins are produced by different neuronal cell populations. Alternatively, if both tPA and plasminogen are synthesized in the same neuronal cell, intracellular damage may be avoided by either translational control of the mRNAs (Huarte et al., 1987), or by sequestration of the protein products in vesicles (Parmer et al., 1997).
In most situations in which tPA has been studied, it requires the presence of plasminogen to be effective, because of the catalytic amplification generated by zymogen activation. The absence of tPA attenuates microglial activation, but the absence of plasminogen does not, indicating that this effect of tPA does not require plasminogen. Microglial activation thus represents the first example of a plasminogen-independent role for tPA in vivo.
Synthesis of tPA and plasminogen in the brain
Synthesis of tPA occurs in many tissues, whereas the production of plasminogen has been thought to occur primarily in the liver (Raum et al., 1980). If the liver were the sole source of plasminogen, then delivery to the brain via the circulation would be necessary; however, access of large molecules from the vasculature to the brain is in general precluded by the BBB, and the presence of plasminogen in the brain would be observed only in pathological situations accompanied by a compromised BBB. Therefore, the hippocampal synthesis of plasminogen and tPA gives credibility to the presumptive functions of a tPA/plasmin proteolytic system in physiological settings in plasticity and remodeling, because all of the components of the cascade are present locally. This may be only one example of a general phenomenon: that the brain parenchyma may require local synthesis of necessary proteins, because its access to the blood-borne material is so restricted.
Various human pathologies involve excitotoxic damage to the brain. The contribution of extracellular proteases to the degeneration pathway provides a target for therapeutic intervention. Because delivery of protease inhibitors protected from excitotoxic neuronal death (Fig. 5) (Tsirka et al., 1996), it seems reasonable to investigate further the effectiveness of protease inhibitors for the therapy of excitotoxic-mediated brain disorders.
Footnotes
This work was supported by fellowships from the International Human Frontier Science Program Organization (S.E.T.), the Danish Medical Research Council (T.H.B.), and the Medical Scientist Training Program (A.D.R.), by an Established Investigator Award from the American Heart Association (J.L.D.), and by grants from National Institutes of Health (S.S., J.L.D.) and the American Cancer Society (S.S.). We are grateful to D. Colflesh for valuable photographic expertise and to R. Hart for generous assistance. We also thank the following for providing reagents, equipment, advice, and/or helpful discussion: D. G. Amaral, R. Burwell, S. Degen, J. Engebrecht, M. Frohman, A. Gualandris, B. Hitzemann, P. Rapp, A. Jorgen-Relo, E. Reich, T. Rosenquist, F. Sallés, A. Verrotti, and J. Wells.
Correspondence should be addressed to Sidney Strickland, Department of Pharmacology, Basic Science Tower, T8, Room 125, State University of New York at Stony Brook, Stony Brook, NY 11794-8651.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}