

Abstract
Evidence indicates that neuropeptide gene expression is tightly coupled to biosynthesis and secretion. Moreover, rhythmic gene expression often accompanies rhythmic secretion. Luteinizing hormone-releasing hormone (LHRH) neurosecretion, which regulates gonadal function, is pulsatile, with interpulse intervals of ∼1 hr and pulse decays of <30 min in rats. As a basis for a rapid fall in peptide secretion, we hypothesize that LHRH mRNA levels rapidly decay. To address this hypothesis, we examined LHRH mRNA turnover in primary postnatal LHRH neurons maintained in long-term hypothalamic/preoptic area slice explant cultures, using in situ hybridization histochemistry (ISHH). Relative LHRH mRNA content per cell was quantitated by single-cell analysis after transcription inhibition with 5,6-dichloro-1-d-ribofuranosyl-benzimidazole (DRB) or actinomycin D. Cultures were maintained in serum-free medium with tetrodotoxin to suppress spontaneous electrical activity and hence assess only intrinsic cellular activity. A plot of LHRH mRNA level per cell versus DRB treatment time showed a rapid initial decay of LHRH mRNA (t½, 5–13 min), followed by a slower decay rate (t½, 329–344 hr). LHRH cell number after drug treatment as determined by immunocytochemistry did not change. Comparison of mammalian LHRH mRNA 3′-untranslated regions showed two conserved regions. These data indicate that, in primary LHRH neurons, LHRH mRNA has an intrinsically high rate of turnover and a mRNA stabilization component. Foremost, decay of LHRH mRNA, the fastest reported for a neuropeptide to date, corresponds to the decay of LHRH peptide pulses.
Messenger RNA (mRNA) stabilization mechanisms regulate many aspects of neuroendocrine function. Expression of early response genes (i.e., c-fosand c-myc) after stimulation by neurotransmitters, cytokines, or growth factors is quelled by rapid mRNA decay (Greenberg and Belasco, 1993). In cells derived from peripheral tissues, stimulation of β-adrenergic receptors decreases the receptor mRNA half-life (Hadcock et al., 1989), whereas stimulation by glucagon-like peptide-1 increases gene expression of insulin by mRNA stabilization (Wang et al., 1995). The mRNAs of biosynthetic enzymes tyrosine hydroxylase and peptidylglycine α-amidating mono-oxygenase stabilize in response to hypoxia (Czyzyk-Krzeska et al., 1994b) and hyperthyroidism (Fraboulet et al., 1996), respectively.In vivo, follicle-stimulating hormone (FSH) regulates expression of cAMP-responsive element modulator (CREM) by altering transcript stability (Foulkes et al., 1993). Likewise, the suprachiasmatic nucleus, which exhibits rhythmic expression of many neuropeptide mRNAs (Albers et al., 1990; Larsen et al., 1994; Yang et al., 1994; Ban et al., 1997), may maintain vasopressin mRNA rhythmicity (Cagampang et al., 1994) by changes in vasopressin transcript stability (Robinson et al., 1988; Carter and Murphy, 1989).
Luteinizing hormone-releasing hormone (LHRH) neurons, dispersed within the preoptic area/hypothalamus and projecting to the median eminence, regulate gonadal function by controlling luteinizing hormone (LH) and FSH release from the pituitary (Silverman et al., 1994). Neurosecretion of LHRH into hypophysial portal circulation from median eminence nerve terminals is unique in two ways. First, in mammals, ovulation and the preovulatory rise in circulating LH and FSH are preceded by a surge of LHRH (Sarkar et al., 1976; Levine and Ramirez, 1982). Second, to maintain reproductive function, release of LHRH from its neurons must be pulsatile (Belchetz et al., 1978), with interpulse intervals of 30–70 min and peak decays of 10–24 min in rats (Levine and Ramirez, 1980, 1982; Dluzen and Ramirez, 1987). Interestingly, peptide secretion often is paralleled by changes in gene expression (Young and Zoeller, 1987; Stachowiak et al., 1990; MacArthur et al., 1992; Wang et al., 1995). Therefore, we hypothesize that LHRH mRNA has a rapid decay rate to accommodate its pulsatile profile. Although investigation of the molecular basis of LHRH pulsatility and surges in vivo is confounded by the scant number (∼1300 in rat) and scattered distribution of LHRH neurons (Wray and Hoffman, 1986a,b), severalin vivo studies suggest a halving of LHRH gene expression in 1–2.5 hr (Kim et al., 1993; Seong et al., 1993; Leonhardt et al., 1995). Similarly, immortalized rat T-cells express an LHRH mRNA isoform (Wilson et al., 1995) with an apparent half-life of <1 hr. However, GT-1 cells, immortalized hypothalamic neurons that secrete LHRH in a pulsatile manner (Krsmanovic et al., 1992; Martinez de la Escalera et al., 1992; Wetsel et al., 1992), have a rather prolonged LHRH mRNA half-life [22 hr, Bruder and Wierman (1994); 32.5 hr, Lei and Rao (1994); 30 hr, Gore et al. (1997)]. Dissonant observations between immortalized and primary LHRH neurons likely reflect the divergence between mitotic and postmitotic cells, trans-synaptic effects, and/or secondary effects of humoral or serum-containing factors.
To circumvent the complexities of immortalized cell models and dispersed in vivo populations, our laboratory has developed a slice explant culture method to study primary postnatal LHRH neurons (Wray et al., 1988) under defined conditions (Wray et al., 1991). In general, organotypic cultures retain features of their in vivo counterparts, including cytoarchitecture (Wray et al., 1988;Maurer and Wray, 1997), spontaneous electrical activity (Gähwiler and Herrling, 1981), voltage-dependent calcium conductances (Mouginot et al., 1997), rhythmic neurosecretion (Shinohara et al., 1994; Tominaga et al., 1994), and rhythmic mRNA fluctuations (Carter and Murphy, 1989). LHRH-containing explants maintain LH peptide expression in pituitary cocultures, indicating patent LHRH secretion (Wray et al., 1988). Using long-term slice explant cultures, the present investigation calculates basal intrinsic LHRH mRNA turnover in primary postnatal LHRH neurons maintained under defined conditions. We report that LHRH mRNA has an inherently high rate of turnover (t1/2, <15 min) as well as a stabilization component. These findings indicate that the unique secretory properties of LHRH neurons are tightly coupled to the stability of LHRH mRNA.
MATERIALS AND METHODS
Materials.5,6-Dichloro-1-d-ribofuranosyl-benzimidazole (DRB), actinomycin D, tetrodotoxin, dimethyl sulfoxide (DMSO),d-glucose, apo-transferrin, putrescine, sodium selenite, bovine insulin, and l-ascorbic acid were purchased from Sigma (St. Louis, MO). Eagle’s basal medium, Earle’s balanced salt solution, Ham’s F-12 nutrient mixture, l-glutamine, penicillin–streptomycin–neomycin antibiotic mixture, and horse serum were purchased from Life Technologies (Grand Island, NY). Boehringer Mannheim (Indianapolis, IN) was the supplier of bovine serum albumin.
Organotypic cultures. Tissue was cultured as slice explants by the roller-tube method as previously described (Wray et al., 1988;Horvath et al., 1992; Rossi et al., 1992). Briefly, brains from 5-d-old rat pups were removed, and the preoptic area/hypothalami were blocked and sectioned at 400 μm on a McIlwain tissue slicer. Coronal slices (4 total) containing LHRH neurons in preoptic/anterior hypothalamus (see Fig. 1) were separated, placed in Gey’s balanced salt solution enriched with glucose, and refrigerated for at least 1 hr. Slices were adhered onto glass coverslips by a plasma/thrombin clot, placed in 15 ml Falcon tubes, and rotated in a Bellco roller drum. For optimal thinning, cultures were grown initially in serum-containing media consisting of 25% heat-inactivated horse serum, 50% Eagle’s basal medium, 25% Earle’s balanced salt solution supplemented with 7.5 mg/ml glucose, 2 mm glutamine, 12.5 μg/ml penicillin, 12.5 μg/ml streptomycin, and 25 μg/ml neomycin (Wray et al., 1988). Seven days before experimentation, cultures were transferred to defined media composed of 50% Eagle’s basal medium and 50% Ham’s F-12 nutrient mixture supplemented with 10 mg/ml bovine serum albumin, 100 μm putrescine, 5 μg/ml insulin, 100 μg/ml transferrin, 2 mm glutamine, 7.5 mg/ml glucose, 12.5 μg/ml penicillin, 12.5 μg/ml streptomycin, and 25 μg/ml neomycin (Wray et al., 1989). Slice explants were fed every 2 d with defined media, and on culture day 15 the medium was supplemented with 1 μm tetrodotoxin to inhibit Na+channels and, hence, trans-synaptic interactions (Wray et al., 1991). After 18 d in culture, the slice explants were treated with vehicle (0.1% DMSO), 150 μm DRB, or 4 μmactinomycin D to inhibit gene transcription in the continued presence of 1 μm tetrodotoxin. At the times indicated, cultures were fixed and prepared for immunocytochemistry or in situhybridization histochemistry (ISHH) (see Fig. 2).

LHRH neurons examined in slice explant cultures. Shown is an LHRH-immunostained parasagittal section of preoptic/hypothalamic area of a neonatal rat brain (top panel). Arrows indicate location of three immunopositive neurons. Rostral is to the left. The positions of the four 400 μm slices, labeled 2, 3, 4, and 5, used for culturing are indicated ventrally. Coronal views of representative vibratome sections (100 μm) from explants on the day of culturing are shown below the parasagittal section, immunostained for LHRH, and labeled 2–5 (scale bar, 1000 μm). AC, Anterior commissure;AH, anterior hypothalamus; ARC, arcuate nucleus; DA, dorsal area of the hypothalamus;DBB, diagonal band of Broca; DM, dorsomedial nucleus of the hypothalamus; F, fornix;MPO, medial preoptic area; PVN, paraventricular nucleus; rPVN, rostral paraventricular nucleus of the hypothalamus; SCN, suprachiasmatic nucleus; VM, ventromedial nucleus of the hypothalamus.
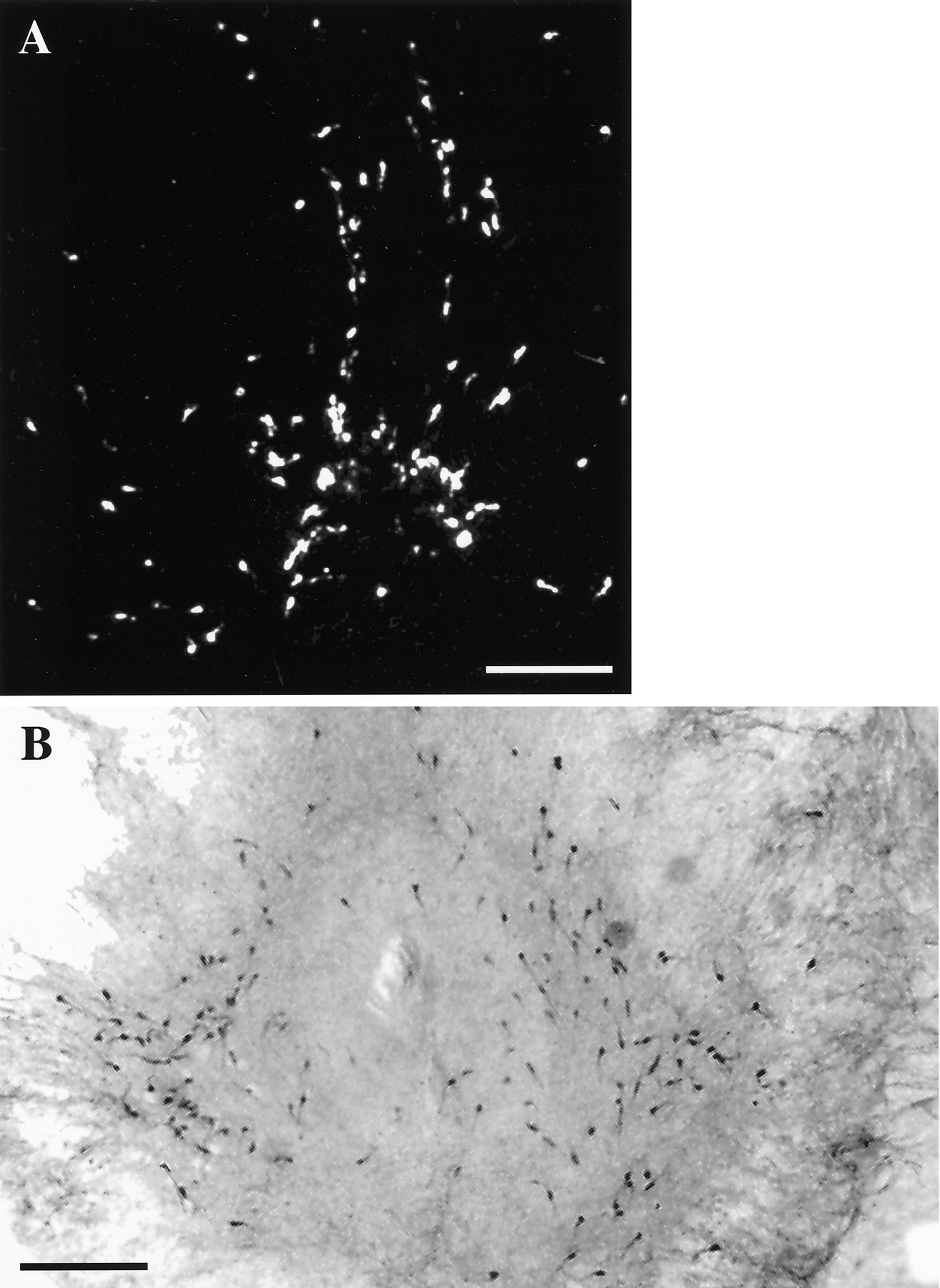
LHRH-expressing cells maintained in slice explant culture for 18 d in vitro. A, Dark-field photomicrograph of a slice 3 culture processed for ISHH, using a synthetic deoxynucleotide antisense probe for LHRH mRNA.B, Bright-field photomicrograph of a slice 4 culture immunocytochemically stained for LHRH. Note the bilateral distribution of LHRH neurons in culture. Scale bar, 500 μm.
Immunocytochemistry. After treatment, slice explants on coverslips were fixed with 4% formaldehyde in PBS for 1 hr and then washed several times with PBS. Cultures were blocked for 1 hr in 10% NGS/0.3% Triton X-100, washed in PBS, and incubated in LHRH antibody (SW1, 1:3000; Wray et al., 1988) overnight at 4°C. The next day the cultures were washed in PBS and incubated in biotinylated secondary antibody (1:500; Vector, Burlingame, CA) in PBS/0.3% Triton X-100 for 1 hr. The cultures were washed with PBS, incubated with avidin–biotin–horseradish peroxidase complex (Elite 1:600; Vector) in PBS/0.3% Triton X-100 for 1 hr, and rinsed with PBS; the complex was visualized by using 3′3-diaminobenzidine and glucose oxidase (Wray et al., 1988). After the reaction the cultures were counterstained with 0.5% methyl green, dehydrated in ethanol, cleared in xylene, and mounted. All cell counts were performed by one investigator, and slides were coded so that the treatment group of a culture was unknown during analysis.
In situ hybridization histochemistry. ISHH was performed as previously described (Wray et al., 1991) with slight modifications. Briefly, slice explants were fixed in 4% formaldehyde, rinsed in PBS, permeabilized in 0.3% Triton X-100/0.05 mEDTA/0.1 m Tris buffer, rinsed in Tris Buffer, washed in 0.25% acetic anhydride/0.1 m triethanolamine hydrochloride/0.9% NaCl, rinsed in 2× SSC, dehydrated through ethanol, delipidated in chloroform, rinsed in ethanol, and air-dried. A 48 oligonucleotide probe (5 pmol), complementary to the coding region of rat LHRH precursor within exon 2 (bases 102–149; Grima et al., 1985), was 3′-end-labeled with [35S]dATP (specific activity, 1000–1500 Ci/mmol; DuPont-NEN, Boston, MA), 100 U of terminal deoxynucleotidyl transferase (Boehringer Mannheim) and 5× tailing buffer (Life Technologies) to a specific activity of 10,000–18,000 Ci/mmol. Labeled probe (500,000 cpm) was applied to each culture in 25 μl of hybridization buffer [4× SSC, 50% formamide, 10% dextran sulfate, 250 μg/ml yeast tRNA, 500 μg/ml sheared single stranded salmon sperm DNA, 1× Denhardt’s solution, and 100 mm dithiothreitol (DTT)]. Cultures were hybridized overnight in humid chambers at 37°C. The next day the cultures were rinsed in 1× SSC/65 mm DTT, washed at high stringency in 2× SSC/50% formamide/41 mm DTT at 45°C, followed by 2× SSC/50% formamide at 45°C without DTT, and washed in 1× SSC at room temperature. Then the cultures were rinsed in water, dehydrated in ethanol, dried, and placed against film. After x-ray film exposure the cultures were dipped in NTB3 (Eastman Kodak, Rochester, NY) and exposed for 21 d. Emulsion-covered cultures were developed in Dektol (Eastman Kodak) at 15–17°C, rinsed in water, and fixed with Kodak fixer and then counterstained with 0.5% methyl green, dehydrated in ethanol, cleared in xylene, and mounted with Permount. Frozen rat brain sections were used as positive controls and were treated by identical ISHH procedures in parallel with the cultures. A second probe generated against mouse LHRH cDNA (bases 1651–1700; Grima et al., 1985) produced similar results (data not shown).
Quantitation and statistical analyses of single-cell data.Images were digitized by an image analysis system consisting of a Sony CCD video camera module model XC-77, Power Macintosh 7100/80, Zeiss upright microscope, and National Institutes of Health Image software (Wayne Rasband, National Institutes of Health, Bethesda, MD). Statistical comparisons of nonparametric and parametric data were calculated with StatView (Abacus Concepts, Berkeley, CA) and Prism (GraphPad Software, San Diego, CA).
Quantitation of mRNA was performed as previously described (Maurer and Wray, 1997). All single-cell analysis was performed by one investigator, and slides were coded so that the treatment group of the culture was unknown to the investigator during analysis. LHRH mRNA levels within single cells were determined by measuring integrated densities of silver grains over a cell and the cell area enclosing silver grains; within an individual culture, all discernible single cells were analyzed. Silver grains deposited on labeled cells, initially detected under dark field, were digitized under bright field, and mean optical density (O.D.) measurements (15% above field background) per cell area, expressed as O.D./μm2, were calculated for single cells and local background. Then the value was multiplied by the highlighted cell area to obtain a total LHRH mRNA level per cell (O.D./cell). Local background multiplied by the measured background cell area was subtracted from each cell measurement to obtain a corrected LHRH mRNA level per single cell: (Areacell × Meancell)−(Areabackground × Meanbackground) =mRNA level/cell. (1)
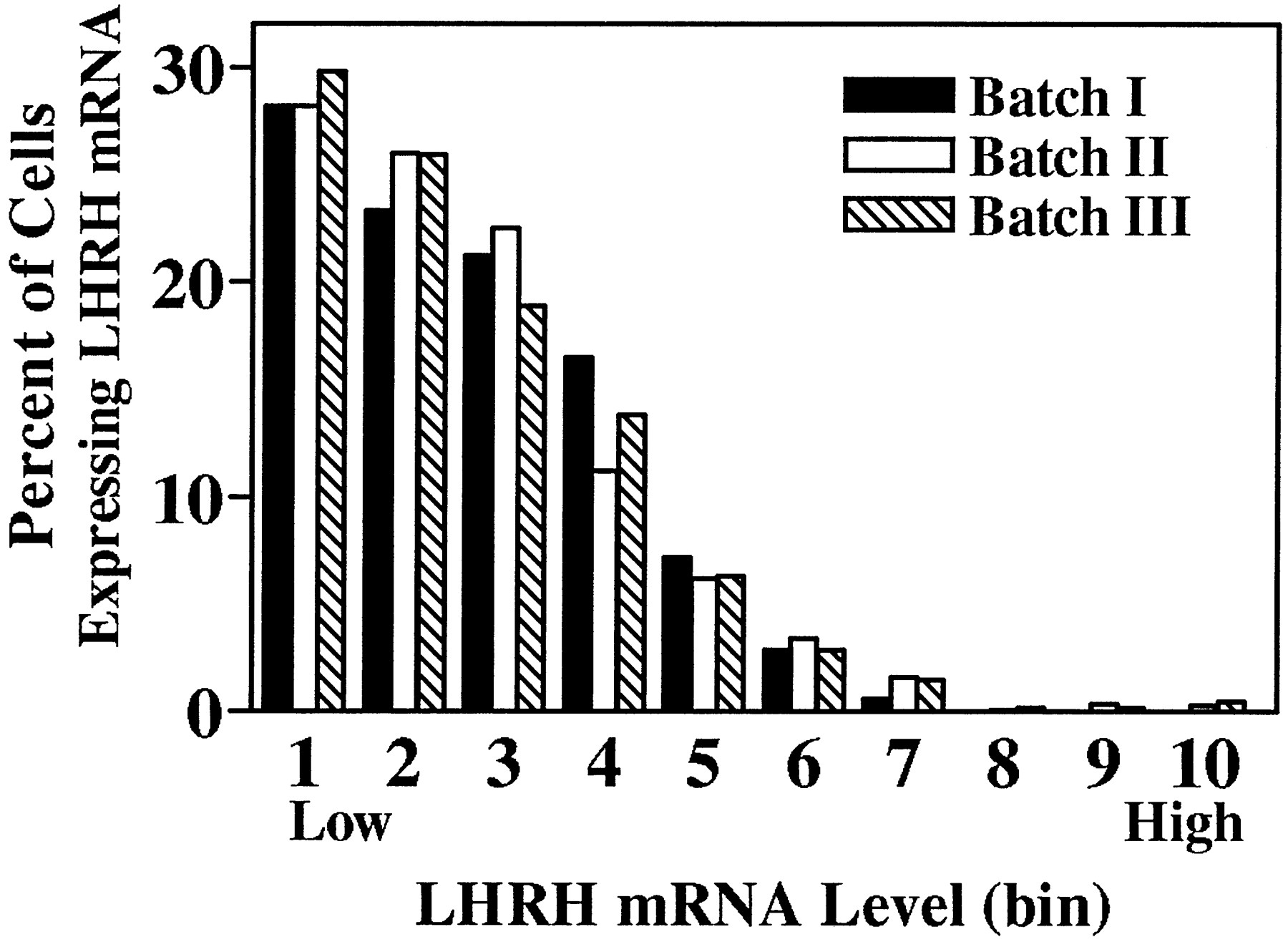
All discernible single cells within an individual slice explant were analyzed, allowing for tabulation of the total number of radiolabeled LHRH neurons observed per culture. Among control cultures from batches I, II, and III no significant differences were noted in the mean number of cells per slice explant (p > 0.05, ANOVA). However, because slice explant cultures were generated on different dates and processed for ISHH separately (batches I, II, and III), mRNA levels per cell from batches II and III were normalized to those of batch I by multiplying values in batches II and III by the ratio of control mean batch I/control mean batches II or III (ratios = 2.22 and 0.52, respectively). For a population of cells within a given treatment group, a range of mRNA levels per cell was observed, with the frequency distribution being positively skewed (Zoeller et al., 1988; Wray et al., 1989; also Figures 3, 5); therefore, significance was determined by the Kolmogorov–Smirnov test for nonparametric data at the p < 0.001 level.

Frequency distributions of hybridization signal intensities (LHRH mRNA level per cell) of individual labeled cells from batches I, II, and III control cultures hybridized with a probe against LHRH mRNA. Because slice explant cultures were generated on different dates and processed for ISHH separately (batches I,II, and III), mRNA levels per cell from batches II and III were normalized to those of batch I by multiplying values in batches II and III by the ratio of control mean batch I/control mean batches II or III (ratios = 2.22 and 0.52, respectively). The total range of LHRH mRNA level per cell values was divided into 10 bins with each bin representing, arbitrarily, 9007 O.D. units; bin 1 contains cells with the lowest levels of LHRH mRNA, whereas bin 10 contains those with the highest levels. The frequency distribution of LHRH mRNA levels of single cells in control cultures from each batch was not significantly different from that of the others (p > 0.05, Kolmogorov–Smirnov).

Significant reduction in LHRH mRNA levels after treatment with DRB. Treatment of explant cultures with 150 μm DRB for 0.5 or 8 hr significantly reduced the LHRH mRNA level per cell (p < 0.001, Kolmogorov–Smirnov).
Treatment with DRB had no effect on LHRH cell number in batch I or III cultures (p > 0.05, ANOVA), but in batch II slice explants significant differences were observed. Batch II control cells had notably lower LHRH mRNA levels than batches I or III (45 and 23%, respectively), and further reduction of LHRH mRNA signal using DRB resulted in cells being below the level of detectability. Therefore, to avoid selection bias, we removed DRB-treated cultures from batch II from the O.D. analysis and evaluated them solely by cell number. For batch II, all discernible LHRH mRNA-containing cells were counted in cultured slices 2–5, and values were meaned within slices and treatment groups. Within individual slices 2–5, DRB treatment consistently reduced the observed cell number per culture. To determine whether this trend was significant, we summed the means from slices within a treatment group, propagated the SE, and calculated and plotted the mean cell number per “whole animal” ±SE for each group to determine LHRH mRNA turnover.
Nonlinear regression curves of LHRH mRNA turnover were fit to mean O.D. and mean cell number per whole animal versus DRB treatment time with a two-phase exponential decay equation (Prism, GraphPad): Y = Span1·eK1X +Span2·eK2X + Plateau. (2)
Y = Span1+ Span2 + Plateau atx = 0. Y decays to Plateau with fast and slow rate components (K1 andK2). These are first-order decay constants and, as such, the half-life is t1/2 = 0.6932/K1 and 0.6932/K2. The fast, initial component was used to determine thet1/2. In addition, LHRH mRNA turnover was calculated by using median O.D. values (see Table 1; Maurer and Wray, 1997). The median, defined as the value in which 50% of all values of a population fall above and below, is an alternative measure of central tendency in skewed distributions.
DRB treatment reduces LHRH mRNA levels in single cells as determined by ISHH
Nucleotide sequence analyses. Nucleotide sequences were compared by the Genetics Computer Group Wisconsin Sequence Analysis Package (v. 8.1) GAP and Bestfit programs. Energetically favorable 3′-untranslated region (3′-UTR) secondary structures were predicted, using m-fold (v. 2.3) by Zucker and Turner with Turner group energy parameters athttp://www.ibc.wustl.edu/~zuker/rna/form1.cgi.
RESULTS
A parasagittal section of the preoptic area/hypothalamic region from a neonatal rat brain illustrating the distribution of LHRH neurons is shown in Figure 1. The positions of the four 400 μm slices labeled 2, 3, 4, and 5 that were used for culturing are indicated ventrally. Examples of in vivocoronal sections, before culturing, immunocytochemically stained for LHRH, and labeled 2–5 are shown. Slice explants maintained for 18 d in vitro (Fig. 2) retained bilateral distribution of LHRH neurons in vivo (Fig. 1,bottom). Cell counts from the ISHH data indicate that 15–25% of the primary LHRH neurons survived in organotypic culture; the distribution of LHRH neurons in slice cultures 2–5 is proportional to that observed in vivo and similar to that previously reported in vitro (Wray et al., 1988).
O.D. levels (corresponding to LHRH mRNA levels) were quantitated. As seen in Figure 3, the frequency distribution of LHRH mRNA levels of single cells in control cultures from each batch were not significantly different from those of the others. LHRH mRNA levels among control slices 2–5 also were not significantly different, as reported previously (Wray et al., 1989) and as seen in vivo (Porkka-Heiskanen et al., 1994). LHRH mRNA levels per cell of all cells within each treatment group were normalized (see Materials and Methods) to their respective controls and pooled to create frequency distributions of single-cell LHRH mRNA levels for each treatment group. DRB and actinomycin D, which act at pharmacologically discrete steps of transcription (Sobell, 1973;Marshall and Price, 1995), were used to block mRNA transcription. As compared with controls, LHRH neurons within slice explant cultures treated with 150 μm DRB had fewer silver grains deposited over individual neurons, indicating a decrease in LHRH mRNA levels (Fig. 4). Note that, although a relatively large proportion of LHRH RNA resides within the nucleus (20–40%; Jakubowski and Roberts, 1994; Yeo et al., 1996), in organotypic culture in which entire LHRH neurons are present, clear nuclear areas lacking silver grains are visible in many cells (Fig. 4,circled cells). Thus cytoplasmic, not nuclear RNA, is detected. Single-cell analysis of individual neurons showed that DRB significantly reduced LHRH mRNA levels at all time points (Fig.5, Table1). Treatment of slice explants with 4 μm actinomycin D also significantly reduced LHRH mRNA levels (data not shown).

Single cells expressing LHRH mRNA were visualized by ISHH. Explant cultures (A) were treated with 150 μm DRB for 8 hr (B), fixed, and processed for ISHH, using an LHRH mRNA probe. Circles on panels indicate single LHRH neurons with clear labeling of cytoplasmic, not nuclear, mRNA (clusters of silver grains 15% more than background). Both panels show batch I, slice 3 cultures at the level of the organum vasculosum lamina terminalis. In the explants,bottom is ventral and center is the third ventricle. Scale bar, 100 μm.
LHRH mRNA degradation or turnover was calculated by using DRB-treated cultures. A plot of LHRH mRNA level (mean O.D. units) versus DRB treatment time (hours) showed a rapid initial decay of LHRH mRNA, followed by a much slower decay rate (Fig.6, top panel). Data were fit by using a two-phase exponential decay (R2 = 0.91). The initial decay occurred with a t1/2 of 0.21 hr (i.e., 13 min), whereas the slow component decayed with a t1/2 of 344 hr. Because extreme values are weighted more heavily than those near the mean in a skewed distribution, the median of each group, a measure of central tendency, also was used to estimate LHRH mRNA turnover. A curve fit (R2 = 0.91) of LHRH mRNA level (median O.D. units) versus DRB treatment time (hours) calculated an initial t1/2 of 0.086 hr (i.e., 5 min), with a t1/2 of 329 hr for the slower component (Table 1). In temporally parallel experiments that used ISHH with single-cell analysis in hypothalamic explant cultures, tyrosine hydroxylase mRNA turnover ranged from 6 to 23 hr (Maurer and Wray, 1997), demonstrating that a generalized rapid destabilization of mRNA was not induced by our experimental method; furthermore, the observed plateau in LHRH mRNA levels at the longer DRB treatment time points was well above our lower limit of detection.

Explant cultures were treated with 150 μm DRB for 0, 0.5, 2, 4, or 8 hr and processed for ISHH, using an LHRH probe. Top panel, LHRH mRNA levels per cell (O.D./cell) were measured, values from slices 2–5 were pooled, and means ± SE of each treatment group were plotted to calculate LHRH mRNA turnover. Bottom panel, For all control and batch II slice explants, all discernible LHRH mRNA-containing cells were counted in slices 2–5; values were meaned within slices and treatment groups; means from slices within a treatment group were summed, the SE propagated, and the mean cell number per whole animal ±SE for each group calculated and plotted to determine LHRH mRNA turnover. DRB had a significant effect on cell number (p < 0.001, ANOVA), and the zero point cell number was significantly different from that of each DRB time point (Bonferroni’s Multiple Comparison Test, p < 0.05).
Batch II ISHH cultures, omitted from the above O.D. analysis (see Materials and Methods), showed a significant decrease in the number of labeled cells with DRB treatment. However, by tabulating the decreasing number of radiolabeled LHRH cells per culture in DRB-treated batch II cultures, we generated a simple estimate of LHRH mRNA turnover. At each time point the number of cells per slice culture 2–5 was summed to generate the number of radiolabeled cells per whole animal (Fig. 6,bottom panel). Data were fit by using a one-phase exponential decay (R2 = 0.91). Thet1/2 was 0.76 hr (i.e., 46 min) as calculated by this method, despite the absence of an 0.5 hr time point. The number of radiolabeled cells per whole animal appeared to plateau at ∼60 cells. Treatment of batch II cultures with DRB or actinomycin D for 16 hr did not decrease this number further (72.4 ± 16.5 and 56.3 ± 8.5, respectively). The cell number data, therefore, verify the single-cell O.D. data in that LHRH mRNA has a rapid turnover and LHRH mRNA levels stabilize thereafter with prolonged transcription inhibitor treatment.
Slice explants treated with DRB were immunostained for LHRH to determine whether the treatments eliminated LHRH neurons and whether cultures remained viable. Figure 7 shows no overt change in LHRH immunostaining after treatment with DRB for 16 hr. Table 2 shows that no significant change in LHRH cell number occurred after DRB treatment for 4 or 16 hr. Similar treatment with actinomycin D for 4 hr produced no significant reduction in cell number in slices 3 or 4 [31.0 ± 11.5 (3) and 22.8 ± 8.9 (4), respectively].

DRB does not reduce LHRH cell number, as determined by immunocytochemistry. Explant cultures (A) were treated with 150 μm DRB for 16 hr (B), fixed, and immunostained for LHRH. Scale bar, 25 μm.
Transcription inhibitor treatment does not reduce LHRH cell number as determined by immunocytochemistrya
DISCUSSION
The present investigation shows that LHRH mRNA has an initial rapid rate of decay (t1/2, 5–13 min), followed by a much slower rate (t1/2, 329–344 hr). No change in cell number was detected with immunocytochemistry. The observed initial decay rate of LHRH mRNA is the most rapid turnover reported for a neuropeptide mRNA to date. This places LHRH mRNA with mRNAs for histone, developmentally regulated transcripts, cytokines, interferons, inflammation mediators, and proto-oncogenes (i.e., c-fos, c-myc, and c-jun), which display half-times of <30 min (for review, see Atwater et al., 1990; Greenberg and Belasco, 1993; Surdej et al., 1994; Jacobson and Peltz, 1996). Furthermore, LHRH mRNA decays in the presence of tetrodotoxin and transcription inhibitors, indicating that the decay mechanism is present in the absence of cellular activation. We propose that rapid turnover of LHRH mRNA is necessary for appropriate neuroendocrine function.
LHRH mRNA decay has a slow component in addition to an initial rapid phase. In LHRH neurons analyzed for mRNA levels by single-cell analysis, LHRH mRNA O.D. levels plateaued well above those previously observed for tyrosine hydroxylase mRNA turnover in anterior hypothalamic and arcuate region explant cultures (both ≲5000 O.D. units; Maurer and Wray, 1997), and cell number data exhibited no subsequent decrease with prolonged DRB or actinomycin D treatment. Therefore, the observed plateau does not appear to result from the loss of assay sensitivity. Our previous experience with DRB and actinomycin D (Maurer and Wray, 1997), as well as evidence in the literature (Attardi and Winters, 1993; Czyzyk-Krzeska et al., 1994b; Yeo et al., 1996), indicates that the observed plateau in LHRH mRNA turnover does not reflect incomplete pharmacological inhibition of transcription. In support of this, in the batch II experimental group (which had a low hybridization signal to background) the number of labeled cells, like the O.D. level in the other two experimental groups, plateaued at 40% of control. Therefore, LHRH mRNA stabilization, not methodological phenomena, appears to generate the observed two-phase decay rate. Because subpopulations of LHRH neurons exist (Lee et al., 1990;Porkka-Heiskanen et al., 1994), each phase of decay could be attributed to a separate cell population. However, even after five half-lives (i.e., 65 min), O.D. level data displayed no significant decrease in cell number, indicating that a rapidly decaying LHRH population did not disappear. In addition, all frequency distributions were unimodal, strengthening the argument for a single population of LHRH neurons with rapid decay and then stabilization of LHRH mRNA. Although our results in TTX-treated LHRH neurons indicate that the entire population possesses the same intrinsic LHRH mRNA decay mechanisms, this does not preclude the possibility that trans-synaptic influences may alter decay rates within LHRH subpopulations.
The two-phase LHRH mRNA decay could be explained by several mechanisms. First, as recently reported for sympathetic neurons (Muslimov et al., 1997), newly synthesized LHRH mRNA could relocalize rapidly to dendrites or axons. Hypothalamic neuropeptide mRNAs for oxytocin and vasopressin mRNAs localize in posterior pituitary terminals (Murphy et al., 1989; Jirikowski et al., 1990). However, no increase in process-like mRNA labeling was observed in organotypic cultures after transcription inhibitor treatment, and, in vivo, LHRH mRNA labeling in the median eminence has not been reported. Second, a transcription-dependent factor that is needed for rapid decay could be depleted quickly after transcription inhibition. A labile transcription-dependent, translation-independent molecule appears to regulate decay of estrogen receptor mRNA (Ree et al., 1992). In GT1 cells phorbol ester-induced turnover of cytoplasmic LHRH mRNA proceeds via a transcription and translation-dependent mechanism (Yeo et al., 1997). Third, a relatively limited nonlabile factor could protect 40% of LHRH mRNA from decay. Stabilizing proteins alter decay of tyrosine hydroxylase (Czyzyk-Krzeska et al., 1994a), GAP-43 (Kohn et al., 1996), apolipoprotein II (Margot and Williams, 1996), and transferrin receptor (Klausner et al., 1993) mRNAs. Association and localization of the protein products from clock genes timeless andperiod (Sehgal et al., 1995) also may reflect a concentration-dependent mechanism controlling gene expression. At present, the latter two mechanisms are attractive explanations for the observed shift of LHRH mRNA decay kinetics, but further investigation is required.
Most mRNA decay appears to proceed via a few general pathways (for review, see Peltz and Jacobson, 1992). Indeed, poly(A+) tail shortening, an indicator of mRNA destabilization, recently was shown to coincide with phorbol ester-induced cytoplasmic LHRH mRNA turnover (Gore et al., 1997). In the present experiments, altered stability of LHRH mRNA likely results from cellular changes in a factor or factors specific for LHRH mRNA degradation, because relatively long mRNA half-lives for other phenotypic markers have been observed by using DRB and actinomycin D in this laboratory (6–23 hr; Maurer and Wray, 1997).Trans-acting factors often interact withcis-acting sequences in the 3′-UTR to mediate mRNA stability (Peltz and Jacobson, 1992). Within the 3′-UTR of rapidly degraded early-response genes and cytokines, AU-rich elements, including the AUUUA motif, impart mRNA instability (for review, see Greenberg and Belasco, 1993). The 3′-UTR of rat LHRH mRNA, however, contains no such region; the 3′-UTR up to and including polyadenylation signal AAUAAA is only 53.1% AU.
The importance of mRNA secondary structure in mRNA stabilization is exemplified by apolipoprotein II mRNA (Binder et al., 1989; Margot and Williams, 1996). Endonucleolytic cleavage of apolipoprotein II mRNA primarily occurs at AAU and UAA elements in single-stranded loop structures in the 3′-UTR (Binder et al., 1989). Estrogen treatment destabilizes apolipoprotein II mRNA (Gordon et al., 1988) and induces formation of eight proteins that bind to its 3′-UTR (Margot and Williams, 1996). Estrogen-induced assembly of a 3′-UTR multiprotein messenger ribonucleoprotein complex ultimately may prevent or facilitate nuclease access cleavage sites (Margot and Williams, 1996). Subsequently, we compared the 128 nucleotide portion of rat LHRH 3′-UTR with other published mammalian LHRH 3′-UTRs and uncovered a high degree of interspecies homology. Compared with rat, 3′-UTRs for mouse, human, pig, and tree shrew maintained homologies of 85.2, 61.7, 53.7, and 65.1%, respectively. These homologies differed only slightly from a similar comparison among coding regions—respectively 92.7, 78.1, 77.4, and 76.0%. Therefore, the 3′-UTR of mammalian LHRH mRNA, like that of the coding region, is highly conserved. Alignment of mammalian 3′-UTRs revealed several conserved regions, and predictions of energetically favorable secondary structures consistently placed two such regions within single-stranded loop domains. With IUPAC–IUB nomenclature, these conserved motifs are ASAAS and WURCM. Conserved 3′-UTR motifs implicate the presence of factors that bind these regions and preclude or promote LHRH mRNA decay.
Mathematical modelling of neuropeptide homeostasis reveals that, as mRNA half-life becomes more rapid, changes in transcription rate quickly reflect gene expression levels (Fitzsimmons et al., 1992). Evidence indicates that peptide gene expression is tightly coupled to biosynthesis and secretion (Young and Zoeller, 1987; Stachowiak et al., 1990; MacArthur et al., 1992; Wang et al., 1995). This relationship also has been observed in the LHRH system. Hypothalamic slices superfused with phorbol ester in vitro exhibit increased LHRH mRNA levels and peptide release (Lee et al., 1990), whereas tissues taken from NMDA-pretreated rats show decreased progesterone-induced LHRH gene expression and peptide release (Seong et al., 1993). In vivo, in response to antiestrogen, elevated LHRH mRNA in mPOA cell bodies is followed by increased LHRH peptide levels in the median eminence (Petersen et al., 1989). Although LHRH neurons release only a small percentage of their total stores (Negro-Vilar et al., 1979; Sherwood et al., 1980; Levine et al., 1985), LHRH gene expression may be integral to ongoing LHRH secretion, if, like other neuroendocrine secretion, newly synthesized secretory granules release their contents before older stores (for review, seePickering, 1978; Gainer and Wray, 1994). With an axonal transport rate of 5.8 mm/hr reported for neurophysin peptide within the hypothalamus (Gainer, 1982), transport of LHRH (∼4 mm in neonate, Fig. 1parasagittal; 6.4 mm in adult, Wray and Hoffman, 1986a) from preoptic area cell bodies to median eminence nerve terminals could replenish the readily releasible peptide stores after one pulse.
Rhythmic gene expression often accompanies rhythmic events. Parallel, circadian fluctuations of enzyme activity and gene expression have been demonstrated for pineal serotoninN-acetyltransferase (Bernard et al., 1997) and liver cholesterol 7α-hydroxylase (Noshiro et al., 1990). In vivo, the mammalian suprachiasmatic nucleus shows rhythmicity of vasoactive intestinal peptide and vasopressin levels (Gillette and Reppert, 1987; Reppert and Uhl, 1987; Shinohara et al., 1993) and their mRNAs (Robinson et al., 1988; Albers et al., 1990; Ban et al., 1997). These rhythms also are maintained in organotypic cultures of suprachiasmatic nuclei in vitro (Carter and Murphy, 1989;Shinohara et al., 1994; Tominaga et al., 1994). Although LHRH peptide rhythms exhibit pulse intervals of 30–70 min and peak decays of 10–24 min (Levine and Ramirez, 1980, 1982; Dluzen and Ramirez, 1987), not the 12–24 hr patterns cited for circadian fluctuations, the rapid LHRH mRNA half-life clearly coincides with decay of LHRH peptide pulses. With rapid LHRH mRNA turnover and an apparent high intrinsic basal transcription rate (Gore et al., 1995; Yeo et al., 1996), LHRH gene expression could accommodate the LHRH secretory profile. Parallel increases in primary transcript and cytoplasmic LHRH mRNA (Gore and Roberts, 1995) indicate both transcription and stabilization of LHRH mRNA during the surge. However, to maintain pulsatile mRNA levels, some kinetic parameter of gene expression must change in a pulsatile manner. LHRH mRNA synthesis could pulse while decay remained constant; conversely, transcription could be constant and the LHRH mRNA decay rate could fluctuate, or both parameters could change concurrently. Our data, showing rapid decay and then stabilization of LHRH mRNA, are consistent with the latter two mechanisms regulating LHRH gene expression under basal conditions.
The relationship between LHRH transcription and pulsatility currently is unknown. During proestrous, transcriptional events such as increasedfos immunostaining (Lee et al., 1990) and LHRH primary transcript levels (Gore and Roberts, 1995) coincide with the LH surge. Evidence from GT-1 cells (Krsmanovic et al., 1992; Martinez de la Escalera et al., 1992; Wetsel et al., 1992) and primates (Saitoh et al., 1995) strongly suggests that hourly LHRH pulses are endogenous. The protein products of endogenous time-keeping genes periodand timeless in Drosophila (Myers et al., 1995) and frequency in Neurospora (Aronson et al., 1994) negatively regulate their own transcription. Similarly, CREM, which is central to development and regulation of hypothalamic–pituitary functions (Foulkes et al., 1992, 1993, 1996), autoregulates (Molina et al., 1993) and intergenically regulates (Foulkes et al., 1996) gene expression by the repressor activity of its gene product ICER (i.e., inducible cAMP early repressor). Such feedback regulation may not occur directly on LHRH gene transcription because, in GT1 cells, phorbol ester-induced LHRH secretion is accompanied by a seemingly paradoxical repression of LHRH gene transcription (Wierman et al., 1995). However, ICER feedback downregulation of adrenocorticotropin hormone release from corticotrophs recently was demonstrated to act by decreasing gene expression of PC1, a post-translational processing enzyme, and not by inhibiting transcription of the hormone precursor directly (Lamas et al., 1997). Therefore, a timekeeping feedback regulation, although acting on transcriptional processes (Sassone-Corsi, 1994), could affect LHRH gene transcription directly or that of a gene regulating transport, synthesis, or stabilization of LHRH mRNA or peptide. Continued investigation of the LHRH system at all levels—gene expression, peptide synthesis, and secretory pathway—likely will be required to fully elucidate the elusive basis of LHRH surges and pulsatility.
Footnotes
J.A.M., a Pharmacology Research Associate Fellow, was supported by the National Institute of General Medical Sciences, National Institutes of Health. We thank Drs. Harold Gainer and Martin Zatz for helpful comments on this manuscript and Dr. James D. Malley for consultation on statistical analyses.
Correspondence should be addressed to Dr. Susan Wray, Section Chief, Cellular and Developmental Neurobiology, Laboratory of Neurochemistry, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Building 36, Room 4D10, Bethesda, MD 20892.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}